
Sphingosine-1 Phosphate Lyase Regulates Sensitivity of Pancreatic Beta-Cells to Lipotoxicity



Abstract
:1. Introduction
2. Results
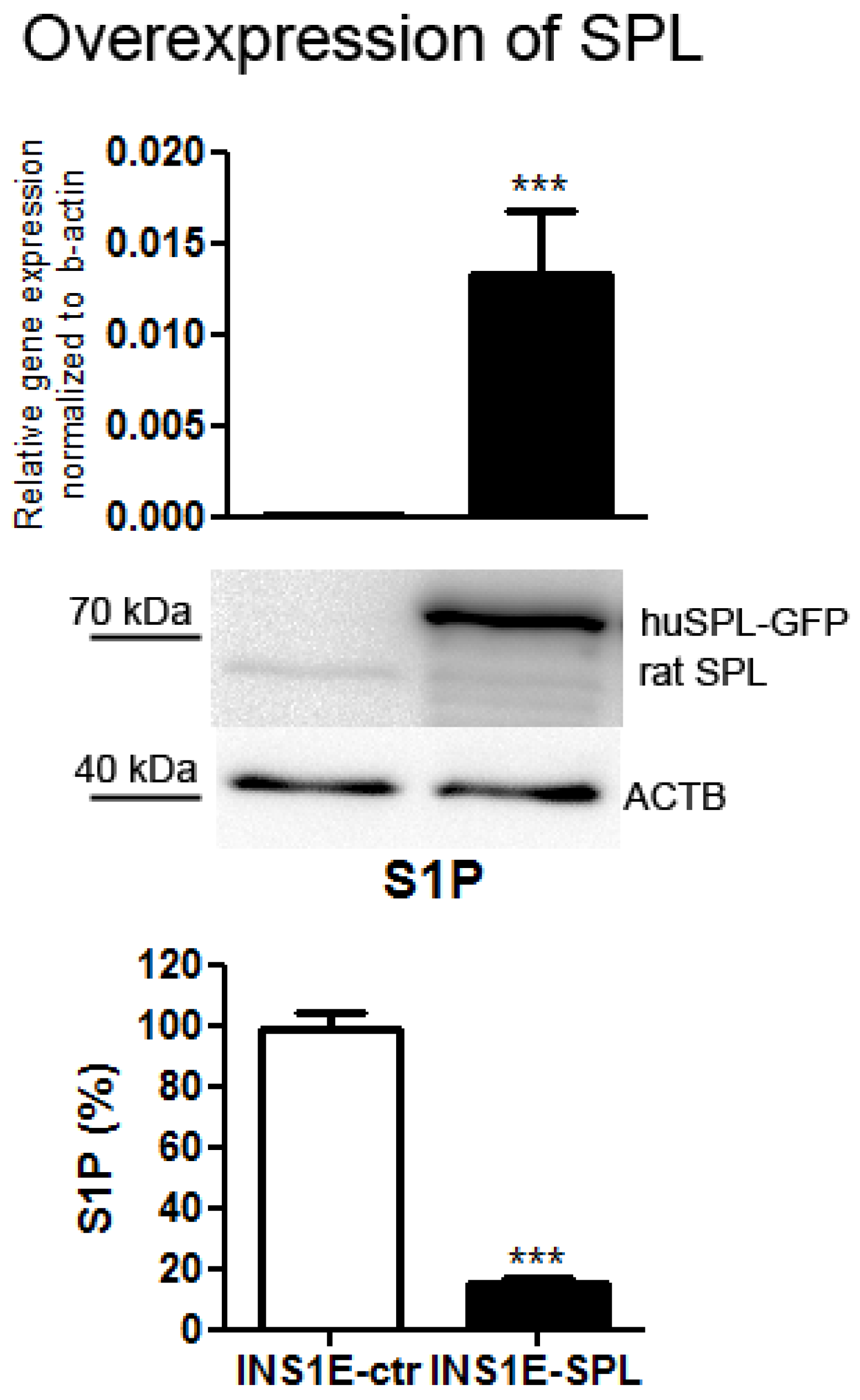
2.1. SPL Overexpression in Insulin-Secreting INS1E Cells Downregulates Intracellular S1P
2.2. SPL Overexpression Sensitizes Insulin-Secreting INS1E Cells to FFA-Mediated Viability Loss
2.3. SPL Overexpression Potentiates FFA-Mediated Proliferation Inhibition
2.4. SPL Overexpression Potentiates FFA-Mediated Oxidative Stress
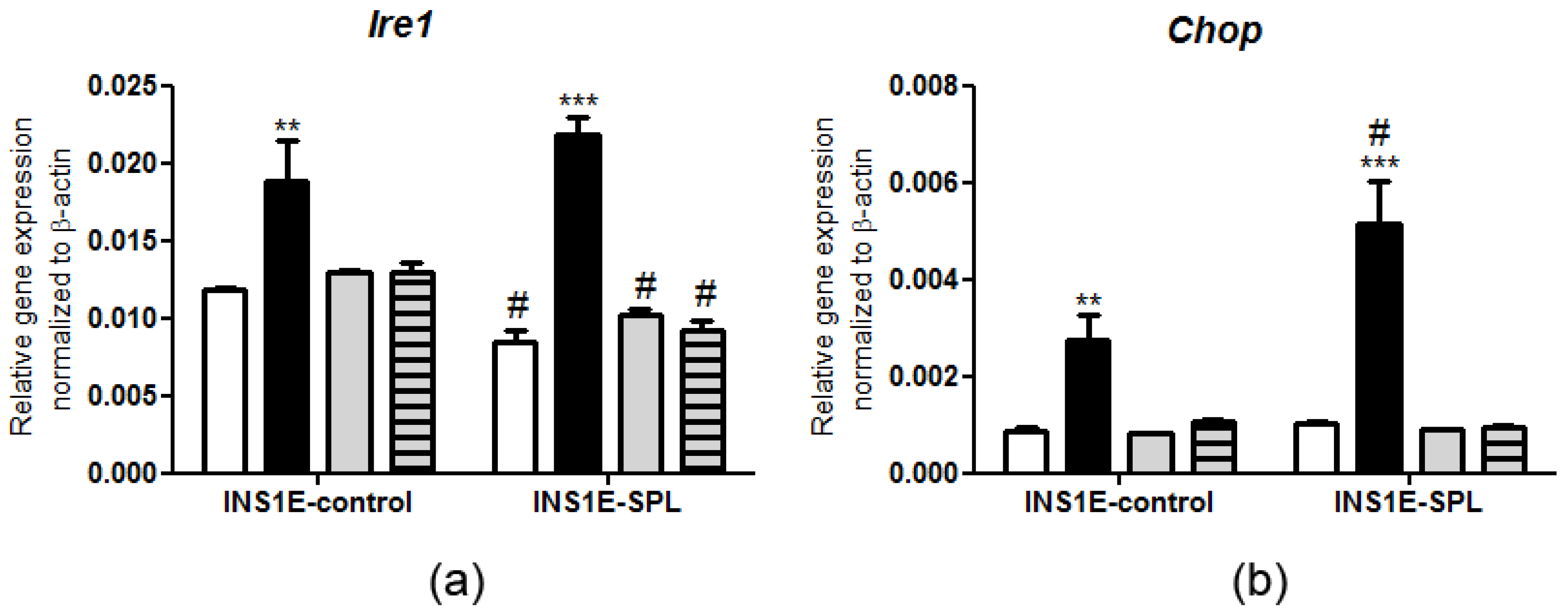
2.5. SPL Overexpression Regulates the ER Stress Response under PA Challenge
2.6. SPL Overexpression Regulates the Expression of Mitochondrial Chaperones in Response to FFAs
2.7. SPL Overexpression Inhibits Lipid Droplet Formation
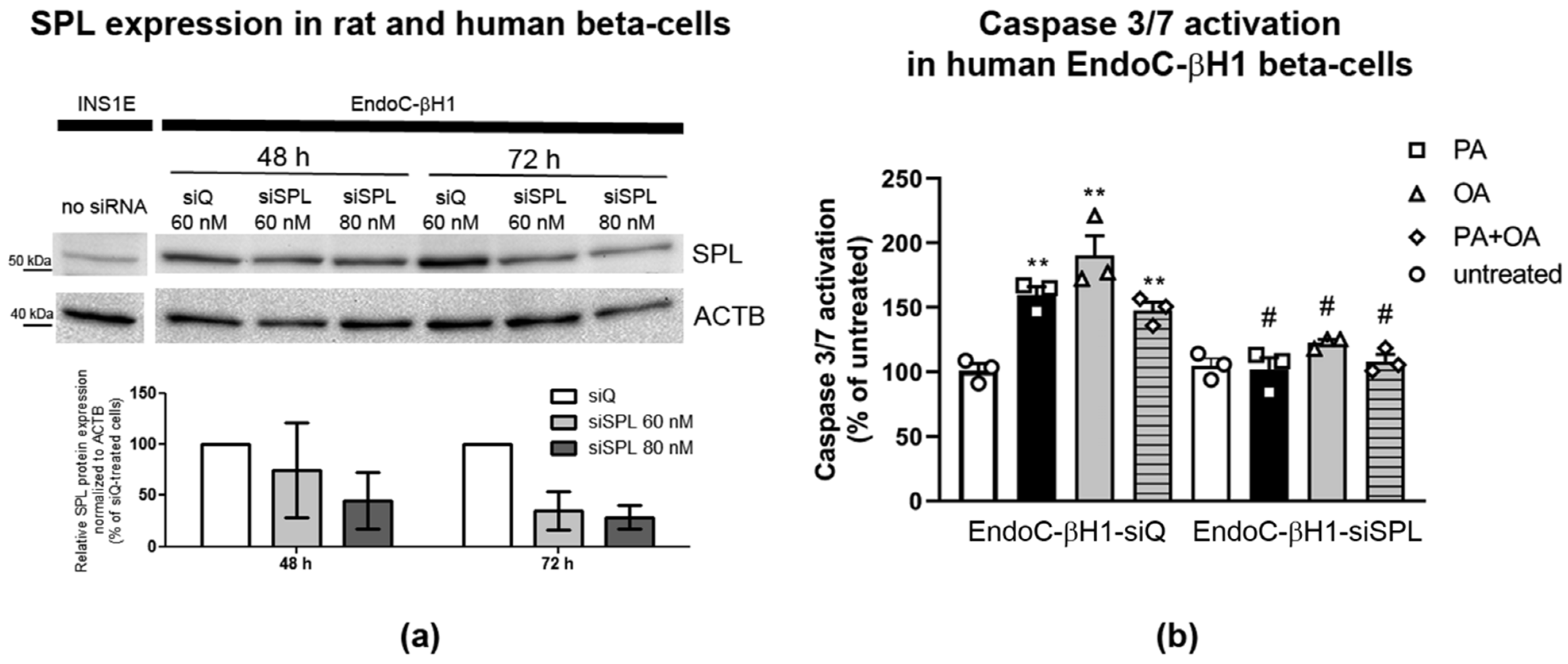
2.8. SPL Knockdown Protects Human EndoC-βH1 Cells from FFA-Mediated Toxicity
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and FFA Incubations
4.3. Overexpression of Human SPL in Rat Insulin-Secreting INS1E Cells
4.4. Suppression of Human SPL in EndoC-βH Beta-Cells
4.5. Detection of S1P by ESI-LC–MS/MS
4.6. Viability Assay
4.7. Proliferation Assay
4.8. Detection of Caspase-3/7 Activation
4.9. RNA Isolation, cDNA Synthesis and Real-Time RT-PCR
4.10. Western Blot Analysis
4.11. Reactive Species Detection by DCF Fluorescence
4.12. Lipid Droplet Detection
4.13. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franks, P.W.; Merino, J. Gene-lifestyle interplay in type 2 diabetes. Curr. Opin. Genet. Dev. 2018, 50, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrin. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, W.; Elsner, M.; Lenzen, S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic beta-cells. Diabetes Obes. Metab. 2010, 12, 149–158. [Google Scholar] [CrossRef]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P.; et al. Glucolipotoxicity impairs ceramide flow from the endoplasmic reticulum to the Golgi apparatus in INS-1 beta-cells. PLoS ONE 2014, 9, e110875. [Google Scholar] [CrossRef]
- Lytrivi, M.; Anne-Laure, C.; Poitout, V.; Cnop, M. Recent insights into mechanisms of beta-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 2019, 432, 1514–1534. [Google Scholar] [CrossRef]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological beta-cell endoplasmic reticulum stress in type 2 diabetes: Current evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef]
- Vela-Guajardo, J.E.; Garza-Gonzalez, S.; Garcia, N. Glucolipotoxicity-induced oxidative stress is related to mitochondrial dysfunction and apoptosis of pancreatic beta-cell. Curr. Diabetes Rev. 2021, 17, e031120187541. [Google Scholar] [CrossRef]
- Oshima, M.; Pechberty, S.; Bellini, L.; Gopel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2020, 63, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Jörns, A.; Tarsi-Chen, L.; Vernea, F.; Aharon-Hananel, G.; Saada, A.; Lenzen, S.; Raz, I. Dietary copper supplementation restores beta-cell function of Cohen diabetic rats: A link between mitochondrial function and glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1023–E1034. [Google Scholar] [CrossRef] [Green Version]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Lupi, R.; Del Guerra, S.; Bugliani, M.; Marselli, L.; Boggi, U. The beta-cell in human type 2 diabetes. Adv. Exp. Med. Biol. 2010, 654, 501–514. [Google Scholar]
- Plötz, T.; Krümmel, B.; Laporte, A.; Pingitore, A.; Persaud, S.J.; Jörns, A.; Elsner, M.; Mehmeti, I.; Lenzen, S. The monounsaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 2017, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Plötz, T.; von Hanstein, A.S.; Krümmel, B.; Laporte, A.; Mehmeti, I.; Lenzen, S. Structure-toxicity relationships of saturated and unsaturated free fatty acids for elucidating the lipotoxic effects in human EndoC-betaH1 beta-cells. Biochim. Biophys. Acta Mol. Bas. Dis. 2019, 1865, 165525. [Google Scholar] [CrossRef]
- Gehrmann, W.; Wurdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism Between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Cantrell Stanford, J.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Ozcan, S. Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef] [Green Version]
- Hla, T.; Dannenberg, A.J. Sphingolipid signaling in metabolic disorders. Cell Metab. 2012, 16, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, C.F.; Bonder, C.S.; Pitson, S.M.; Coates, P.T. The sphingolipid rheostat: A potential target for improving pancreatic islet survival and function. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 262–272. [Google Scholar] [CrossRef]
- Veret, J.; Bellini, L.; Giussani, P.; Ng, C.; Magnan, C.; Le Stunff, H. Roles of sphingolipid metabolism in pancreatic beta cell dysfunction induced by lipotoxicity. J. Clin. Med. 2014, 3, 646–662. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbe, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 beta-cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 beta-cell survival. Biochim. Biophys. Acta 2013, 1831, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.A.; Allegood, J.C.; Sims, K.; Wang, E.W.; Sullards, M.C.; Merrill, A.H., Jr. Quantitation of fatty acyl-coenzyme As in mammalian cells by liquid chromatography-electrospray ionization tandem mass spectrometry. J. Lip. Res. 2008, 49, 1113–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.; Tyka, K.; Saba, J.D.; Lenzen, S.; Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 2017, 292, 20292–20304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; Tonelli, F.; Lim, K.G.; Long, J.S.; Edwards, J.; Pyne, S. Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans. 2012, 40, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzym. Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H.; Peterson, C.; Liu, H.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta 2002, 1582, 8–17. [Google Scholar] [CrossRef]
- Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D. The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell Signal. 2011, 23, 1144–1152. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D.; Hecht, S.S. The sphingolipid degradation product trans-2-hexadecenal forms adducts with DNA. Biochem. Biophys. Res. Commun. 2012, 424, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurgul-Convey, E. Sphingolipids in type 1 diabetes: Focus on beta-bells. Cells 2020, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef]
- Ng, M.L.; Wadham, C.; Sukocheva, O.A. The role of sphingolipid signalling in diabetesassociated pathologies (Review). Int. J. Mol. Med. 2017, 39, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Kanazawa, I.; Sugimoto, T. Visceral fat accumulation is associated with increased plasma sphingosine-1-phosphate levels in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2018, 143, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Furuya, H.; Ng, M.L.; Friedemann, M.; Menschikowski, M.; Tarasov, V.V.; Chubarev, V.N.; Klochkov, S.G.; Neganova, M.E.; Mangoni, A.A.; et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol. Ther. 2020, 207, 107464. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.L.; Yarla, N.S.; Menschikowski, M.; Sukocheva, O.A. Regulatory role of sphingosine kinase and sphingosine-1-phosphate receptor signaling in progenitor/stem cells. World J. Stem Cells 2018, 10, 119–133. [Google Scholar] [CrossRef]
- Schwiebs, A.; Faqar-Uz-Zaman, F.; Herrero San Juan, M.; Radeke, H.H. S1P lyase regulates intestinal stem cell quiescence via Ki-67 and FOXO3. Int. J. Mol. Sci. 2021, 22, 5682. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Chen, J.; Lay, A.; Don, A.; Vadas, M.; Xia, P. Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic beta-cell death in diet-induced obese mice. FASEB J. 2013, 27, 4294–4304. [Google Scholar] [CrossRef]
- Ravichandran, S.; Finlin, B.S.; Kern, P.A.; Ozcan, S. Sphk2(−/−) mice are protected from obesity and insulin resistance. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 570–576. [Google Scholar] [CrossRef]
- Song, Z.; Wang, W.; Li, N.; Yan, S.; Rong, K.; Lan, T.; Xia, P. Sphingosine kinase 2 promotes lipotoxicity in pancreatic beta-cells and the progression of diabetes. FASEB J. 2019, 33, 3636–3646. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Ladriere, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. Diabetes Obes. Metab. 2010, 12, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.R.; Meikle, P.J.; Biden, T.J. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Diakogiannaki, E.; Morgan, N.G. Differential regulation of the ER stress response by long-chain fatty acids in the pancreatic beta-cell. Biochem. Soc. Trans. 2008, 36, 959–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W.L. Prohibitin 2: At a communications crossroads. IUBMB Life 2015, 67, 239–254. [Google Scholar] [CrossRef]
- Supale, S.; Thorel, F.; Merkwirth, C.; Gjinovci, A.; Herrera, P.L.; Scorrano, L.; Meda, P.; Langer, T.; Maechler, P. Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes 2013, 62, 3488–3499. [Google Scholar] [CrossRef] [Green Version]
- Baltrusch, S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 2016, 59, 683–687. [Google Scholar] [CrossRef] [Green Version]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Plötz, T.; Hartmann, M.; Lenzen, S.; Elsner, M. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr. Metab. 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, J.; Klint, C.; Wierup, N.; Strom, K.; Larsson, S.; Sundler, F.; Lupi, R.; Marchetti, P.; Xu, G.; Kimmel, A.; et al. Perilipin is present in islets of Langerhans and protects against lipotoxicity when overexpressed in the beta-cell line INS-1. Endocrinology 2009, 150, 3049–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Ding, Y.; Chen, Y.; Zhang, S.; Huo, C.; Wang, Y.; Yu, J.; Zhang, P.; Na, H.; Zhang, H.; et al. The proteomics of lipid droplets: Structure, dynamics, and functions of the organelle conserved from bacteria to humans. J. Lip. Res. 2012, 53, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahn, T.H.; Zhang, Y.; Lee, M.J.; Sommer, A.G.; Mostoslavsky, G.; Fried, S.K.; Greenberg, A.S.; Puri, V. FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem. Biophys. Res. Commun. 2013, 432, 296–301. [Google Scholar] [CrossRef] [Green Version]
- von Hanstein, A.S.; Lenzen, S.; Plötz, T. Toxicity of fatty acid profiles of popular edible oils in human EndoC-betaH1 beta-cells. Nutr. Diabetes 2020, 10, 5. [Google Scholar] [CrossRef]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Kaminski, M.T.; Lenzen, S. Physiological characterization of the human EndoC-betaH1 beta-cell line. Biochem. Biophys. Res. Commun. 2015, 464, 13–19. [Google Scholar] [CrossRef]
- Tyka, K.; Jörns, A.; Dunst, A.; Tang, Y.; Bryde, T.H.; Mehmeti, I.; Walentinsson, A.; Marselli, L.; Cnop, M.; Tyrberg, B.; et al. MCPIP1 is a novel link between diabetogenic conditions and impaired insulin secretory capacity. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166199. [Google Scholar] [CrossRef]
- Thomas, P.; Leslie, K.A.; Welters, H.J.; Morgan, N.G. Long-chain saturated fatty acid species are not toxic to human pancreatic beta-cells and may offer protection against pro-inflammatory cytokine induced beta-cell death. Nutr. Metab. 2021, 18, 9. [Google Scholar] [CrossRef]
- Lytrivi, M.; Ghaddar, K.; Lopes, M.; Rosengren, V.; Piron, A.; Yi, X.; Johansson, H.; Lehtio, J.; Igoillo-Esteve, M.; Cunha, D.A.; et al. Combined transcriptome and proteome profiling of the pancreatic beta-cell response to palmitate unveils key pathways of beta-cell lipotoxicity. BMC Genom. 2020, 21, 590. [Google Scholar] [CrossRef]
- Morgan, N.G.; Dhayal, S.; Diakogiannaki, E.; Welters, H.J. The cytoprotective actions of long-chain mono-unsaturated fatty acids in pancreatic beta-cells. Biochem. Soc. Trans. 2008, 36, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Mauvoisin, D.; Mounier, C. Hormonal and nutritional regulation of SCD1 gene expression. Biochimie 2011, 93, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Olson, L.K. Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic beta-cells by stearoyl-CoA desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E640–E649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [Green Version]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef]
- Fyrst, H.; Saba, J.D. Sphingosine-1-phosphate lyase in development and disease: Sphingolipid metabolism takes flight. Biochim. Biophys. Acta 2008, 1781, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Saba, J.D. Lyase to live by: Sphingosine phosphate lyase as a therapeutic target. Expert Opin. Ther. Targets 2009, 13, 1013–1025. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Oskouian, B.; Fyrst, H.; Zhang, M.; Paris, F.; Saba, J.D. S1P lyase regulates DNA damage responses through a novel sphingolipid feedback mechanism. Cell Death Dis. 2011, 2, e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzanares-Estreder, S.; Pascual-Ahuir, A.; Proft, M. Stress-Activated Degradation of Sphingolipids Regulates Mitochondrial Function and Cell Death in Yeast. Oxid. Med. Cell. Longev. 2017, 2017, 2708345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroi, D.N.; Karunakaran, I.; Graler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Karaca, I.; Tamboli, I.Y.; Glebov, K.; Richter, J.; Fell, L.H.; Grimm, M.O.; Haupenthal, V.J.; Hartmann, T.; Gräler, M.H.; van Echten-Deckert, G.; et al. Deficiency of sphingosine-1-phosphate lyase impairs lysosomal metabolism of the amyloid precursor protein. J. Biol. Chem. 2014, 289, 16761–16772. [Google Scholar] [CrossRef] [Green Version]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [Green Version]
- Deevska, G.M.; Nikolova-Karakashian, M.N. The expanding role of sphingolipids in lipid droplet biogenesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1155–1165. [Google Scholar] [CrossRef]
- Conte, M.; Franceschi, C.; Sandri, M.; Salvioli, S. Perilipin 2 and age-related metabolic diseases: A new perspective. Trends Endocrinol. Metab. 2016, 27, 893–903. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Beverly, L.J.; Siskind, L.J. Sphingolipids and mitochondrial apoptosis. J. Bioenerg. Biomembr. 2016, 48, 153–168. [Google Scholar] [CrossRef]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell 2019, 177, 1536–1552 e1523. [Google Scholar] [CrossRef]
- Hong, S.-W.; Lee, J.; Kwon, H.; Park, S.-E.; Rhee, E.-J.; Park, C.-Y.; Oh, K.-W.; Park, S.-W.; Lee, W.-Y. Deficiency of sphingosine-1-phosphate reduces the expression of prohibitin and causes β-cell impairment via mitochondrial dysregulation. Endocrinol. Metab. (Seoul) 2018, 33, 403–412. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Allard, C.; Alvarez-Mercado, A.I.; Fuselier, T.; Kim, J.H.; Coons, L.A.; Hewitt, S.C.; Urano, F.; Korach, K.S.; Levin, E.R.; et al. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep. 2018, 24, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yang, J.; Liu, W.; Yao, G.; Xu, F.; Hayashi, T.; Onodera, S.; Ikejima, T. Attenuating effect of silibinin on palmitic acid-induced apoptosis and mitochondrial dysfunction in pancreatic beta-cells is mediated by estrogen receptor alpha. Mol. Cell Biochem. 2019, 460, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Engel, N.; Lisec, J.; Piechulla, B.; Nebe, B. Metabolic profiling reveals sphingosine-1-phosphate kinase 2 and lyase as key targets of (phyto-) estrogen action in the breast cancer cell line MCF-7 and not in MCF-12A. PLoS ONE 2012, 7, e47833. [Google Scholar] [CrossRef] [PubMed]
- Slopien, R.; Wender-Ozegowska, E.; Rogowicz-Frontczak, A.; Meczekalski, B.; Zozulinska-Ziolkiewicz, D.; Jaremek, J.D.; Cano, A.; Chedraui, P.; Goulis, D.G.; Lopes, P.; et al. Menopause and diabetes: EMAS clinical guide. Maturitas 2018, 117, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Weigel, C.; Huttner, S.S.; Ludwig, K.; Krieg, N.; Hofmann, S.; Schroder, N.H.; Robbe, L.; Kluge, S.; Nierhaus, A.; Winkler, M.S.; et al. S1P lyase inhibition protects against sepsis by promoting disease tolerance via the S1P/S1PR3 axis. EBioMedicine 2020, 58, 102898. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | FW | REV |
|---|---|---|
| Rat B-Act | GAACACGGCATTGTAACCAACTGG | GGCCACACGCAGCTCATTGTA |
| Rat Chop | CCAGCAGAGGTCACAAGCAC | CGCACTGACCACTCTGTTTC |
| Rat Ire1 | TTCTACATCTGGCAGCGGGAGG | TTCCACTTGGTGATGCGCCC |
| Rat Mnf2 | TCCAAGGTCAGGGGAATCAGCG | TGGTGGTGTGGCCAATCCCA |
| Rat Phb2 | AAGGAGTCATGGTGCCAAA | GTGTCCGGCATCCACG |
| Rat Plin2 | TCGTCTCTCAGCTCTCCTGT | TAGGTGGAGCTCACCAAGGG |
| Human SPL | ACGGCCTGGTGGCATTA | CTGACAATTGGGGATTCCC |
| Human B-Act | ATGGATGATGATATCGCCGC | TTCTGACCCATGCCCACCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Plötz, T.; Gräler, M.H.; Gurgul-Convey, E. Sphingosine-1 Phosphate Lyase Regulates Sensitivity of Pancreatic Beta-Cells to Lipotoxicity. Int. J. Mol. Sci. 2021, 22, 10893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910893
Tang Y, Plötz T, Gräler MH, Gurgul-Convey E. Sphingosine-1 Phosphate Lyase Regulates Sensitivity of Pancreatic Beta-Cells to Lipotoxicity. International Journal of Molecular Sciences. 2021; 22(19):10893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910893
Chicago/Turabian StyleTang, Yadi, Thomas Plötz, Markus H. Gräler, and Ewa Gurgul-Convey. 2021. "Sphingosine-1 Phosphate Lyase Regulates Sensitivity of Pancreatic Beta-Cells to Lipotoxicity" International Journal of Molecular Sciences 22, no. 19: 10893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910893