
A Survey of Autoencoder Algorithms to Pave the Diagnosis of Rare Diseases


{kind=link}
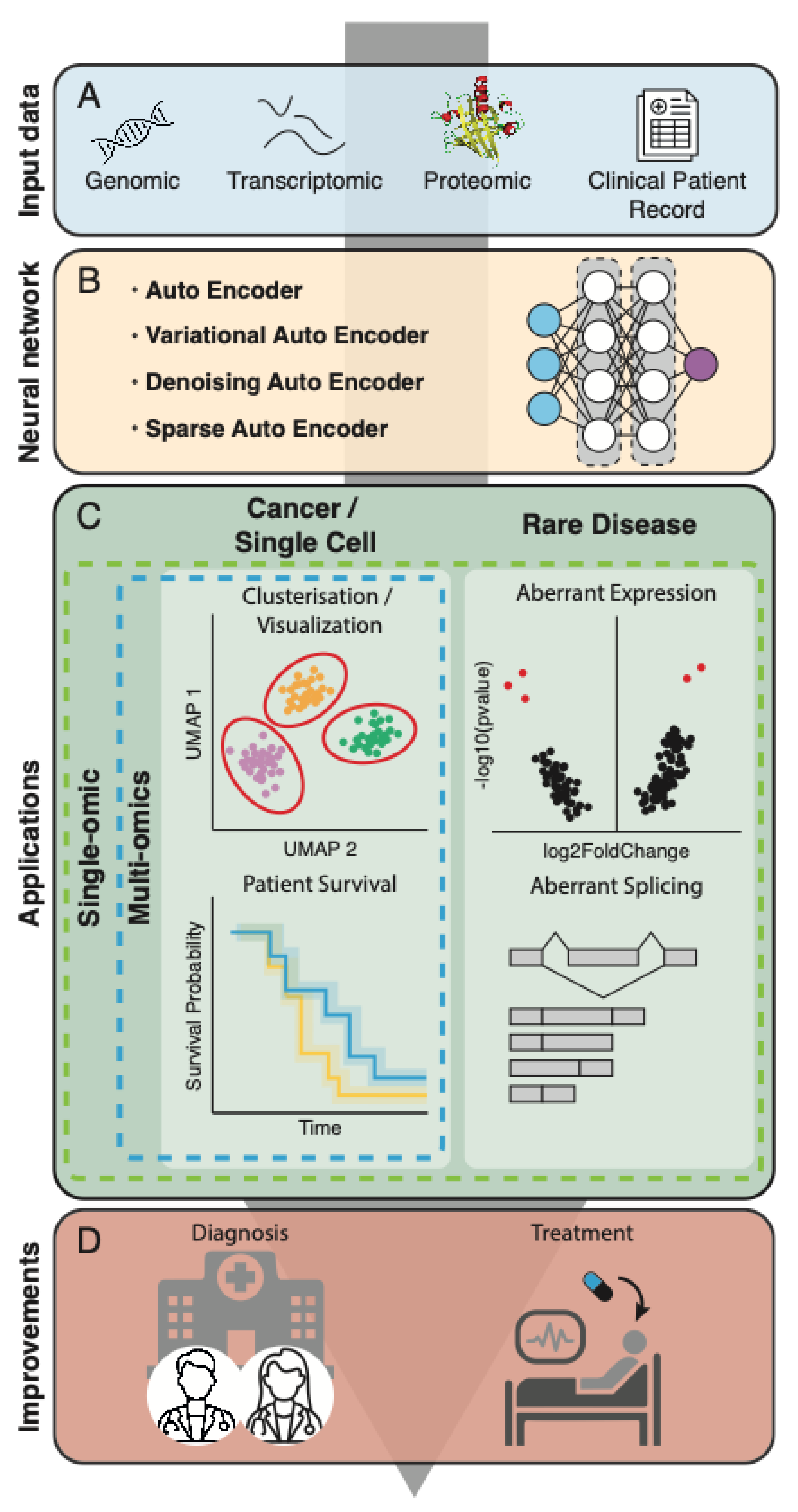{kind=link}
Abstract
:1. Introduction
1.1. RDs and Their Diagnosis
1.2. Omics and Multi-Omics Approaches for RD Diagnosis
2. Artificial Intelligence Methods for Biology
Basis of the AE Algorithm and Its Variant
3. AE Applications in Biological and Medical Contexts beyond RD
3.1. AE Applications in Single Cell
3.2. AE Applications in Cancer
3.2.1. Drug Response Prediction
3.2.2. Cancer Classification and Stratification
3.3. VAEs Structure for Data Integration
3.4. AE Applications in RDs
4. Discussion and Conclusions: Open Challenges and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Labory, J.; Fierville, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Paquis-Flucklinger, V.; Bottini, S. Multi-Omics Approaches to Improve Mitochondrial Disease Diagnosis: Challenges, Advances, and Perspectives. Front. Mol. Biosci. 2020, 7, 590842. [Google Scholar] [CrossRef] [PubMed]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2014, arXiv:1312.6114. [cs, stat]. [Google Scholar]
- Rezende, D.J.; Mohamed, S.; Wierstra, D. Stochastic Backpropagation and Approximate Inference in Deep Generative Models. arXiv 2014, arXiv:1401.4082. [cs, stat]. [Google Scholar]
- Bengio, Y.; Courville, A.; Vincent, P. Representation Learning: A Review and New Perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Wolf, F.A.; Theis, F.J. scGen predicts single-cell perturbation responses. Nat. Methods 2019, 16, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.; Rana, P.; Cruz, A.; Calderón, V.; Azevedo, V.; Ramos, R.; Ghosh, P. Performance Comparison of Deep Learning Autoencoders for Cancer Subtype Detection Using Multi-Omics Data. Cancers 2021, 13, 2013. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.N.; Johansen, J.; Allesøe, R.L.; Sønderby, C.K.; Armenteros, J.J.A.; Grønbech, C.H.; Jensen, L.J.; Nielsen, H.B.; Petersen, T.N.; Winther, O.; et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021, 39, 555–560. [Google Scholar] [CrossRef]
- Simidjievski, N.; Bodnar, C.; Tariq, I.; Scherer, P.; Andres-Terre, H.; Shams, Z.; Jamnik, M.; Liò, P. Variational Autoencoders for Cancer Data Integration: Design Principles and Computational Practice. Front. Genet. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Bao, F.; Dai, Q.; Wu, L.F.; Altschuler, S.J. Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat. Methods 2019, 16, 311–314. [Google Scholar] [CrossRef]
- Grønbech, C.H.; Vording, M.F.; Timshel, P.N.; Sønderby, C.K.; Pers, T.H.; Winther, O. scVAE: Variational auto-encoders for single-cell gene expression data. Bioinformatics 2020, 36, 4415–4422. [Google Scholar] [CrossRef]
- Belkin, M.; Niyogi, P. Laplacian Eigenmaps for Dimensionality Reduction and Data Representation. Neural Comput. 2003, 15, 1373–1396. [Google Scholar] [CrossRef] [Green Version]
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes: Global Report on Birth Defects, the Hidden Toll of Dying and Disabled Children. In March of Dimes: Global Report on Birth Defects, the Hidden Toll of Dying and Disabled Children; March of Dimes Birth Defects Foundation: Arlington, VA, USA, 2005. [Google Scholar]
- Baird, P.A.; Anderson, T.W.; Newcombe, H.B.; Lowry, R.B. Genetic disorders in children and young adults: A population study. Am. J. Hum. Genet. 1988, 42, 677–693. [Google Scholar]
- DI Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Ng, S.B.; Turner, E.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nat. Cell Biol. 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Ku, C.-S.; Naidoo, N.; Pawitan, Y. Revisiting Mendelian disorders through exome sequencing. Qual. Life Res. 2011, 129, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.-H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2013, 16, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Liew, W.K.M.; Ben-Omran, T.; Darras, B.T.; Prabhu, S.P.; De Vivo, D.C.; Vatta, M.; Yang, Y.; Eng, C.M.; Chung, W.K. Clinical Application of Whole-Exome Sequencing. JAMA Neurol. 2013, 70, 788–791. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. npj Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; Ahmed, W. Beyond Metabolomics: A Review of Multi-Omics-Based Approaches. In Microbial Metabolomics; Springer: Cham, Switzerland, 2016; pp. 289–312. [Google Scholar]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Cunningham, P.; Cord, M.; Delany, S.J. Supervised Learning. In Machine Learning Techniques for Multimedia: Case Studies on Organization and Retrieval; Cord, M., Cunningham, P., Eds.; Cognitive Technologies; Springer: Berlin/Heidelberg, Germany, 2008; pp. 21–49. ISBN 9783540751717. [Google Scholar]
- Greene, D.; Cunningham, P.; Mayer, R. Unsupervised Learning and Clustering. In Machine Learning Techniques for Multimedia: Case Studies on Organization and Retrieval; Cord, M., Cunningham, P., Eds.; Cognitive Technologies; Springer: Berlin/Heidelberg, Germany, 2008; pp. 51–90. ISBN 9783540751717. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020, arXiv:1802.03426. [cs, stat]. [Google Scholar]
- Cheplygina, V.; de Bruijne, M.; Pluim, J.P.W. Not-so-supervised: A survey of semi-supervised, multi-instance, and transfer learning in medical image analysis. Med Image Anal. 2019, 54, 280–296. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Vincent, P.; LaRochelle, H.; Bengio, Y.; Manzagol, P.-A. Extracting and composing robust features with denoising autoencoders. In Proceedings of the 25th international conference on Machine learning-ICML ’08, Helsinki, Finland, 5–9 July 2008; Association for Computing Machinery: New York, NY, USA; pp. 1096–1103. [Google Scholar]
- Coates, A.; Ng, A.; Lee, H. An Analysis of Single-Layer Networks in Unsupervised Feature Learning. In Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, Fort Lauderdale, FL, USA, 11–13 April 2011; pp. 215–223. [Google Scholar]
- Makhzani, A.; Frey, B. K-Sparse Autoencoders. arXiv 2014, arXiv:1312.5663. [cs]. [Google Scholar]
- Ferles, C.; Papanikolaou, Y.; Naidoo, K.J. Denoising Autoencoder Self-Organizing Map (DASOM). Neural Networks 2018, 105, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gu, J. VASC: Dimension Reduction and Visualization of Single-cell RNA-seq Data by Deep Variational Autoencoder. Genom. Proteom. Bioinform. 2018, 16, 320–331. [Google Scholar] [CrossRef]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Wang, H.; Ganapathiraju, M. Learning structure in gene expression data using deep architectures, with an application to gene clustering. In Proceedings of the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Washington, DC, USA, 12–19 November 2015; IEEE: Piscataway, NJ, USA, 2015; pp. 1328–1335. [Google Scholar]
- Amodio, M.; van Dijk, D.; Srinivasan, K.; Chen, W.S.; Mohsen, H.; Moon, K.R.; Campbell, A.; Zhao, Y.; Wang, X.; Venkataswamy, M.; et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods 2019, 16, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Cai, C.; Gao, Y.; Su, S.; Wu, J. Variational Autoencoder for Low Bit-Rate Image Compression. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition Workshops, Salt Lake City, UT, USA, 18–22 June 2018; pp. 2617–2620. [Google Scholar]
- Tan, J.; Ung, M.; Cheng, C.; Greene, C.S. Unsupervised Feature Construction and Knowledge Extraction from Genome-Wide Assays of Breast Cancer with Denoising Autoencoders. Pac. Symp. Biocomput. 2015, 20, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Poirion, O.B.; Chaudhary, K.; Garmire, L.X. Deep Learning data integration for better risk stratification models of bladder cancer. AMIA Jt. Summits Transl. Sci. Proc. 2018, 2017, 197–206. [Google Scholar]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lv, C.; Jin, Y.; Cheng, G.; Fu, Y.; Yuan, D.; Tao, Y.; Guo, Y.; Ni, X.; Shi, T. Deep Learning-Based Multi-Omics Data Integration Reveals Two Prognostic Subtypes in High-Risk Neuroblastoma. Front. Genet. 2018, 9, 477. [Google Scholar] [CrossRef] [Green Version]
- Rampášek, L.; Hidru, D.; Smirnov, A.; Haibe-Kains, B.; Goldenberg, A. Dr.VAE: Improving drug response prediction via modeling of drug perturbation effects. Bioinformatics 2019, 35, 3743–3751. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Hammond, J.H.; Hogan, D.A.; Greene, C.S. ADAGE-Based Integration of Publicly Available Pseudomonas aeruginosa Gene Expression Data with Denoising Autoencoders Illuminates Microbe-Host Interactions. mSystems 2016, 1, 00025-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Doing, G.; Lewis, K.A.; Price, C.E.; Chen, K.M.; Cady, K.C.; Perchuk, B.; Laub, M.T.; Hogan, D.A.; Greene, C.S. Unsupervised Extraction of Stable Expression Signatures from Public Compendia with an Ensemble of Neural Networks. Cell Syst. 2017, 5, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xie, X.; Shi, J.; He, W.; Chen, Q.; Chen, L.; Gu, W.; Zhou, T. Denoising Autoencoder, A Deep Learning Algorithm, Aids the Identification of a Novel Molecular Signature of Lung Adenocarcinoma. Genom. Proteom. Bioinform. 2020, 18, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cai, C.; Chen, V.; Lu, X. Learning a hierarchical representation of the yeast transcriptomic machinery using an autoencoder model. BMC Bioinform. 2016, 17, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Miotto, R.; Li, L.; Kidd, B.; Dudley, J.T. Deep Patient: An Unsupervised Representation to Predict the Future of Patients from the Electronic Health Records. Sci. Rep. 2016, 6, 26094. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nat. Cell Biol. 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kolodziejczyk, A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.; Marioni, J.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Kim, J.K.; Kolodziejczyk, A.; Illicic, T.; Teichmann, S.; Marioni, J.C. Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat. Commun. 2015, 6, 8687. [Google Scholar] [CrossRef] [Green Version]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.; Marioni, J.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Gomes, T.; Teichmann, S.A.; Talavera-López, C. Immunology Driven by Large-Scale Single-Cell Sequencing. Trends Immunol. 2019, 40, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Talwar, D.; Mongia, A.; Sengupta, D.; Majumdar, A. AutoImpute: Autoencoder based imputation of single-cell RNA-seq data. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Jain, S.; Kim, H.; Bar-Joseph, Z. Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res. 2017, 45, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Kang, H.M.; Subramaniam, M.; Targ, S.; Nguyen, M.; Maliskova, L.; McCarthy, E.; Wan, E.; Wong, S.; Byrnes, L.; Lanata, C.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89–94. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef]
- Alessandri, L.; Cordero, F.; Beccuti, M.; Licheri, N.; Arigoni, M.; Olivero, M.; Di Renzo, M.F.; Sapino, A.; Calogero, R. Sparsely-connected autoencoder (SCA) for single cell RNAseq data mining. npj Syst. Biol. Appl. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Trong, T.N.; Mehtonen, J.; González, G.; Kramer, R.; Hautamäki, V.; Heinäniemi, M. Semisupervised Generative Autoencoder for Single-Cell Data. J. Comput. Biol. 2020, 27, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Zuo, C.; Chen, L. Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data. Briefings Bioinform. 2020, 22. [Google Scholar] [CrossRef] [PubMed]
- Gayoso, A.; Steier, Z.; Lopez, R.; Regier, J.; Nazor, K.L.; Streets, A.; Yosef, N. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 2021, 18, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Minoura, K.; Abe, K.; Nam, H.; Nishikawa, H.; Shimamura, T. ScMM: Mixture-of-Experts Multimodal Deep Generative Model for Single-Cell Multiomics Data Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dincer, A.B.; Celik, S.; Hiranuma, N.; Lee, S.-I. DeepProfile: Deep Learning of Cancer Molecular Profiles for Precision Medicine. bioRxiv 2018. [Google Scholar] [CrossRef]
- Chiu, Y.-C.; Chen, H.-I.H.; Zhang, T.; Zhang, S.; Gorthi, A.; Wang, L.-J.; Huang, Y.; Chen, Y. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med. Genom. 2019, 12, 18. [Google Scholar] [CrossRef]
- Way, G.P.; Greene, C.S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. Pac. Symp. Biocomput. 2018, 23, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Baptista, D.; Ferreira, P.; Rocha, M. Deep learning for drug response prediction in cancer. Brief. Bioinform. 2021, 22, 360–379. [Google Scholar] [CrossRef]
- Rami-Porta, R.; Crowley, J.; Goldstraw, P. Review the Revised TNM Staging System for Lung Cancer. Ann. Thorac. Cardiovasc. Surg. 2009, 15, 4–9. [Google Scholar]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review the Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Współczesna Onkologia 2015, 1A, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wu, J.; Lin, Z.; Zhao, X. A semi-supervised deep learning method based on stacked sparse auto-encoder for cancer prediction using RNA-seq data. Comput. Methods Programs Biomed. 2018, 166, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xing, Y.; Sun, K.; Guo, Y. OmiEmbed: A Unified Multi-Task Deep Learning Framework for Multi-Omics Data. Cancers 2021, 13, 3047. [Google Scholar] [CrossRef] [PubMed]
- Hira, M.T.; Razzaque, M.A.; Angione, C.; Scrivens, J.; Sawan, S.; Sarker, M. Integrated multi-omics analysis of ovarian cancer using variational autoencoders. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Chen, H.-I.H.; Chiu, Y.-C.; Zhang, T.; Zhang, S.; Huang, Y.; Chen, Y. GSAE: An autoencoder with embedded gene-set nodes for genomics functional characterization. BMC Syst. Biol. 2018, 12, 142. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Zhang, A. Integrate multi-omics data with biological interaction networks using Multi-view Factorization AutoEncoder (MAE). BMC Genom. 2019, 20, 944. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; He, K.; Shi, C.; Guan, D.; Tian, Y.; Al-Dhelaan, A.; Al-Dhelaan, M. Multi-view network embedding with node similarity ensemble. World Wide Web 2020, 23, 2699–2714. [Google Scholar] [CrossRef]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef] [Green Version]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019, 104, 466–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Network, U.D.; Huang, A.Y.; Wang, L.-K.; Bs, A.J.Y.; Bs, G.R.; Eskin, A.; Ms, R.H.S.; Ms, N.D.; Bs, S.N.-R.; et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet. Med. 2020, 22, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H.; Yépez, V.A. How Machine Learning and Statistical Models Advance Molecular Diagnostics of Rare Disorders Via Analysis of RNA Sequencing Data. Front. Mol. Biosci. 2021, 8, 473. [Google Scholar] [CrossRef]
- Brechtmann, F.; Mertes, C.; Matusevičiūtė, A.; Yépez, V.A.; Avsec, Ž.; Herzog, M.; Bader, D.M.; Prokisch, H.; Gagneur, J. OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data. Am. J. Hum. Genet. 2018, 103, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The GTEx Consortium; Ardlie, K.G.; DeLuca, D.S.; Segre, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.; Sobczak, K. Intrinsic Regulatory Role of RNA Structural Arrangement in Alternative Splicing Control. Int. J. Mol. Sci. 2020, 21, 5161. [Google Scholar] [CrossRef] [PubMed]
- Mertes, C.; Scheller, I.F.; Yépez, V.A.; Çelik, M.H.; Liang, Y.; Kremer, L.S.; Gusic, M.; Prokisch, H.; Gagneur, J. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Greene, C.S. Parameter tuning is a key part of dimensionality reduction via deep variational autoencoders for single cell RNA transcriptomics. Pac. Symp. Biocomput. 2019, 24, 362–373. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratella, D.; Ait-El-Mkadem Saadi, S.; Bannwarth, S.; Paquis-Fluckinger, V.; Bottini, S. A Survey of Autoencoder Algorithms to Pave the Diagnosis of Rare Diseases. Int. J. Mol. Sci. 2021, 22, 10891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910891
Pratella D, Ait-El-Mkadem Saadi S, Bannwarth S, Paquis-Fluckinger V, Bottini S. A Survey of Autoencoder Algorithms to Pave the Diagnosis of Rare Diseases. International Journal of Molecular Sciences. 2021; 22(19):10891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910891
Chicago/Turabian StylePratella, David, Samira Ait-El-Mkadem Saadi, Sylvie Bannwarth, Véronique Paquis-Fluckinger, and Silvia Bottini. 2021. "A Survey of Autoencoder Algorithms to Pave the Diagnosis of Rare Diseases" International Journal of Molecular Sciences 22, no. 19: 10891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910891