
Complex Transposon Insertion as a Novel Cause of Pompe Disease

 , , , , and
, , , , and 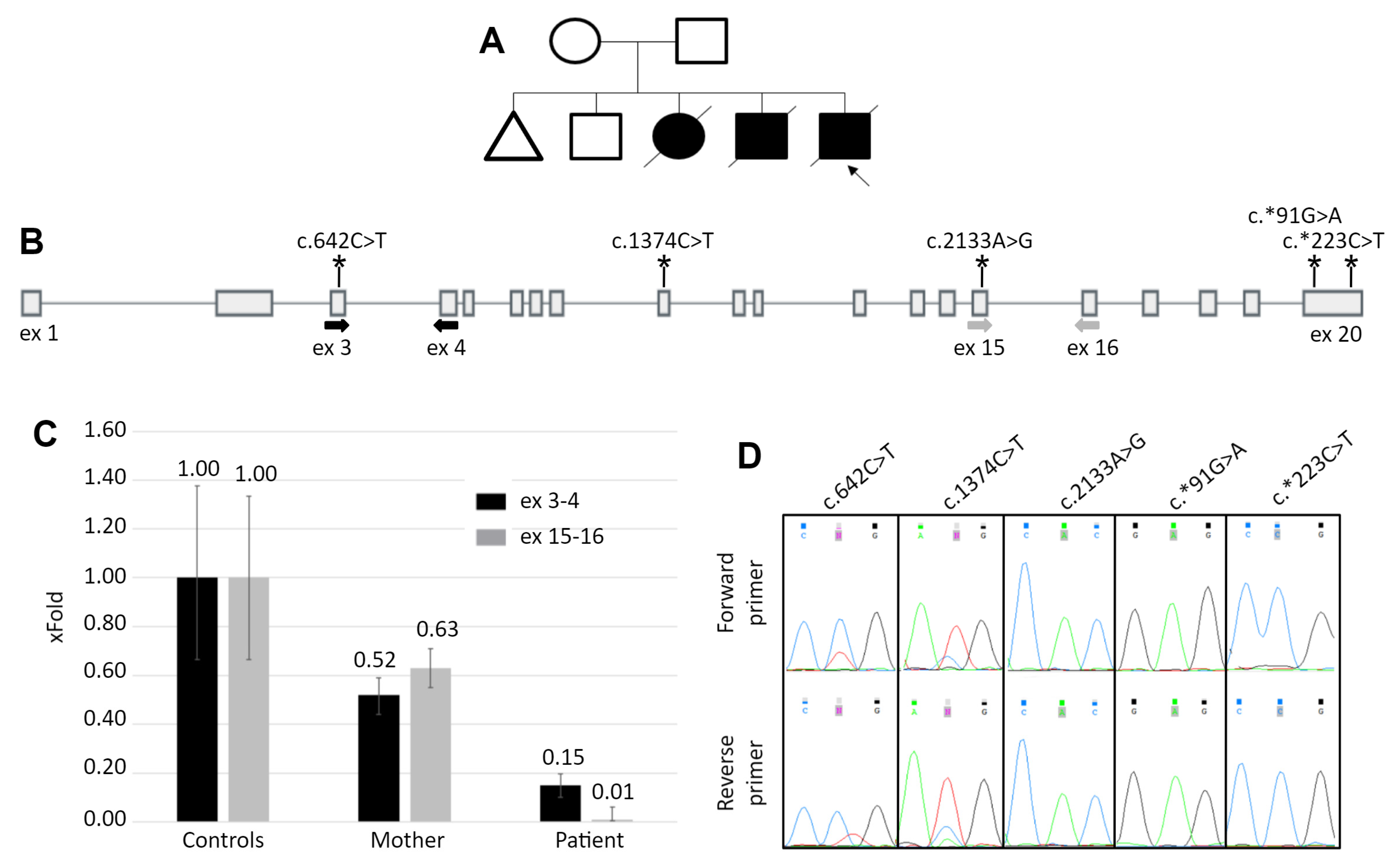{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biochemical Analysis
2.2. DNA Analysis
2.3. Bisulfite Sequencing
2.4. RNA Analysis
2.5. Rapid Amplification of cDNA Ends
2.6. Real-Time PCR
3. Results
3.1. Patient’s Summary
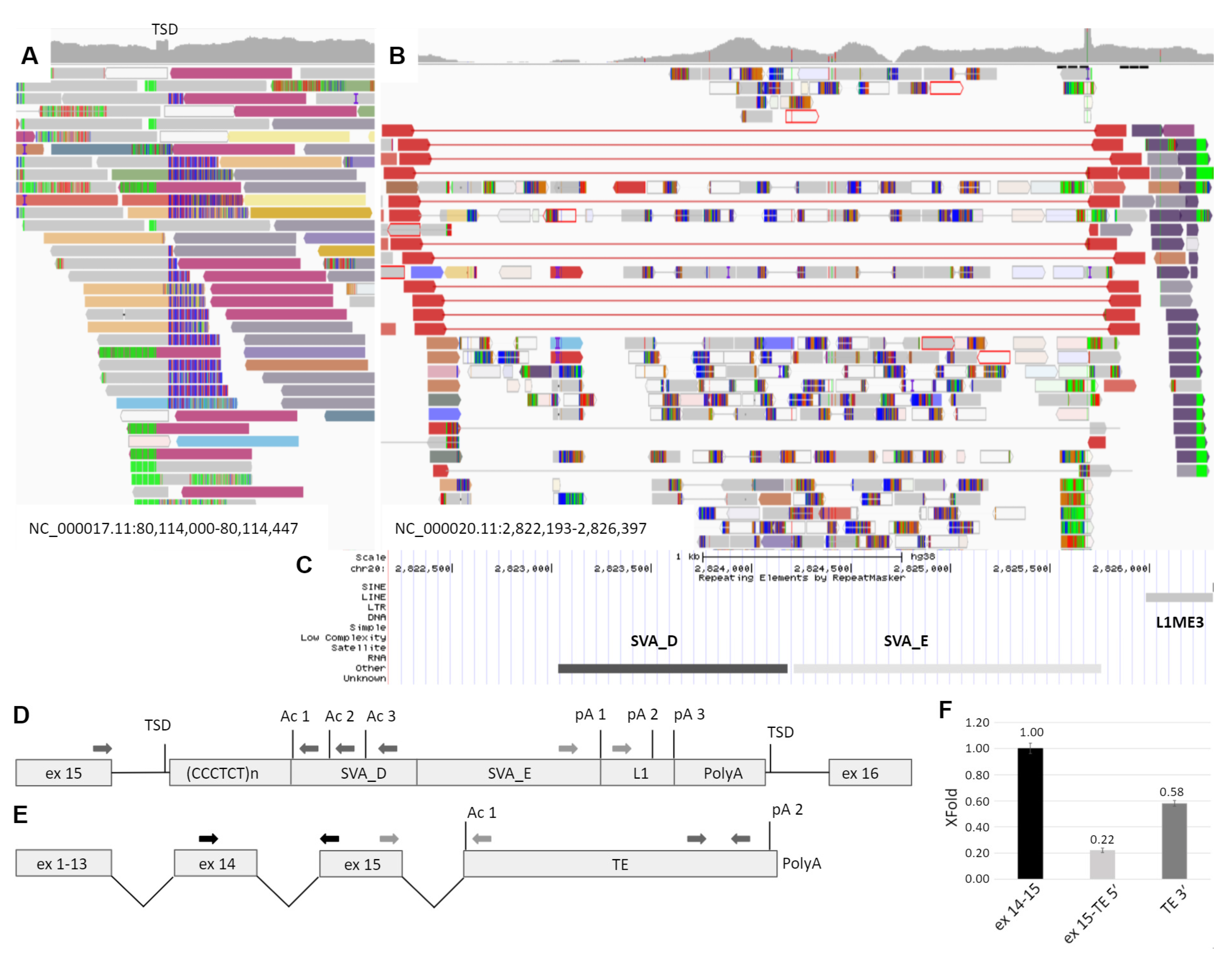
3.2. Molecular Genetic Analysis
3.3. Study of Mechanism of the TE Insertion Molecular Pathogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bodamer, O.A.; Scott, C.R.; Giugliani, R.; on behalf of the Pompe Disease Newborn Screening Working Group. Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [Green Version]
- Hers, H.G. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. [Google Scholar] [CrossRef]
- Griffin, J.L. Infantile acid maltase deficiency. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984, 45, 37–50. [Google Scholar] [CrossRef]
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018, 15, 928–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güngör, D.; Reuser, A. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. Part A 2013, 161, 399. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.; Bailey, L. Pompe Disease. GeneReviews®. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1261/ (accessed on 15 July 2021).
- Lin, N.; Huang, J.; Violante, S.; Orsini, J.J.; Caggana, M.; Hughes, E.E.; Stevens, C.; DiAntonio, L.; Liao, H.C.; Hong, X.; et al. Liquid Chromatography—Tandem Mass Spectrometry Assay of Leukocyte Acid α-Glucosidase for Post-Newborn Screening Evaluation of Pompe Disease. Clin. Chem. 2017, 63, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef]
- Niño, M.Y.; Groen, S.L.I.; Bergsma, A.J.; Van Der Beek, N.A.; Kroos, M.; Hoogeveen-Westerveld, M.; Van Der Ploeg, A.T.; Pijnappel, W.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 119. [Google Scholar] [CrossRef]
- Craig, N.L. (Ed.) Mobile DNA III; ASM Press: Washington, DC, USA, 2020. [Google Scholar]
- Hancks, D.C.; Kazazian, H.H. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2008, 41, 209–219. [Google Scholar] [CrossRef]
- Matz, M.; Shagin, D.; Bogdanova, E.; Britanova, O.; Lukyanov, S.; Diatchenko, L.; Chenchik, A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999, 27, 1558–1560. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bennett, E.A.; Coleman, L.E.; Tsui, C.; Pittard, W.S.; Devine, S.E. Natural Genetic Variation Caused by Transposable Elements in Humans. Genetics 2004, 168, 933–951. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.K. Different integration site structures between L1 protein-mediated retrotransposition in cis and retrotransposition in trans. Mob. DNA 2010, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open 4.0; Institute for Systems Biology: Washington, DC, USA, 2015; Available online: https://www.repeatmasker.org/ (accessed on 15 July 2021).
- Conley, A.B.; Piriyapongsa, J.; Jordan, I.K. Retroviral promoters in the human genome. Bioinformatics 2008, 24, 1563–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conley, A.B.; Jordan, I.K. Epigenetic regulation of human cis -natural antisense transcripts. Nucleic Acids Res. 2012, 40, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, R.; Palazzo, A.; Lorusso, P.; Viggiano, L.; Massimiliano Marsano, R. “What you need, baby, I got it”: Transposable elements as suppliers of cis-operating sequences in drosophila. Biology 2020, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, J.N.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2020, 49, D1046–D1057. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and Evolution of Human microRNAs From Transposable Elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.; Ballester, B.; Gonçalves, Â.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of Retrotransposon Expansion Remodel Genome Organization and CTCF Binding in Multiple Mammalian Lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Molaro, A.; Malik, H.S. Hide and seek: How chromatin-based pathways silence retroelements in the mammalian germline. Curr. Opin. Genet. Dev. 2016, 37, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 System Contributes to the Early Embryonic Establishment of Site-Specific DNA Methylation Patterns Maintained during Development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef] [Green Version]
- Payer, L.M.; Steranka, J.P.; Yang, W.R.; Kryatova, M.; Medabalimi, S.; Ardeljan, D.; Liu, C.; Boeke, J.D.; Avramopoulos, D.; Burns, K.H. Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E3984–E3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Norris, E.T.; Jordan, I.K. Human Retrotransposon Insertion Polymorphisms Are Associated with Health and Disease via Gene Regulatory Phenotypes. Front. Microbiol. 2017, 8, 1418. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, E.M.; Goodier, J.L.; Zhang, Y.; Kazazian, H.H., Jr. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 2003, 73, 1444–1451. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xing, J.; Grover, D.; Hedges, D.J.; Han, K.; Walker, J.A.; Batzer, M.A. SVA Elements: A Hominid-specific Retroposon Family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef]
- Damert, A.; Raiz, J.; Horn, A.V.; Löwer, J.; Wang, H.; Xing, J.; Batzer, M.A.; Löwer, R.; Schumann, G.G. 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 2009, 19, 1992–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Wang, H.; Belancio, V.P.; Cordaux, R.; Deininger, P.; Batzer, M.A. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 17608–17613. [Google Scholar] [CrossRef] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol. 2010, 20, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Han, J.S.; Szak, S.T.; Boeke, J. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 2004, 429, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ustyugova, S.V.; Lebedev, Y.B.; Sverdlov, E.D. Long L1 insertions in human gene introns specifically reduce the content of corresponding primary transcripts. Genetica 2006, 128, 261–272. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bychkov, I.; Baydakova, G.; Filatova, A.; Migiaev, O.; Marakhonov, A.; Pechatnikova, N.; Pomerantseva, E.; Konovalov, F.; Ampleeva, M.; Kaimonov, V.; et al. Complex Transposon Insertion as a Novel Cause of Pompe Disease. Int. J. Mol. Sci. 2021, 22, 10887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910887
Bychkov I, Baydakova G, Filatova A, Migiaev O, Marakhonov A, Pechatnikova N, Pomerantseva E, Konovalov F, Ampleeva M, Kaimonov V, et al. Complex Transposon Insertion as a Novel Cause of Pompe Disease. International Journal of Molecular Sciences. 2021; 22(19):10887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910887
Chicago/Turabian StyleBychkov, Igor, Galina Baydakova, Alexandra Filatova, Ochir Migiaev, Andrey Marakhonov, Nataliya Pechatnikova, Ekaterina Pomerantseva, Fedor Konovalov, Maria Ampleeva, Vladimir Kaimonov, and et al. 2021. "Complex Transposon Insertion as a Novel Cause of Pompe Disease" International Journal of Molecular Sciences 22, no. 19: 10887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910887