Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants

 , , and
, , and 
Abstract
:1. Introduction
2. Drug-Induced Chromatin Modifications
3. Nanobodies for Exploration and Modification of Histone Marks
4. Direct Gene Manipulation for Genome-Wide Chromatin Modulation
4.1. The Limitations of Knock-Out and Knock-Down Mutant Lines for Mark Function Analyses
4.2. Expressing Mutant Histones Carrying Non-Modifiable Residues
4.3. Editing Histones and Chromatin Factors: New Perspectives Brought by the CRISPR-Cas Tool
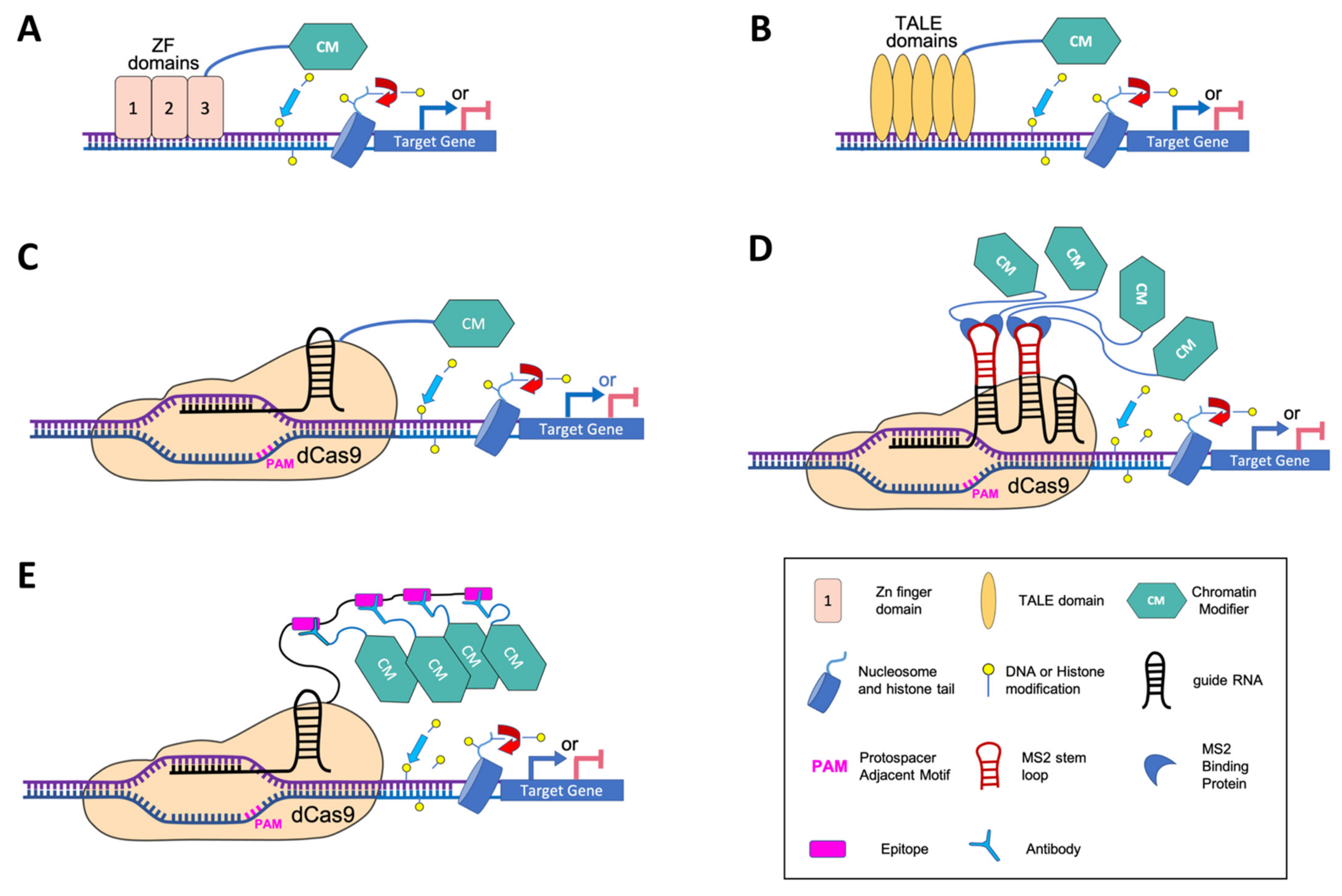
5. Programmable DNA-Binding Platforms to Modify Chromatin at Specific Loci
5.1. Zinc Finger (ZF) Engineered Proteins
5.2. Transcription Activator-Like Effectors (TALE)-Based Editing Tools
6. Targeted Epigenetic Editing with dCas9
6.1. The First Generation of dCas9 Targeting Tested with ATFs
6.2. Chimeric dCas9 Systems with Enhanced Targeting and Transcriptional Effector Capacity
6.3. dCas9-Based Epigenetic Editing
6.3.1. Editing DNA Methylation
6.3.2. Editing Histone Modifications
6.3.3. Pending Concerns with the dCas9 System
7. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nat. Cell Biol. 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-Five Years of the Nucleosome, Fundamental Particle of the Eukaryote Chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nat. Cell Biol. 2010, 466, 388–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, Z.; Gutierrez, C. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nat. Cell Biol. 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Seki, M. Histone Modifications Form Epigenetic Regulatory Networks to Regulate Abiotic Stress Response. Plant Physiol. 2020, 182, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated Nucleosome Mobility and the Histone Code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef]
- Ingouff, M.; Berger, F. Histone3 variants in plants. Chromosoma 2010, 119, 27–33. [Google Scholar] [CrossRef]
- Stroud, H.; Otero, S.; Desvoyes, B.; Ramírez-Parra, E.; Jacobsen, S.E.; Gutierrez, C. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2012, 109, 5370–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollmann, H.; Holec, S.; Alden, K.; Clarke, N.D.; Jacques, P.-É.; Berger, F. Dynamic Deposition of Histone Variant H3.3 Accompanies Developmental Remodeling of the Arabidopsis Transcriptome. PLoS Genet. 2012, 8, e1002658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, H.; Nakamura, M.; Siretskiy, A.; Borghi, L.; Moraes, I.; Wildhaber, T.; Gruissem, W.; Hennig, L. Arabidopsis replacement histone variant H3.3 occupies promoters of regulated genes. Genome Biol. 2014, 15, R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S. Histone modifications: Combinatorial complexity or cumulative simplicity? Proc. Natl. Acad. Sci. USA 2005, 102, 5308–5309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, L.; Sung, Z.R. PcG and trxG in plants–friends or foes. Trends Genet. 2015, 31, 252–262. [Google Scholar] [CrossRef]
- Shafiq, S.; Berr, A.; Shen, W.-H. Combinatorial functions of diverse histone methylations in Arabidopsis thaliana flowering time regulation. New Phytol. 2014, 201, 312–322. [Google Scholar] [CrossRef]
- Engelhorn, J.; Blanvillain, R.; Carles, C.C. Gene activation and cell fate control in plants: A chromatin perspective. Cell. Mol. Life Sci. 2014, 71, 3119–3137. [Google Scholar] [CrossRef]
- Berr, A.; Shafiq, S.; Shen, W.-H. Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta 2011, 1809, 567–576. [Google Scholar] [CrossRef]
- Mio, C.; Bulotta, S.; Russo, D.; Damante, G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Wiuf, A.; Kristensen, L.H.; Kristensen, O.; Dorosz, J.; Jensen, J.; Gajhede, M. Structure and binding properties of a cameloid nanobody raised against KDM5B. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; Helma, J.; Schneider, A.F.L.; Leonhardt, H.; Hackenberger, C.P.R. Nanobodies: Chemical Functionalization Strategies and Intracellular Applications. Angew. Chem. Int. Ed. 2018, 57, 2314–2333. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.-M.; Morgan, M.; Gao, X.; Jackson, J.; Rickels, R.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014, 345, 1065–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [Green Version]
- Gehre, M.; Bunina, D.; Sidoli, S.; Lübke, M.J.; Diaz, N.; Trovato, M.; Garcia, B.A.; Zaugg, J.B.; Noh, K. Lysine 4 of histone H3.3 is required for embryonic stem cell differentiation, histone enrichment at regulatory regions and transcription accuracy. Nat. Genet. 2020, 52, 273–282. [Google Scholar] [CrossRef]
- Sanders, D.; Qian, S.; Fieweger, R.; Lu, L.; Dowell, J.A.; Denu, J.M.; Chen, J. Histone Lysine-to-Methionine Mutations Reduce Histone Methylation and Cause Developmental Pleiotropy. Plant Physiol. 2017, 173, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Mukai, M.; Ueda, J.; Muraki, M.; Stasevich, T.J.; Horikoshi, N.; Kujirai, T.; Kita, H.; Kimura, T.; Hira, S.; et al. Genetically encoded system to track histone modification in vivo. Sci. Rep. 2013, 3, 2436. [Google Scholar] [CrossRef] [Green Version]
- Rajan, M.; Mortusewicz, O.; Rothbauer, U.; Hastert, F.D.; Schmidthals, K.; Rapp, A.; Leonhardt, H.; Cardoso, M.C. Generation of an alpaca-derived nanobody recognizing γ-H2AX. FEBS Open Bio 2015, 5, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Jullien, D.; Vignard, J.; Fedor, Y.; Béry, N.; Olichon, A.; Crozatier, M.; Erard, M.; Cassard, H.; Ducommun, B.; Salles, B.; et al. Chromatibody, a novel non-invasive molecular tool to explore and manipulate chromatin in living cells. J. Cell Sci. 2016, 129, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Zhou, Y.; Li, M.; Fang, Y. Histone 3 lysine 36 to methionine mutations stably interact with and sequester SDG8 in Arabidopsis thaliana. Sci. China Life Sci. 2018, 61, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J.; Słomski, R. CRISPR/Cas9 Immune System as a Tool for Genome Engineering. Arch. Immunol. Ther. Exp. 2016, 65, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Waryah, C.B.; Moses, C.; Arooj, M.; Blancafort, P. Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing. Methods Mol. Biol. 2018, 1767, 19–63. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef] [PubMed]
- Saberianfar, R.; Chin-Fatt, A.; Scott, A.; Henry, K.A.; Topp, E.; Menassa, R. Plant-Produced Chimeric VHH-sIgA Against Enterohemorrhagic E. coli Intimin Shows Cross-Serotype Inhibition of Bacterial Adhesion to Epithelial Cells. Front. Plant Sci. 2019, 10, 270. [Google Scholar] [CrossRef] [Green Version]
- Grimanelli, D.; Roudier, F. Epigenetics and Development in Plants: Green Light to Convergent Innovations. Curr. Top. Dev. Biol. 2013, 104, 189–222. [Google Scholar] [CrossRef]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2014, 31, 1120–1123. [Google Scholar] [CrossRef]
- Jiang, D.; Berger, F. DNA replication–coupled histone modification maintains Polycomb gene silencing in plants. Science 2017, 357, 1146–1149. [Google Scholar] [CrossRef] [Green Version]
- Wollmann, H.; Stroud, H.; Yelagandula, R.; Tarutani, Y.; Jiang, D.; Jing, L.; Jamge, B.; Takeuchi, H.; Holec, S.; Nie, X.; et al. The histone H3 variant H3.3 regulates gene body DNA methylation in Arabidopsis thaliana. Genome Biol. 2017, 18, 94. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.; Bailey, T.L. GT-Scan: Identifying unique genomic targets. Bioinformatics 2014, 30, 2673–2675. [Google Scholar] [CrossRef]
- Zhu, L.J.; Holmes, B.R.; Aronin, N.; Brodsky, M.H. CRISPRseek: A Bioconductor Package to Identify Target-Specific Guide RNAs for CRISPR-Cas9 Genome-Editing Systems. PLoS ONE 2014, 9, e108424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, M.; Jacob, Y.; Susaki, D.; Leblanc, C.; Buendía, D.; Axelsson, E.; Kawashima, T.; Voigt, P.; Boavida, L.C.; Becker, J.; et al. Targeted reprogramming of H3K27me3 resets epigenetic memory in plant paternal chromatin. Nat. Cell Biol. 2020, 22, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Deal, R.B.; Topp, C.N.; McKinney, E.C.; Meagher, R.B. Repression of Flowering in Arabidopsis Requires Activation of FLOWERING LOCUS C Expression by the Histone Variant H2A.Z. Plant Cell 2007, 19, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Park, J.; Kim, J.-S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman-Derr, D.; Zilberman, D. Deposition of Histone Variant H2A.Z within Gene Bodies Regulates Responsive Genes. PLoS Genet. 2012, 8, e1002988. [Google Scholar] [CrossRef] [Green Version]
- Jupe, F.; Rivkin, A.C.; Michael, T.P.; Zander, M.; Motley, S.T.; Sandoval, J.P.; Slotkin, R.K.; Chen, H.; Castanon, R.; Nery, J.R.; et al. The complex architecture and epigenomic impact of plant T-DNA insertions. PLoS Genet. 2019, 15, e1007819. [Google Scholar] [CrossRef] [Green Version]
- Pliatsika, V.; Rigoutsos, I. “Off-Spotter”: Very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct 2015, 10, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Yelagandula, R.; Stroud, H.; Holec, S.; Zhou, K.; Feng, S.; Zhong, X.; Muthurajan, U.M.; Nie, X.; Kawashima, T.; Groth, M.; et al. The Histone Variant H2A.W Defines Heterochromatin and Promotes Chromatin Condensation in Arabidopsis. Cell 2014, 158, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhang, Z.; Hua, K.; Gao, X.; Mao, Y.; Botella, J.R.; Zhu, J.-K. A Highly Efficient Cell Division-Specific CRISPR/Cas9 System Generates Homozygous Mutants for Multiple Genes in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 3925. [Google Scholar] [CrossRef] [Green Version]
- Bourguet, P.; Picard, C.L.; Yelagandula, R.; Pélissier, T.; Lorković, Z.J.; Pouch-Pélissier, M.-N.; Jacobsen, S.E.; Berger, F.; Mathieu, O. The Histone Variant H2A.W Promotes Heterochromatin Accessibility for Efficient DNA Methylation in Arabidopsis. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.W.C.; Emery, L.E.; Miller, K.M. CRISPR/Cas9 Gene Editing of Human Histone H2A Variant H2AX and MacroH2A. Adv. Struct. Saf. Stud. 2018, 1832, 255–269. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Kim, I.S.; Ji, F.; Huebner, A.J.; Di Stefano, B.; Schwarz, B.A.; Charlton, J.; Coffey, A.; Choi, J.; Walsh, R.M.; et al. Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nat. Cell Biol. 2019, 21, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, e00478-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, J.C.; Barnes, K.M.; Dong, L.; Yue, S.; Graber, E.; Rapaport, R.; Dauber, A.; Nilsson, O.; Baron, J. Ezh2 Mutations Found in the Weaver Overgrowth Syndrome Cause a Partial Loss of H3K27 Histone Methyltransferase Activity. J. Clin. Endocrinol. Metab. 2017, 103, 1470–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009, 19, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Nuetzmann, H.-W.; Fischer, J.; Scherlach, K.; Hertweck, C.; Brakhage, A.A. Distinct Amino Acids of Histone H3 Control Secondary Metabolism in Aspergillus nidulans. Appl. Environ. Microbiol. 2013, 79, 6102–6109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Cai, J.; Gao, W.; Meng, X.; Gao, F.; Wu, P.; Duan, C.; Wang, R.; Dinislam, M.; Lin, L.; et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018, 419, 280–290. [Google Scholar] [CrossRef]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Fan, D.; Wang, X.; Tang, X.; Ye, X.; Ren, S.; Wang, D.; Luo, K. Histone H3K9 demethylase JMJ25 epigenetically modulates anthocyanin biosynthesis in poplar. Plant J. 2018, 96, 1121–1136. [Google Scholar] [CrossRef]
- Rheinbay, E.; Louis, D.N.; Bernstein, B.E.; Suvà, M.L. A Tell-Tail Sign of Chromatin: Histone Mutations Drive Pediatric Glioblastoma. Cancer Cell 2012, 21, 329–331. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Li, D.; Jin, L.; Ruan, Y.; Shen, W.-H.; Liu, C. Histone lysine methyltransferases BnaSDG8.A and BnaSDG8.C are involved in the floral transition in Brassica napus. Plant J. 2018, 95, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, G.; Zhang, X.; An, C.; Cheng, C.; Wang, H. Temperature effect on CRISPR-Cas9 mediated genome editing. J. Genet. Genom. 2017, 44, 199–205. [Google Scholar] [CrossRef]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, C.; Zhang, F.; Mendez, J.; Lozano, Y.; Chatpar, K.; Irish, V.F.; Jacob, Y. Increased efficiency of targeted mutagenesis by CRISPR/Cas9 in plants using heat stress. Plant J. 2017, 93, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Malzahn, A.A.; Tang, X.; Lee, K.; Ren, Q.; Sretenovic, S.; Zhang, Y.; Chen, H.; Kang, M.; Bao, Y.; Zheng, X.; et al. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Nandy, S.; Pathak, B.; Zhao, S.; Srivastava, V. Heat-shock-inducible CRISPR/Cas9 system generates heritable mutations in rice. Plant Direct 2019, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jung, Y.-J.; Lee, H.K.; Hong, S.G.; Kim, O.-S. Complete genome sequence of Pedobacter cryoconitis PAMC 27485, a CRISPR-Cas system-containing psychrophile isolated from Antarctica. J. Biotechnol. 2016, 226, 74–75. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Durai, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Madan, S.; Sundar, D. Exploiting the recognition code for elucidating the mechanism of zinc finger protein-DNA interactions. BMC Genom. 2016, 17, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, B.; Schwimmer, L.J.; Fuller, R.P.; Ye, Y.; Asawapornmongkol, L.; Barbas, C.F. Modular system for the construction of zinc-finger libraries and proteins. Nat. Protoc. 2010, 5, 791–810. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Barbas, C.F. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Gräslund, T.; Li, X.; Magnenat, L.; Popkov, M.; Barbas, C.F. Exploring Strategies for the Design of Artificial Transcription Factors: Targeting Sites Proximal to Known Regulatory Regions for the Induction of Gamma-Globin Expression and the Treatment of Sickle Cell Disease. J. Biol. Chem. 2005, 280, 3707–3714. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Dreier, B.; Barbas, C.F. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 2000, 97, 1495–1500. [Google Scholar] [CrossRef] [Green Version]
- Segal, D.J.; Stege, J.T.; Barbas, C.F. Zinc fingers and a green thumb: Manipulating gene expression in plants. Curr. Opin. Plant Biol. 2003, 6, 163–168. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB–Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.; Sánchez-García, I.; Klug, A. In vivo repression by a site-specific DNA-binding protein designed against an oncogenic sequence. Nat. Cell Biol. 1994, 372, 642–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, J.J.; Li, Y.; Hossain, M.; Huang, S.; Bungert, J. Dissecting the function of the adult β-globin downstream promoter region using an artificial zinc finger DNA-binding domain. Nucleic Acids Res. 2014, 42, 4363–4374. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kazemier, H.G.; De Groote, M.L.; Ruiters, M.H.J.; Xu, G.-L.; Rots, M.G. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2013, 42, 1563–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huisman, C.; Van Der Wijst, M.G.P.; Schokker, M.; Blancafort, P.; Terpstra, M.M.; Kok, K.; Van Der Zee, A.G.; Schuuring, E.; Wisman, G.B.A.; Rots, M.G. Re-expression of Selected Epigenetically Silenced Candidate Tumor Suppressor Genes in Cervical Cancer by TET2-directed Demethylation. Mol. Ther. 2016, 24, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falahi, F.; Huisman, C.; Kazemier, H.G.; Van Der Vlies, P.; Kok, K.; Hospers, G.A.P.; Rots, M.G. Towards Sustained Silencing of HER2/neu in Cancer By Epigenetic Editing. Mol. Cancer Res. 2013, 11, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Snowden, A.W.; Gregory, P.D.; Case, C.C.; Pabo, C.O. Gene-Specific Targeting of H3K9 Methylation Is Sufficient for Initiating Repression In Vivo. Curr. Biol. 2002, 12, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Kungulovski, G.; Nunna, S.; Thomas, M.; Zanger, U.M.; Reinhardt, R.; Jeltsch, A. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenet. Chromatin 2015, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nat. Cell Biol. 2014, 507, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.-C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandell, J.G.; Barbas, C.F. Zinc Finger Tools: Custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 2006, 34, W516–W523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayakanthan, M.; Muthukumaran, J.; Chandrasekar, S.; Chawla, K.; Punetha, A.; Sundar, D. ZifBASE: A database of zinc finger proteins and associated resources. BMC Genom. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurt, J.A.; Thibodeau, S.A.; Hirsh, A.S.; Pabo, C.O.; Joung, J.K. Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc. Natl. Acad. Sci. USA 2003, 100, 12271–12276. [Google Scholar] [CrossRef] [Green Version]
- Durai, S.; Bosley, A.; Abulencia, A.B.; Chandrasegaran, S.; Ostermeier, M. A Bacterial One-Hybrid Selection System for Interrogating Zinc Finger- DNA Interactions. Comb. Chem. High Throughput Screen. 2006, 9, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voytas, D.F. Plant Genome Engineering with Sequence-Specific Nucleases. Annu. Rev. Plant Biol. 2013, 64, 327–350. [Google Scholar] [CrossRef]
- Grimmer, M.R.; Stolzenburg, S.; Ford, E.; Lister, R.; Blancafort, P.; Farnham, P.J. Analysis of an artificial zinc finger epigenetic modulator: Widespread binding but limited regulation. Nucleic Acids Res. 2014, 42, 10856–10868. [Google Scholar] [CrossRef]
- Jankele, R.; Svoboda, P. TAL effectors: Tools for DNA Targeting. Brief. Funct. Genom. 2014, 13, 409–419. [Google Scholar] [CrossRef]
- Miller, J.C.; Zhang, L.; Xia, D.F.; Campo, J.J.; Ankoudinova, I.V.; Guschin, D.; Babiarz, J.E.; Meng, X.; Hinkley, S.J.; Lam, S.C.; et al. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat. Chem. Biol. 2015, 12, 465–471. [Google Scholar] [CrossRef]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Zhou, R.; Kuo, Y.-C.; Cunniff, M.M.; Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012, 3, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfouz, M.M.; Li, L.; Piatek, M.; Fang, X.; Mansour, H.; Bangarusamy, D.K.; Zhu, J.-K. Targeted transcriptional repression using a chimeric TALE-SRDX repressor protein. Plant Mol. Biol. 2011, 78, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Lowder, L.G.; Zhou, J.; Zhang, Y.; Malzahn, A.; Zhong, Z.; Hsieh, T.-F.; Voytas, D.F.; Zhang, Y.; Qi, Y. Robust Transcriptional Activation in Plants Using Multiplexed CRISPR-Act2.0 and mTALE-Act Systems. Mol. Plant 2018, 11, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, T.; Prange, A.; Hoppe, T.; Tissier, A. Split-TALE: A TALE-Based Two-Component System for Synthetic Biology Applications in Planta. Plant Physiol. 2019, 179, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.M.; Brigham, M.D.; Trevino, A.E.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nat. Cell Biol. 2013, 500, 472–476. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.N.-S.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The Crystal Structure of TAL Effector PthXo1 Bound to Its DNA Target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [Green Version]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; VanDyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, W117–W122. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.C.; McNulty, M.S.; Poshusta, T.L.; Campbell, J.M.; Martínez-Gálvez, G.; Argue, D.P.; Lee, H.B.; Urban, M.D.; Bullard, C.E.; Blackburn, P.R.; et al. FusX: A Rapid One-Step Transcription Activator-Like Effector Assembly System for Genome Science. Hum. Gene Ther. 2016, 27, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neff, K.L.; Argue, D.P.; Ma, A.C.; Lee, H.B.; Clark, K.J.; Ekker, S.C. Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinform. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjana, N.E.; Cong, L.; Zhou, Y.; Cunniff, M.M.; Feng, G.; Zhang, F. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 2012, 7, 171–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubik, G.; Schmidt, M.J.; Penner, J.E.; Summerer, D. Programmable and Highly Resolved In Vitro Detection of 5-Methylcytosine by TALEs. Angew. Chem. Int. Ed. 2014, 53, 6002–6006. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [Green Version]
- Howe, F.S.; Russell, A.J.C.; Lamstaes, A.R.; El Sagheer, A.H.; Nair, A.; Brown, T.; Mellor, J. CRISPRi is not strand-specific at all loci and redefines the transcriptional landscape. eLife 2017, 6, e29878. [Google Scholar] [CrossRef]
- Brocken, D.J.; Tark-Dame, M.; Dame, R.T. dCas9: A Versatile Tool for Epigenome Editing. Curr. Issues Mol. Biol. 2018, 26, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W.; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Vilar, M.; Bernabé-Orts, J.M.; Fernández-Del-Carmen, A.; Ziarsolo, P.; Blanca, J.; Granell, A.; Orzaez, D. A modular toolbox for gRNA–Cas9 genome engineering in plants based on the GoldenBraid standard. Plant Methods 2016, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Piatek, A.A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.A.H.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef]
- Gentzel, I.N.; Park, C.H.; Bellizzi, M.; Xiao, G.; Gadhave, K.R.; Murphree, C.; Yang, Q.; LaMantia, J.; Redinbaugh, M.G.; Balint-Kurti, P.; et al. A CRISPR/dCas9 toolkit for functional analysis of maize genes. Plant Methods 2020, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.-F. A potent Cas9-derived gene activator for plant and mammalian cells. Nat. Plants 2017, 3, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nat. Cell Biol. 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Mong, E.F.; Yang, Y.; Akat, K.M.; Canfield, J.; VanWye, J.; Lockhart, J.; Tsibris, J.C.M.; Schatz, F.; Lockwood, C.J.; Tuschl, T.; et al. Chromosome 19 microRNA cluster enhances cell reprogramming by inhibiting epithelial-to-mesenchymal transition. Sci. Rep. 2020, 10, 3029. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 activators in multiple species. Nat. Methods 2016, 13, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandelakis, M.; Delgado, E.; Ebrahimkhani, M.R. CRISPR-Based Synthetic Transcription Factors In Vivo: The Future of Therapeutic Cellular Programming. Cell Syst. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Qi, L.S. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Kunii, A.; Hara, Y.; Takenaga, M.; Hattori, N.; Fukazawa, T.; Ushijima, T.; Yamamoto, T.; Sakuma, T. Three-Component Repurposed Technology for Enhanced Expression: Highly Accumulable Transcriptional Activators via Branched Tag Arrays. CRISPR J. 2018, 1, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-J.; Dempewolf, E.; Zhang, W.; Wang, Z.-Y. RNA-guided transcriptional activation via CRISPR/dCas9 mimics overexpression phenotypes in Arabidopsis. PLoS ONE 2017, 12, e0179410. [Google Scholar] [CrossRef] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Selma, S.; Bernabé-Orts, J.M.; Vazquez-Vilar, M.; Diego-Martin, B.; Ajenjo, M.; Garcia-Carpintero, V.; Granell, A.; Orzaez, D. Strong gene activation in plants with genome-wide specificity using a new orthogonal CRISPR/Cas9-based programmable transcriptional activator. Plant Biotechnol. J. 2019, 17, 1703–1705. [Google Scholar] [CrossRef] [Green Version]
- Young, J.K.; Gasior, S.L.; Jones, S.; Wang, L.; Navarro, P.; Vickroy, B.; Barrangou, R. The repurposing of type I-E CRISPR-Cascade for gene activation in plants. Commun. Biol. 2019, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Chiarella, A.M.; Lu, D.; Hathaway, N.A. Epigenetic Control of a Local Chromatin Landscape. Int. J. Mol. Sci. 2020, 21, 943. [Google Scholar] [CrossRef] [Green Version]
- Brezgin, S.; Kostyusheva, A.; Kostyushev, D.S.; Chulanov, V.P. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019, 20, 6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.V.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Huang, Y.-H.; Goodell, M.A. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9–histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.A.; Halmai, J.; Le, V.M.; Mackay, J.P.; Farnham, P.J.; Segal, D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.Y.; Zhao, Y.-T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Fukushima, H.S.; Takeda, H.; Nakamura, R. Targeted in vivo epigenome editing of H3K27me3. Epigenet. Chromatin 2019, 12, 17. [Google Scholar] [CrossRef]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Gjaltema, R.A.F.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef]
- Sajwan, S.; Mannervik, M. Gene activation by dCas9-CBP and the SAM system differ in target preference. Sci. Rep. 2019, 9, 18104. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Neumann, M.; Duro, D.I.; Schmid, M. CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 2019, 14, e0222778. [Google Scholar] [CrossRef] [PubMed]
- Paixao, J.F.R.; Gillet, F.-X.; Ribeiro, T.P.; Bournaud, C.; Lourenço-Tessutti, I.T.; Noriega, D.D.; De Melo, B.P.; De Almeida-Engler, J.; Grossi-De-Sa, M.F. Improved drought stress tolerance in Arabidopsis by CRISPR/dCas9 fusion with a Histone AcetylTransferase. Sci. Rep. 2019, 9, 8080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.D.; Abdulrazak, A.; Sato, H.; Maqbool, S.B.; Suzuki, M.; Greally, J.M.; Simões-Pires, C.A. A Cellular Stress Response Induced by the CRISPR-dCas9 Activation System Is Not Heritable Through Cell Divisions. CRISPR J. 2020, 3, 188–197. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-based Photoactivatable Transcription System. Chem. Biol. 2015, 22, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.L.; Mariano, N.C.; Bermudez, A.; Arruda, N.L.; Wu, F.; Luo, Y.; Shankar, G.; Jia, L.; Chen, H.; Hu, J.-F.; et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun. 2017, 8, 15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.L.; Chang, E.Y.; Mariano, N.C.; Bermudez, A.; Arruda, N.L.; Wu, F.; Luo, Y.; Shankar, G.; Huynh, S.K.; Huang, C.-C.; et al. CRISPR-Mediated Reorganization of Chromatin Loop Structure. J. Vis. Exp. 2018, e57457. [Google Scholar] [CrossRef]
- Braun, S.M.G.; Kirkland, J.G.; Chory, E.J.; Husmann, D.; Calarco, J.P.; Crabtree, G.R. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 2017, 8, 560. [Google Scholar] [CrossRef]


{kind=link}
| Compound | Target Origin | Assays in Animals | Assays in Plants | |||||
|---|---|---|---|---|---|---|---|---|
| Observed Effects | Techniques Used | Refs. | Observed Effects | Techniques Used | Refs. | |||
| Inhibitors of HDACs | Trichostatin A (TSA) | HDAC Synthetic | Differentiation of tumor cells in mammalian cell culture and rat mammary cancer Increase histone acetylation | Western blot, Immunoprecipitation, HDAC activity assay, Co-IP analysis | [21,22,23] | Affects global levels of histone acetylation and induces somatic embryogenesis in Arabidopsis Produces doubled haploid in wheat | RT-qPCR, microarrays, HDAC activity assay, fluorescent imaging quantification | [24,25,26,27,28] |
| Sirtinol | Sirtuin-type HDAC Synthetic | Apoptotic and autophagic cell death in MCF-7 human breast cancer cells High inhibitor activity in leukemia cells | Western blot, flow cytometry | [29,30,31] | Impacts shoot and root meristems maintenance, affects body axis formation and vascularization in Arabidopsis | GUS activity measurement, RT-qPCR | [32,33,34,35] | |
| Nicotinamide | Sirtuin-type HDAC Natural product of NADH2 oxidation | Inhibitor of the SIRT1 in vitro. Affects H3K9 acetylation in rat brain cells | Western blot, RT-qPCR, MRI | [36,37] | Alters histone acetylation and induces VIN3 expression in Arabidopsis | RT-qPCR, ChIP-PCR | [38] | |
| Ky-2, Ky-14Ky-9, Ky-72 | HDAC Synthetic | Affects inflammatory response in human macrophages. Enhances H3 acetylation in THP-1 cells | Western blot, RT-qPCR | [39,40] | Enhances high salinity stress tolerance in Arabidopsis and tobacco BY-2 cells through increase in H4 acetylation.Enhances H3 acetylation in Arabidopsis | RT-qPCR, Western blot, fluorescent imaging quantification | [26,41,42] | |
| HC toxin | HDAC From Cochliobolus carbonum | High efficiency in intrahepatic cholangiocarcinoma cells by inhibiting HDAC1 in a post-transcriptional manner | Flow cytometry analysis, Western blot, RT-qPCR, immunofluorescence | [43,44] | Leads to hyperacetylation of H4 and all isoforms of H3 histones in maize | HDAC activity assay, Western blot | [45] | |
| Nitrtic oxide | HDAC From bacteria (e.g., Moraxella catarrhalis) | Suppresses the serum-induced histone acetylation and enhanced histone deacetylase (HDAC) activity in human umbilical vein ECs (HUVECs) | Enzymatic activity assay, Western blot, immunofluorescence | [46,47] | Leads to hyperacetylation at specific genes in Arabidopsis | HDAC activity assay, Western blot, ChIPseq | [48] | |
| Depudecin | HDAC From Alternaria brassicicola | Morphological reversion of NIH 3T transformed fibroblasts | Trapoxin Binding Assay, histone acetylation assays | [49] | Has a minor role in virulence on Brassica oleracea but not in Arabidopsis | Discussed in the text but no supporting results | [50] | |
| Inhibitors of HATs | Compound C646, C107 | p300, acetyl-CoA competitor Synthetic | Inhibits histone H4 acetylation in animal cells | FRET, Western blot, RT-qPCR, radioactive assays of acetylation | [51] | Reduces the level of H3K9 acetylation in tobacco BY-2 cells and Arabidopsis | Western blot, fluorescent imaging quantification | [26,52] |
| Curcumin (diféruloylméthane) | p300 From Curcuma longa | Inhibits histone acetylation in mammalian cells | Filter binding, fluorography, Hoechst staining Western blot | [53] | Affects H3 and H4 acetylation in maize and Arabidopsis | ChIP-PCR, RT-PCR, Western blot | [54,55] | |
| MC1626, Anacardic acid (quinolic analogue of anacardic acid) | p300 HAT From cashew nut (Anacardium occidentale) | Reversibly and noncompetitively inhibits HAT activity in Plasmodium falciparum Inhibits the H3 acetylation level in yeast | Western blot, ChIP-PCR, RT-qPCR, microarray | [56,57] | Inhibits UV-B induced deacetylation of H3K9 and H3K14 and the specific induction of UVR8-regulated genes in Arabidopsis | ChIP-PCR, RT-qPCR | [58] | |
| MB-3, Gamma-butyrolactone | mammalian GCN5 Synthetic | Inactivates the GCN5 in mammalian cell lines | RT-qPCR, Western blot, ChIP-PCR | [59] | Causes a decrease in H3K9 and H3K14 acetylation in Arabidopsis | ChIP-PCR, RT-PCR, Western blot, ChIPseq, RNAseq | [52,60] | |
| Inhibitors of HMTs | RDS 3434, 1,5-bis (3-bromo-4-methoxyphenyl) penta-1,4-dien-3-one compound | SAM competitor for EZH2 binding Synthetic | EZH2 inhibitor in human leukemia cells | Western blot, qRT-PCR | [61] | Causes a decrease in H3K27me3 in a dose-dependent manner in Arabidopsis | RT-qPCR, Western blot | [62] |
| BIX-01294 (diazepin-quinazolin-amine derivative) | HMT Synthetic | Affects the H3K9me2 in mammalian cell lines | Immuno-cytochemical assay, RT-qPCR, Western blot, ChIP | [63] | Affects H3K9 methylation, promotes cell reprogramming, totipotency and embryogenesis in Brassica napus and Hordeum vulgare | Colorimetric histone methylation assay, immunofluorescence, RT-qPCR | [64] | |
| Disrupters of methyl supply | DHPA (dihydroxypropyladenine) | SAH hydrolase inhibitor Synthetic | Inhibition of SAHH induced hypomethylation in the p66shc gene promoter in mice | Western blot | [65] | Induces decrease in DNA methylation and H3K9me2 and releases silenced transgenes in Arabidopsis and tobacco | ChIP-PCR, RT-qPCR | [66,67] |
| Sulfamethazine (SMZ) | PABA competitive antagonist Synthetic | Reduces levels of DNA methylation and H3K9me2 in Arabidopsis | ChIP-PCR, RT-qPCR, bisulfite sequencing | [68] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fal, K.; Tomkova, D.; Vachon, G.; Chabouté, M.-E.; Berr, A.; Carles, C.C. Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants. Int. J. Mol. Sci. 2021, 22, 512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020512
Fal K, Tomkova D, Vachon G, Chabouté M-E, Berr A, Carles CC. Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants. International Journal of Molecular Sciences. 2021; 22(2):512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020512
Chicago/Turabian StyleFal, Kateryna, Denisa Tomkova, Gilles Vachon, Marie-Edith Chabouté, Alexandre Berr, and Cristel C. Carles. 2021. "Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants" International Journal of Molecular Sciences 22, no. 2: 512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020512