Genetics and Genomics of SOST: Functional Analysis of Variants and Genomic Regulation in Osteoblasts

 , and
, and
Abstract
:1. Introduction
2. Results
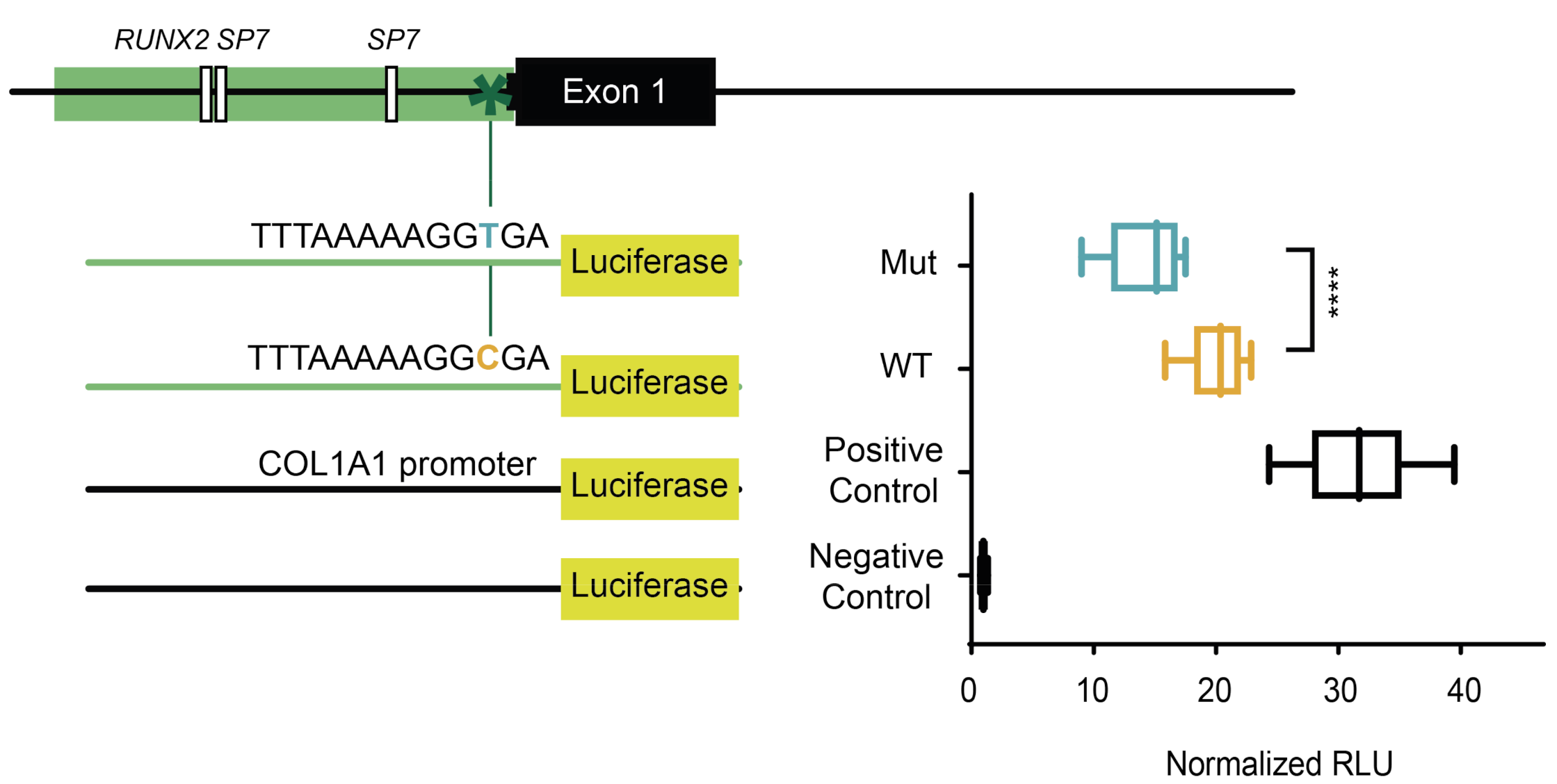
2.1. The Minor Allele of a Variant in the Extended TATA Box of SOST Displays Lower Transcriptional Capacity
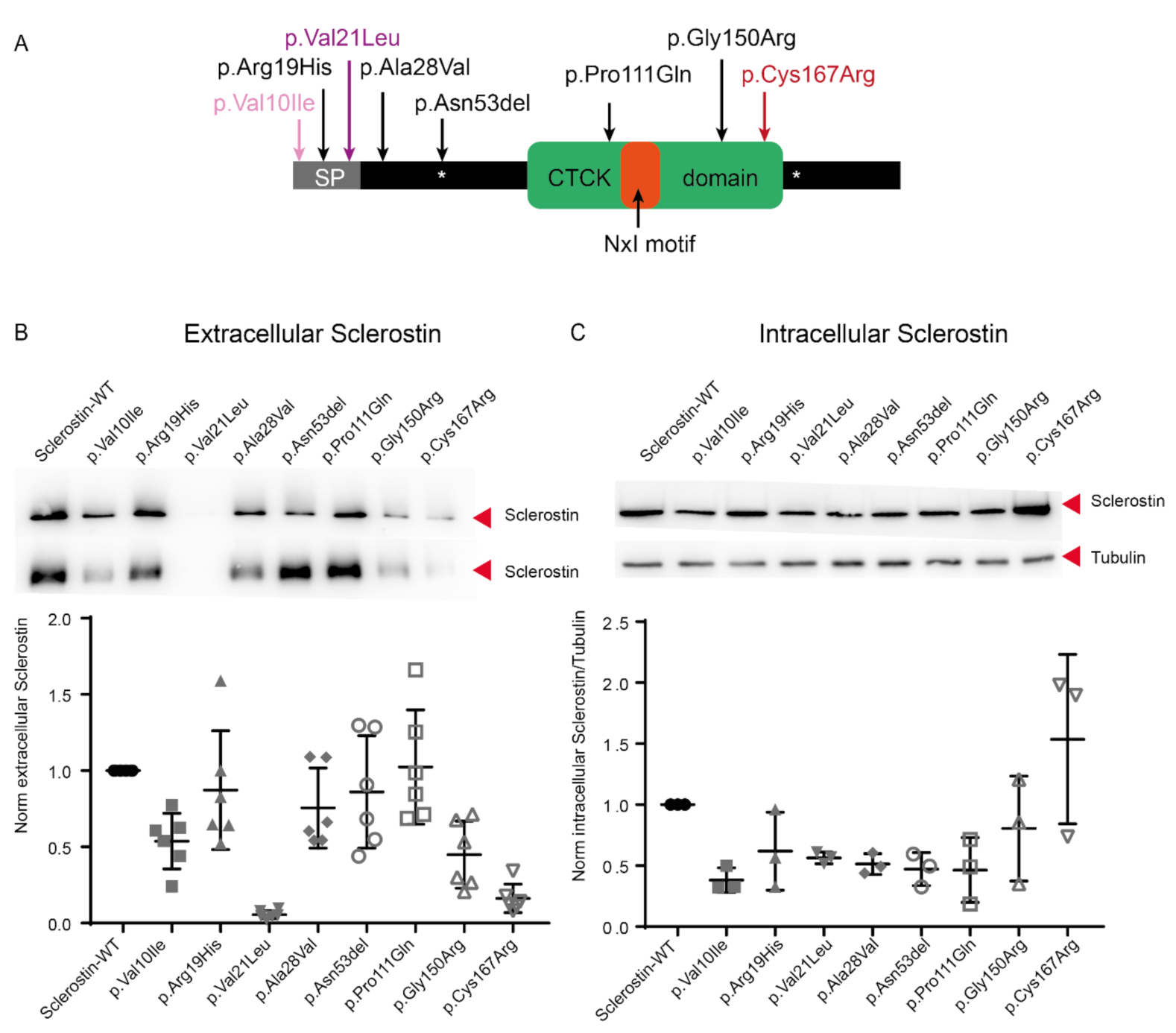
2.2. Lower Protein Abundance for the Minor Allele of rs17882143, Both in the Intracelullar and Extracellular Spaces
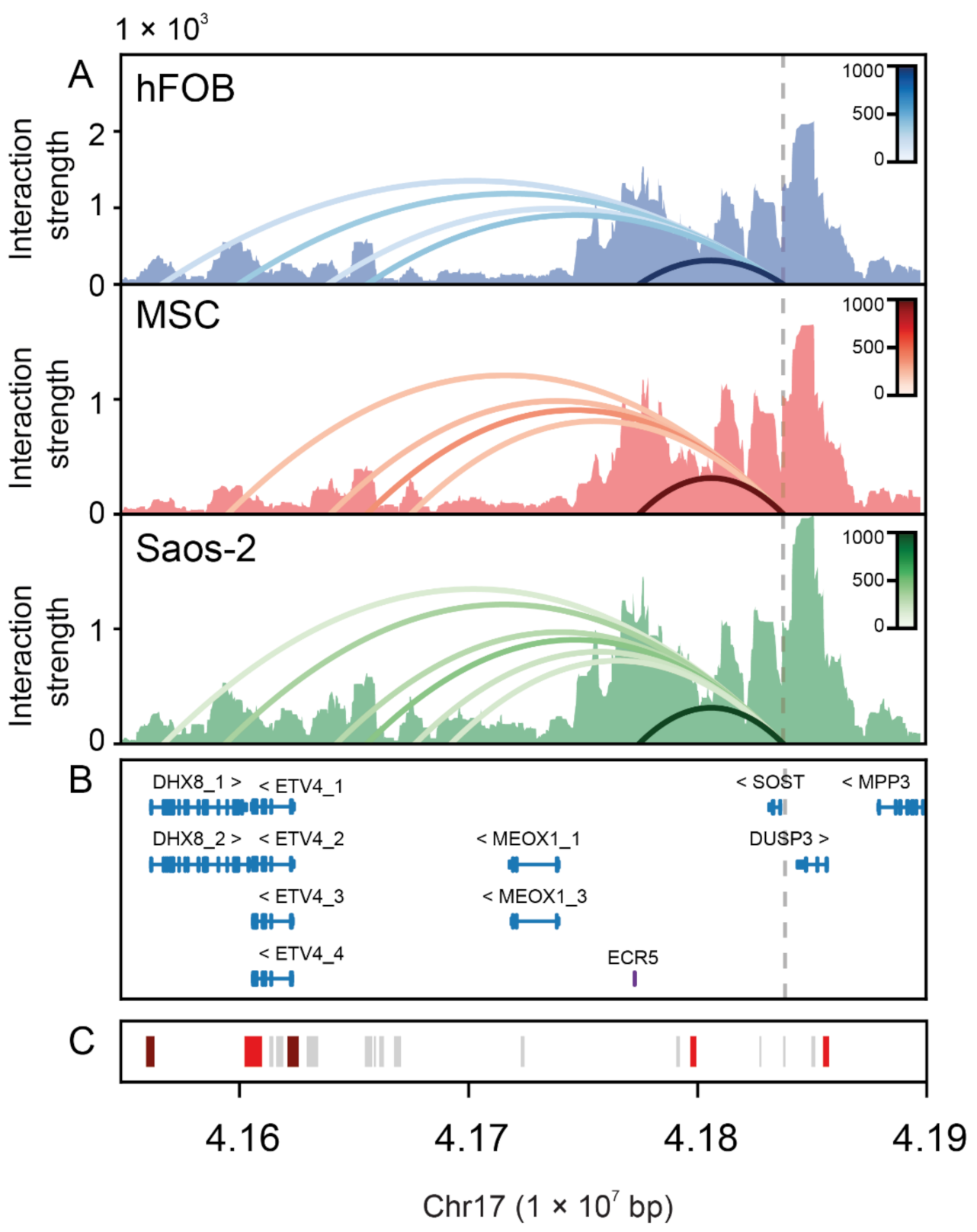
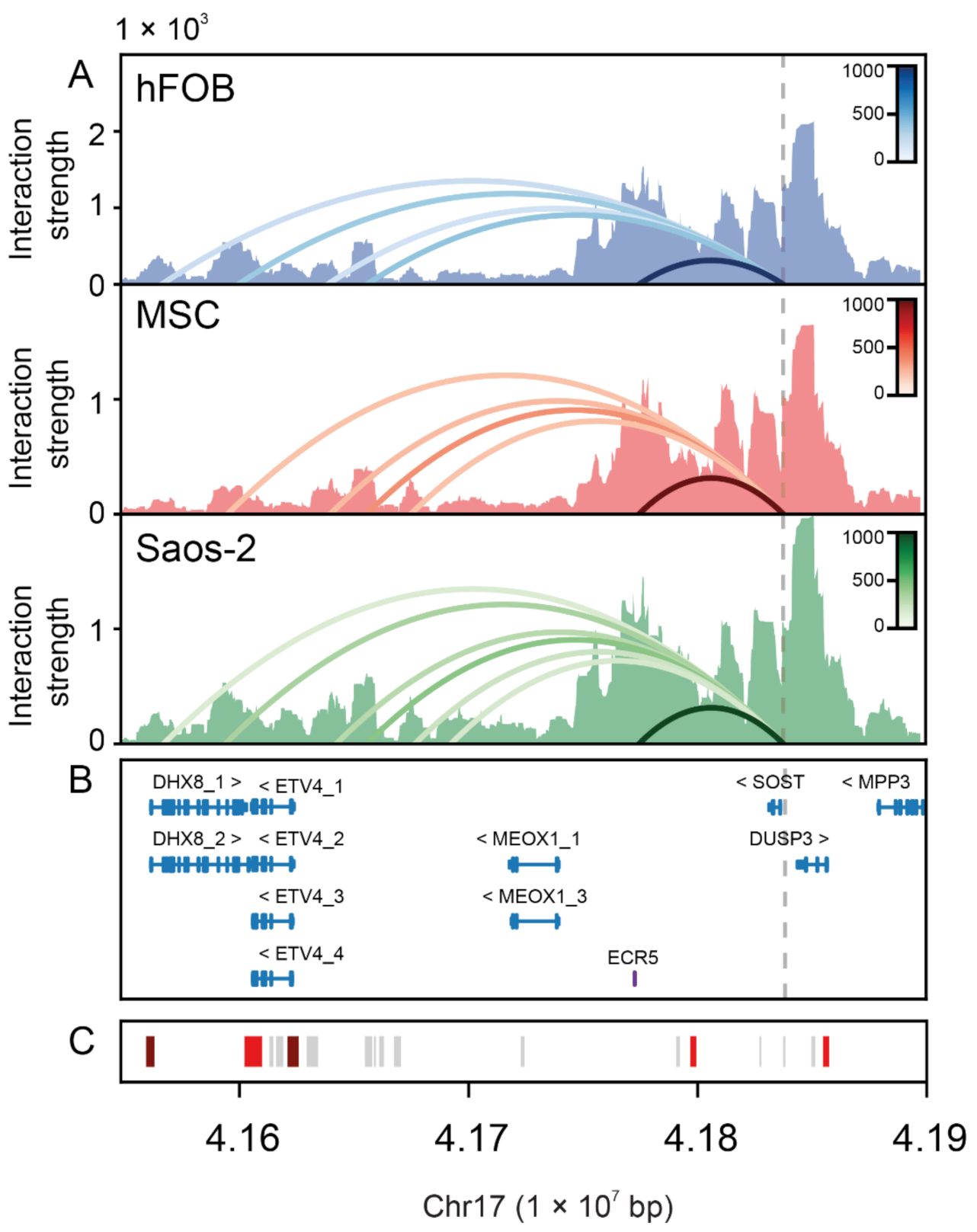
2.3. Physical Interaction between the SOST Proximal Promoter and Several Distal Regulatory Elements in Bone Cells
2.4. The rs17882143 (p.Val10Ile) Variant Present in One HBM Woman
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Luciferase Reporter Constructs, SOST Expression Vectors and Site-Directed Mutagenesis
4.3. Luciferase Gene Reporter Assay
4.4. Western Blot Assay
4.5. PCR Amplification and Sequencing
4.6. Bioinformatic Analysis and in Silico Predictions of the Effect of the Variants
4.7. 4C-Seq
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BMD | Bone mineral density |
| CDD | Craniodiaphyseal dysplasia |
| HBM | High bone mass |
| KO | Knock-out |
References
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semënov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.Y.; Dieckmann, M.; Herz, J.; Niemeier, A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE 2009, 4, e7930. [Google Scholar] [CrossRef]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.R.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.-K.; Kramer, I.; Huber, T.; Kinzel, B.; Guth-Gundel, S.; Leupin, O.; Kneissel, M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc. Natl. Acad. Sci. USA 2014, 111, E5187–E5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veverka, V.; Henry, A.J.; Slocombe, P.M.; Ventom, A.; Mulloy, B.; Muskett, F.W.; Muzylak, M.; Greenslade, K.; Moore, A.; Zhang, L.; et al. Characterization of the structural features and interactions of sclerostin: Molecular insight into a key regulator of Wnt-mediated bone formation. J. Biol. Chem. 2009, 284, 10890–10900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattamuri, C.; Luedeke, D.M.; Nolan, K.; Rankin, S.A.; Greis, K.D.; Zorn, A.M.; Thompson, T.B. Members of the DAN family are BMP antagonists that form highly stable noncovalent dimers. J. Mol. Biol. 2012, 424, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, E.; Wang, W.; Tam, C.; Hwang, J.; Zhang, Y.; Spittler, D.; Huang, O.W.; Gong, Y.; Estevez, A.; Zilberleyb, I.; et al. Wnt antagonists bind through a short peptide to the first β-propeller domain of LRP5/6. Structure 2011, 19, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, B.; Stein, S.; Oshman, D.; Rennert, O.; Thurnau, G.; Wall, J.; Bodensteiner, J.; Brown, O. Dominantly inherited craniodiaphyseal dysplasia: A new craniotubular dysplasia. Clin. Genet. 1986, 30, 381–391. [Google Scholar] [CrossRef]
- Kim, S.J.; Bieganski, T.; Sohn, Y.B.; Kozlowski, K.; Semënov, M.; Okamoto, N.; Kim, C.H.; Ko, A.-R.; Ahn, G.H.; Choi, Y.-L.; et al. Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Hum. Genet. 2011, 129, 497–502. [Google Scholar] [CrossRef]
- Balemans, W.; Van Den Ende, J.; Freire Paes-Alves, A.; Dikkers, F.G.; Willems, P.J.; Vanhoenacker, F.; de Almeida-Melo, N.; Alves, C.F.; Stratakis, C.A.; Hill, S.C.; et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am. J. Hum. Genet. 1999, 64, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Belkhribchia, M.R.; Collet, C.; Laplanche, J.-L.; Hassani, R. Novel SOST gene mutation in a sclerosteosis patient from Morocco: A case report. Eur. J. Med. Genet. 2014, 57, 133–137. [Google Scholar] [CrossRef]
- Whyte, M.P.; Deepak Amalnath, S.; McAlister, W.H.; Pedapati, R.; Muthupillai, V.; Duan, S.; Huskey, M.; Bijanki, V.N.; Mumm, S. Sclerosteosis: Report of type 1 or 2 in three Indian Tamil families and literature review. Bone 2018, 116, 321–332. [Google Scholar] [CrossRef]
- Yagi, H.; Takagi, M.; Hasegawa, Y.; Kayserili, H.; Nishimura, G. Sclerosteosis (craniotubular hyperostosis-syndactyly) with complex hyperphalangy of the index finger. Pediatr. Radiol. 2015, 45, 1239–1243. [Google Scholar] [CrossRef]
- Piters, E.; Culha, C.; Moester, M.; Van Bezooijen, R.; Adriaensen, D.; Mueller, T.; Weidauer, S.; Jennes, K.; de Freitas, F.; Löwik, C.; et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum. Mutat. 2010, 31, E1526–E1543. [Google Scholar] [CrossRef] [Green Version]
- Bhadada, S.K.; Rastogi, A.; Steenackers, E.; Boudin, E.; Arya, A.; Dhiman, V.; Bhansali, A.; Van Hul, W. Novel SOST gene mutation in a sclerosteosis patient and her parents. Bone 2013, 52, 707–710. [Google Scholar] [CrossRef]
- Fayez, A.; Aglan, M.; Esmaiel, N.; El Zanaty, T.; Abdel Kader, M.; El Ruby, M. A Novel Loss-of-Sclerostin Function Mutation in a First Egyptian Family with Sclerosteosis. BioMed Res. Int. 2015, 2015, 517815. [Google Scholar] [CrossRef] [Green Version]
- Fijalkowski, I.; Geets, E.; Steenackers, E.; Van Hoof, V.; Ramos, F.J.; Mortier, G.; Fortuna, A.M.; Van Hul, W.; Boudin, E. A Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone. J. Bone Miner. Res. 2016, 31, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Van Hul, W.; Balemans, W.; Van Hul, E.; Dikkers, F.G.; Obee, H.; Stokroos, R.J.; Hildering, P.; Vanhoenacker, F.; Van Camp, G.; Willems, P.J. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am. J. Hum. Genet. 1998, 62, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- van Lierop, A.H.; Hamdy, N.A.; Hamersma, H.; van Bezooijen, R.L.; Power, J.; Loveridge, N.; Papapoulos, S.E. Patients with sclerosteosis and disease carriers: Human models of the effect of sclerostin on bone turnover. J. Bone Miner. Res. 2011, 26, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- van Lierop, A.H.; Hamdy, N.A.T.; van Egmond, M.E.; Bakker, E.; Dikkers, F.G.; Papapoulos, S.E. Van Buchem disease: Clinical, biochemical, and densitometric features of patients and disease carriers. J. Bone Miner. Res. 2013, 28, 848–854. [Google Scholar] [CrossRef]
- Gregson, C.L.; Wheeler, L.; Hardcastle, S.A.; Appleton, L.H.; Addison, K.A.; Brugmans, M.; Clark, G.R.; Ward, K.A.; Paggiosi, M.; Stone, M.; et al. Mutations in Known Monogenic High Bone Mass Loci Only Explain a Small Proportion of High Bone Mass Cases. J. Bone Miner. Res. 2016, 31, 640–649. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.L.; Gong, G.; Kimberling, W.; Reckér, S.M.; Kimmel, D.B.; Recker, R.B. Linkage of a gene causing high bone mass to human chromosome 11 (11q12-13). Am. J. Hum. Genet. 1997, 60, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Little, R.D.; Carulli, J.P.; Del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.-C.; Eustace, B.; et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef]
- Semenov, M.V.; He, X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J. Biol. Chem. 2006, 281, 38276–38284. [Google Scholar] [CrossRef] [Green Version]
- Ellies, D.L.; Viviano, B.; McCarthy, J.; Rey, J.-P.; Itasaki, N.; Saunders, S.; Krumlauf, R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J. Bone Miner. Res. 2006, 21, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Loots, G.G. Genetics of Sost/SOST in sclerosteosis and van Buchem disease animal models. Metabolism 2018, 80, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin expression and functions beyond the osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ominsky, M.S.; Niu, Q.-T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Krause, C.; Korchynskyi, O.; de Rooij, K.; Weidauer, S.E.; de Gorter, D.J.J.; van Bezooijen, R.L.; Hatsell, S.; Economides, A.N.; Mueller, T.D.; Löwik, C.W.G.M.; et al. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J. Biol. Chem. 2010, 285, 41614–41626. [Google Scholar] [CrossRef] [Green Version]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Rhee, Y.; Allen, M.R.; Condon, K.; Lezcano, V.; Ronda, A.C.; Galli, C.; Olivos, N.; Passeri, G.; O’Brien, C.A.; Bivi, N.; et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 2011, 26, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Collette, N.M.; Genetos, D.C.; Economides, A.N.; Xie, L.; Shahnazari, M.; Yao, W.; Lane, N.E.; Harland, R.M.; Loots, G.G. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc. Natl. Acad. Sci. USA. 2012, 109, 14092–14097. [Google Scholar] [CrossRef] [Green Version]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.-Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recker, R.R.; Benson, C.T.; Matsumoto, T.; Bolognese, M.A.; Robins, D.A.; Alam, J.; Chiang, A.Y.; Hu, L.; Krege, J.H.; Sowa, H.; et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J. Bone Miner. Res. 2015, 30, 216–224. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Devogelaer, J.-P.; Durigova, M.; Goemaere, S.; Hemsley, S.; Jakob, F.; Junker, U.; Ruckle, J.; Seefried, L.; Winkle, P.J. BPS804 Anti-Sclerostin Antibody in Adults With Moderate Osteogenesis Imperfecta: Results of a Randomized Phase 2a Trial. J. Bone Miner. Res. 2017, 32, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Morony, S.; Gong, J.; Cao, J.; Gao, Y.; Shalhoub, V.; Tipton, B.; Haldankar, R.; et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J. Bone Miner. Res. 2009, 24, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Ominsky, M.S.; Vlasseros, F.; Jolette, J.; Smith, S.Y.; Stouch, B.; Doellgast, G.; Gong, J.; Gao, Y.; Cao, J.; Graham, K.; et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J. Bone Miner. Res. 2010, 25, 948–959. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Roca-Ayats, N.; Monistrol-Mula, A.; García-Giralt, N.; Díez-Pérez, A.; Nogués, X.; Mellibovsky, L.; Grinberg, D.; Balcells, S. Common and rare variants of WNT16, DKK1 and SOST and their relationship with bone mineral density. Sci. Rep. 2018, 8, 10951. [Google Scholar] [CrossRef]
- García-Giralt, N.; Enjuanes, A.; Bustamante, M.; Mellibovsky, L.; Nogués, X.; Carreras, R.; Díez-Pérez, A.; Grinberg, D.; Balcells, S. In vitro functional assay of alleles and haplotypes of two COL1A1-promoter SNPs. Bone 2005, 36, 902–908. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Sarrión, P.; Mellibovsky, L.; Urreizti, R.; Civit, S.; Cols, N.; García-Giralt, N.; Yoskovitz, G.; Aranguren, A.; Malouf, J.; Di Gregorio, S.; et al. Genetic analysis of high bone mass cases from the BARCOS cohort of Spanish postmenopausal women. PLoS ONE 2014, 9, e94607. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, M.; Zulian, A.; Cancello, R.; Alberti, L.; Gilardini, L.; Di Blasio, A.M.; Invitti, C. A novel missense mutation in the signal peptide of the human POMC gene: A possible additional link between early-onset type 2 diabetes and obesity. Eur. J. Hum. Genet. 2012, 20, 1290–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohren, S.; Weiskirchen, R. Non-synonymous gene polymorphisms in the secretory signal peptide of human TGF-beta1 affect cellular synthesis but not secretion of TGF-beta1. Biochem. Biophys. Res. Commun. 2009, 379, 1015–1020. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, S.; Lu, C.; Wu, Q.; Li, S.; Fu, H.; Wang, G.; Lv, C.; Nie, L.; Zhang, Y.; et al. Missense mutations in the signal peptide of the porcine GH gene affect cellular synthesis and secretion. Pituitary 2016, 19, 362–369. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Casado-Díaz, A.; Santiago-Mora, R.; Jiménez, R.; Caballero-Villarraso, J.; Herrera, C.; Torres, A.; Dorado, G.; Quesada-Gómez, J.M. Cryopreserved human bone marrow mononuclear cells as a source of mesenchymal stromal cells: Application in osteoporosis research. Cytotherapy 2008, 10, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Miñán, A.; Bessa, J.; Tena, J.J.; Gómez-Skarmeta, J.L. Assay for transposase-accessible chromatin and circularized chromosome conformation capture, two methods to explore the regulatory landscapes of genes in zebrafish. Methods Cell Biol. 2016, 135, 413–430. [Google Scholar] [PubMed]
- van de Werken, H.J.G.; de Vree, P.J.P.; Splinter, E.; Holwerda, S.J.B.; Klous, P.; de Wit, E.; de Laat, W. 4C technology: Protocols and data analysis. Methods Enzymol. 2012, 513, 89–112. [Google Scholar] [PubMed]
- Noordermeer, D.; Leleu, M.; Splinter, E.; Rougemont, J.; De Laat, W.; Duboule, D. The dynamic architecture of Hox gene clusters. Science 2011, 334, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Irastorza-Azcarate, I.; Acemel, R.D.; Tena, J.J.; Maeso, I.; Gómez-Skarmeta, J.L.; Devos, D.P. 4Cin: A computational pipeline for 3D genome modeling and virtual Hi-C analyses from 4C data. PLoS Comput. Biol. 2018, 14, e1006030. [Google Scholar] [CrossRef] [PubMed]
- Smemo, S.; Tena, J.J.; Kim, K.-H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Position | rs Number | Type | MAF (EUR) | MAF (HBM Cohort) | Predicted Functionality |
|---|---|---|---|---|---|
| SOST | |||||
| g.41838229C > T | rs1237278 | 5′ UP | 0.355 (C) | 0.45 (C) | eQTL; TFBS |
| g.41837719G > A | rs851058 | 5′ UP | 0.402 (A) | 0.35 (A) | eQTL; TFBS |
| g.41837510_41837512del | rs10534024 | 5′ UP | 0.352 (TCCTCCT) | 0.45 (TCCTCCT) | eQTL |
| p.Val10Ile | rs17882143 | M | 0.018 (T) | 0.05 (T) | eQTL T; B; LB |
| c.-22G > A | rs17885799 | 5′ UTR | - | 0.05 (A) | TFBS, ESE |
| c.*320C > T | rs17883310 | 3′ UTR | 0.013 (T) | 0.05 (T) | miRNA |
| c.*1004G > A | rs17886183 | 3′ UTR | 0.004 (A) | 0.05 (A) | eQTL miRNA |
| c.*1266_*1267insG | rs17881550 | 3′ UTR | 0.412 (ins) | 0.35 (ins) | eQTL |
| ECR5 | |||||
| g.41774092G > A | rs552004150 | 3′ D | 0.003 (A) | 0.05 (A) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Gil, N.; Roca-Ayats, N.; Cozar, M.; Garcia-Giralt, N.; Ovejero, D.; Nogués, X.; Grinberg, D.; Balcells, S. Genetics and Genomics of SOST: Functional Analysis of Variants and Genomic Regulation in Osteoblasts. Int. J. Mol. Sci. 2021, 22, 489. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020489
Martínez-Gil N, Roca-Ayats N, Cozar M, Garcia-Giralt N, Ovejero D, Nogués X, Grinberg D, Balcells S. Genetics and Genomics of SOST: Functional Analysis of Variants and Genomic Regulation in Osteoblasts. International Journal of Molecular Sciences. 2021; 22(2):489. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020489
Chicago/Turabian StyleMartínez-Gil, Núria, Neus Roca-Ayats, Mónica Cozar, Natàlia Garcia-Giralt, Diana Ovejero, Xavier Nogués, Daniel Grinberg, and Susanna Balcells. 2021. "Genetics and Genomics of SOST: Functional Analysis of Variants and Genomic Regulation in Osteoblasts" International Journal of Molecular Sciences 22, no. 2: 489. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020489