Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
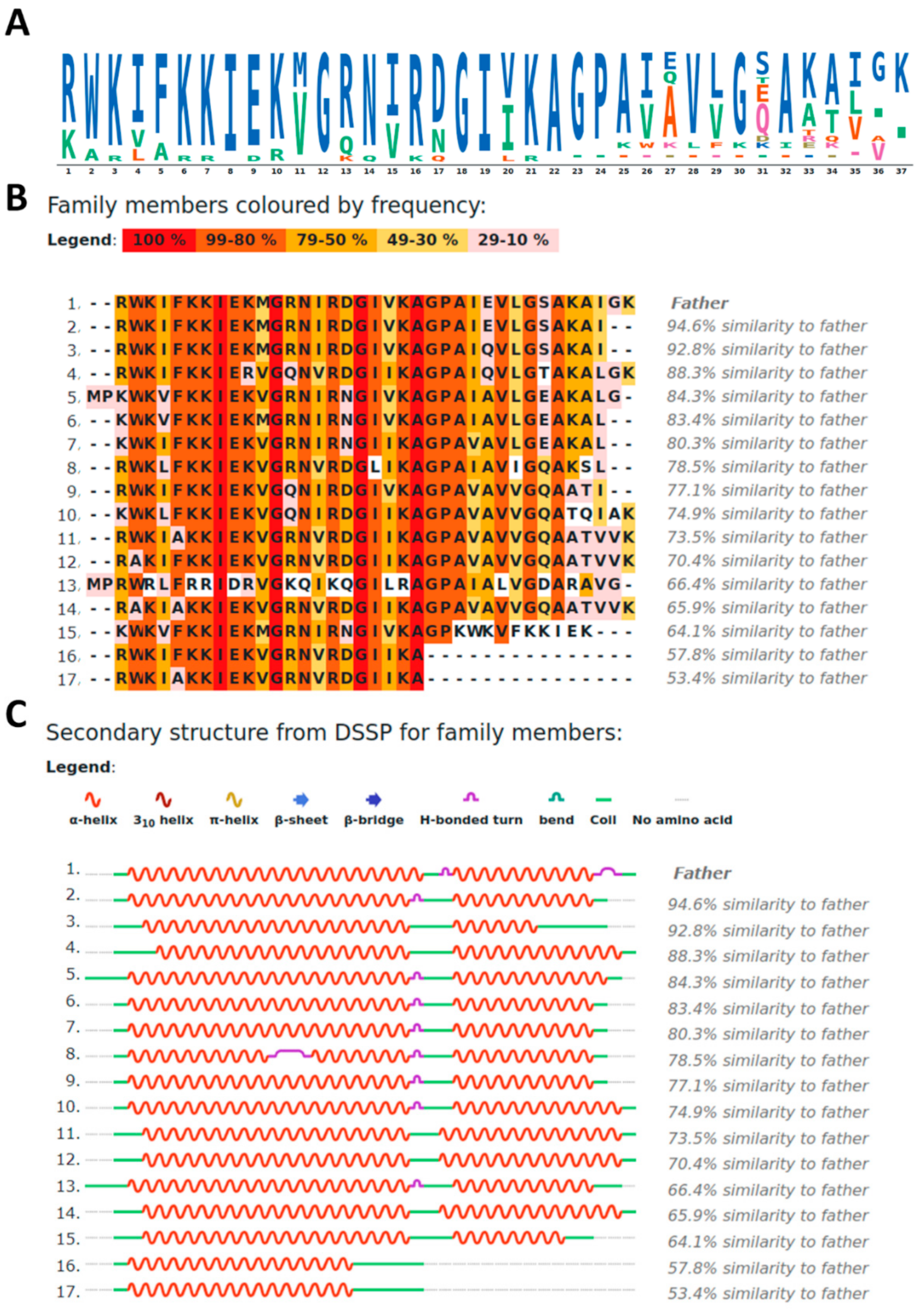
2.1. Property-Sequence Alignment of CXJ
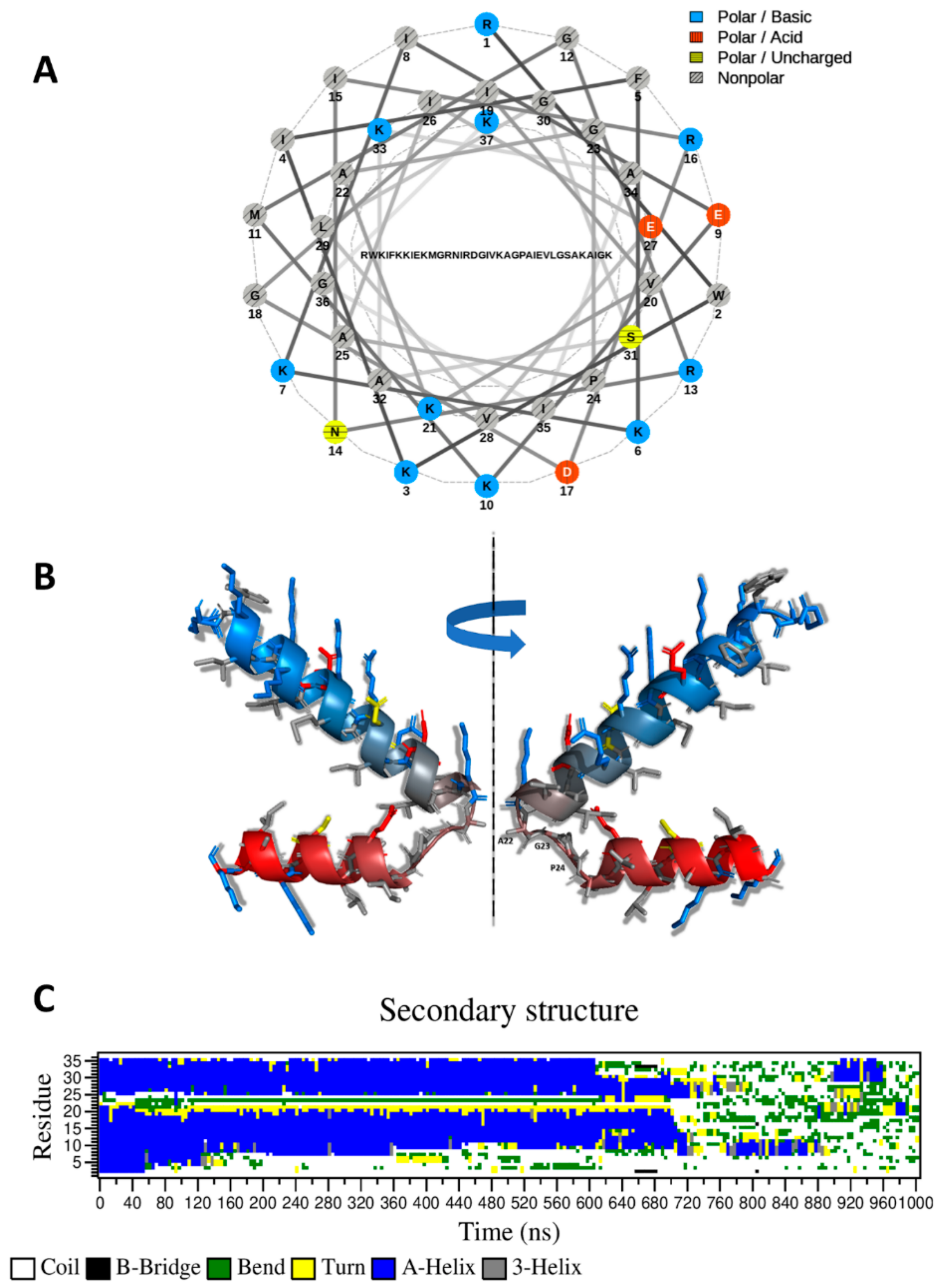
2.2. CXJ Can Form an Amphipathic Helix but Remains Unstructured in Solution
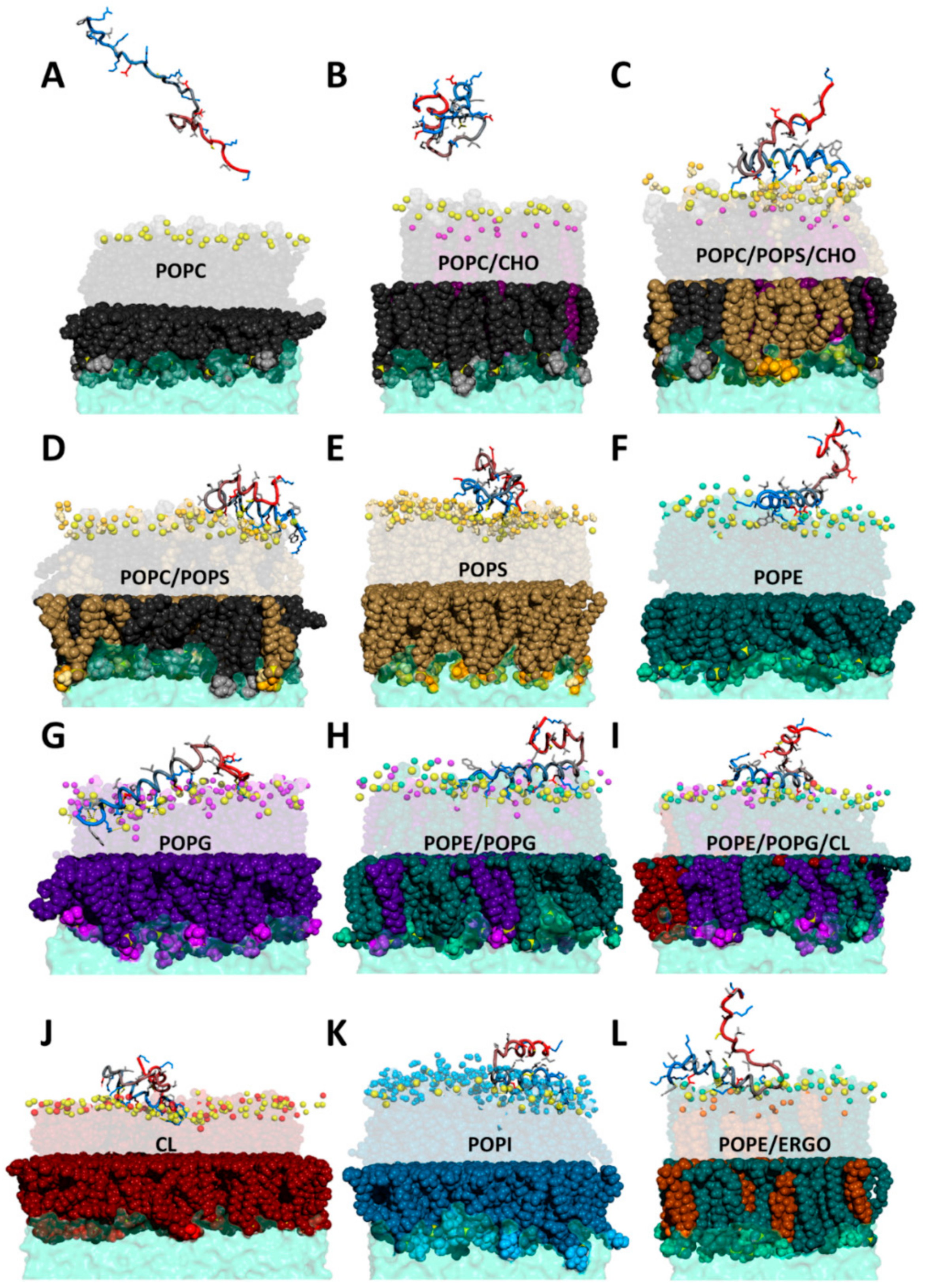
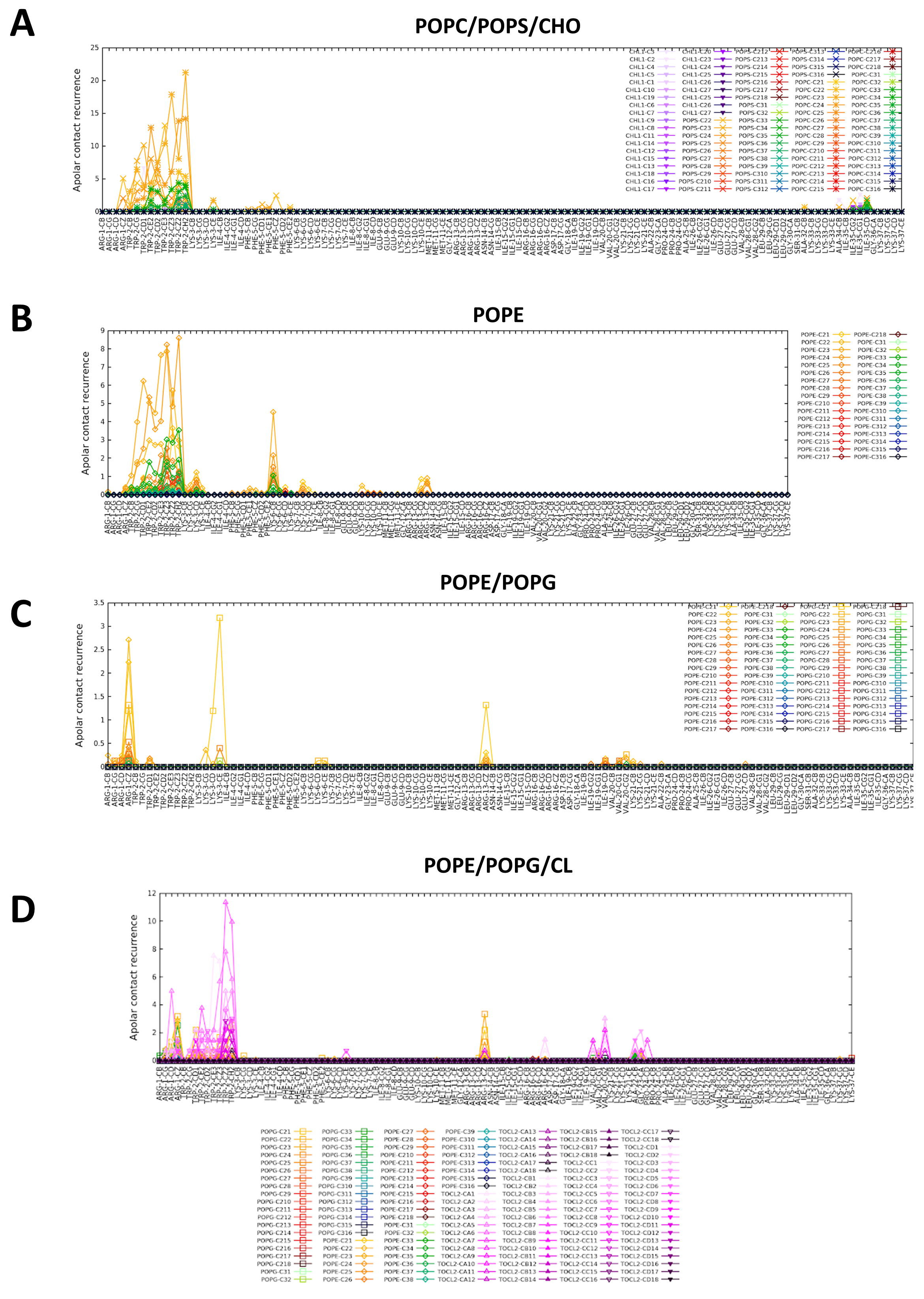
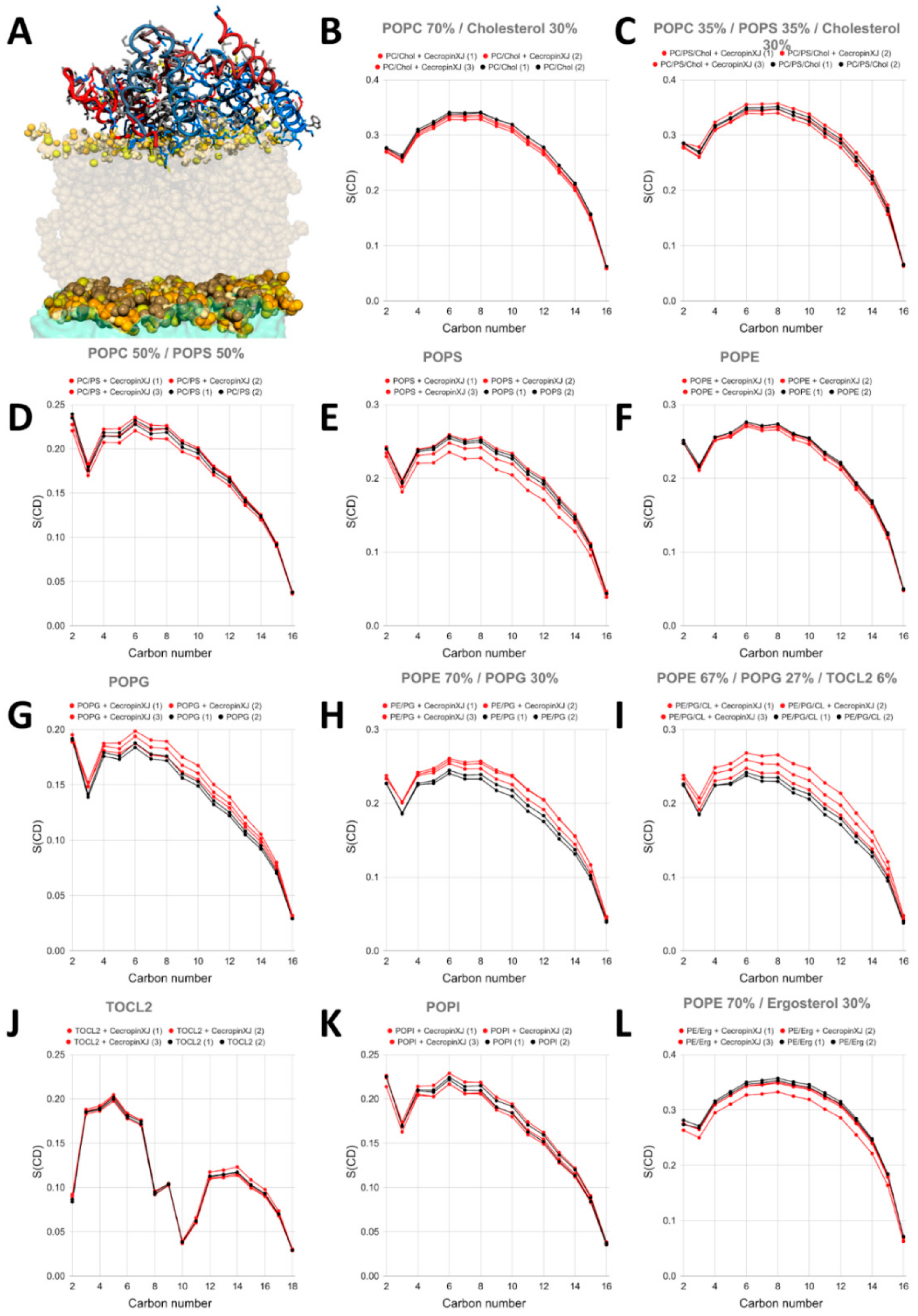
2.3. CXJ Does Not Interact with Phosphatidylcholine Membranes
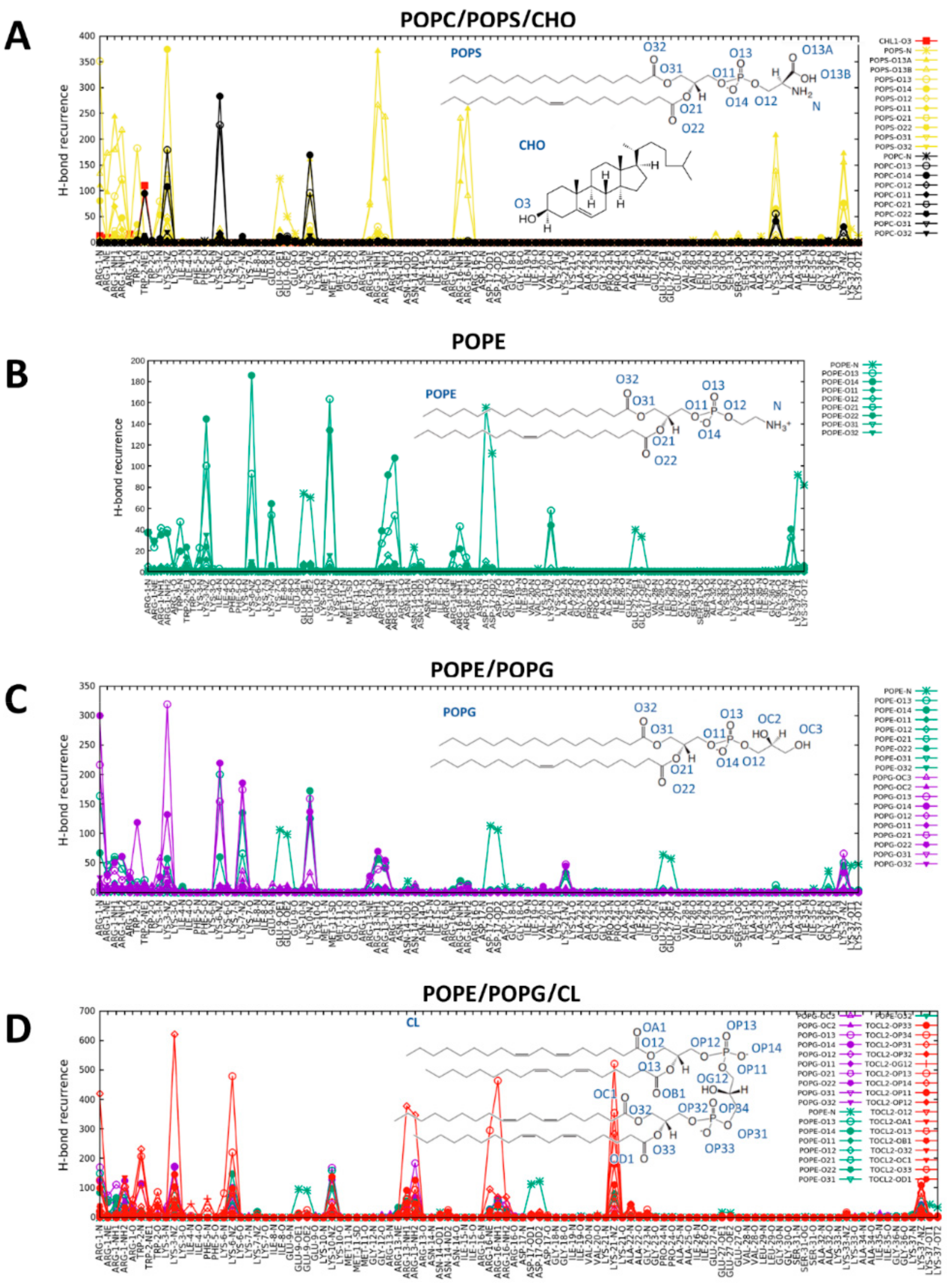
2.4. CXJ Specifically Recognises PS and PE Headgroups, Exposed in Apoptotic Cancer Cells
2.4.1. The Effect of CXJ on PS-Containing Membranes
2.4.2. The Effect of CXJ on PE Membranes
2.5. CXJ Interacts with Bacterial Biomimetic Membranes and Each of Their Pure Components
2.5.1. The Effect of CXJ on PG Membranes
2.5.2. The Effect of CXJ on PE/PG Mixtures Frequently Found in Bacterial Membranes
2.5.3. The Effect of CXJ on PE/PG/CL Mixtures and Pure CL Membranes
2.6. CXJ Interacts with Components of the Fungal Membrane
2.7. The Effect of Concentration in the Activity of CXJ
2.8. The Effect of C-Terminal Amidation in the Activity of CXJ
2.9. Final Remarks on the Internalization of CPPs
3. Materials and Methods
3.1. Sequence Alignment by ADAPTABLE Web Server
3.2. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xia, L.; Zhang, F.; Liu, Z.; Ma, J.; Yang, J. Expression and characterization of cecropinXJ, a bioactive antimicrobial peptide from (Bombycidae, Lepidoptera) in Escherichia coli. Exp. Ther. Med. 2013, 5, 1745–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Liu, Z.; Ma, J.; Sun, S.; Yang, J.; Zhang, F. Expression, purification and characterization of cecropin antibacterial peptide from Bombyx mori in Saccharomyces cerevisiae. Protein Exp. Purif. 2013, 90, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, J.; Li, J.; Xia, L.; Yang, J.; Sun, S.; Ma, J.; Zhang, F. A potential food biopreservative, CecXJ-37N, non-covalently intercalates into the nucleotides of bacterial genomic DNA beyond membrane attack. Food Chem. 2017, 217, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wu, Y.; Kang, S.; Ma, J.; Yang, J.; Zhang, F. CecropinXJ, a silkworm antimicrobial peptide, induces cytoskeleton disruption in esophageal carcinoma cells. Acta Biochim. Biophys. Sin. 2014, 46, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Xia, L.-J.; Li, J.-Y.; Zhang, F.-C. CecropinXJ inhibits the proliferation of human gastric cancer BGC823 cells and induces cell death in vitro and in vivo. Int. J. Oncol. 2015, 46, 2181–2193. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Wu, Y.; Ma, J.I.; Yang, J.; Zhang, F. The antibacterial peptide from cecropinXJ induced growth arrest and apoptosis in human hepatocellular carcinoma cells. Oncol. Lett. 2016, 12, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Kalmouni, M.; Al-Hosani, S.; Magzoub, M. Cancer targeting peptides. Cell. Mol. Life Sci. 2019, 76, 2171–2183. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Translocation Mechanisms of CPPs. In CPP, Cell-Penetrating Peptides; Springer: Singapore, 2019; pp. 359–394. [Google Scholar]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Gao, Y.; Qi, Y.; Chen, L.; Ma, Y.; Li, Y. Peptide-based cancer therapy: Opportunity and challenge. Cancer Lett. 2014, 351, 13–22. [Google Scholar] [CrossRef]
- Constance, J.E.; Lim, C.S. Targeting malignant mitochondria with therapeutic peptides. Ther. Deliv. 2012, 3, 961–979. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, F.R.; di Pietro, M.; Secrier, M.; Moehler, M.; Goepfert, K.; Lima, S.S.C.; Pinto, L.F.R.; Hendricks, D.; Parker, M.I.; Herceg, Z. Molecular landscape of esophageal cancer: Implications for early detection and personalized therapy. Ann. N. Y. Acad. Sci. 2018, 1434, 342–359. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef]
- Eslick, G.D. Epidemiology of esophageal cancer. Gastroenterol. Clin. N. Am. 2009, 38, 17–25, vii. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C. Barrett’s oesophagus and oesophageal adenocarcinoma: How does acid interfere with cell proliferation and differentiation? Gut 2005, 54, i21–i26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.E.; Grandis, J.R.; Ko, A.H. New Strategies in Esophageal Carcinoma: Translational Insights from Signaling Pathways and Immune Checkpoints. Clin. Cancer Res. 2016, 22, 4283–4290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, H. Ivor Lewis Esophagectomy: A Curative Operation for Esophageal Cancer. In Illustrated Abdominal Surgery; Shinohara, H., Ed.; Springer: Singapore, 2020; pp. 133–185. ISBN 9789811517952. [Google Scholar]
- Pericay, C.; Macías-Declara, I.; Arrazubi, V.; Vilà, L.; Marín, M. Treatment in esophagogastric junction cancer: Past, present and future. Cir. Esp. 2019, 97, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Sanz Álvarez, L.; Turienzo Santos, E.; Rodicio Miravalles, J.L.; Moreno Gijón, M.; Amoza Pais, S.; Sanz Navarro, S.; Rizzo Ramos, A. Evidence in follow-up and prognosis of esophagogastric junction cancer. Cir. Esp. 2019, 97, 465–469. [Google Scholar] [CrossRef]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Seyfried, T.; Alfarouk, K.O.; Da Veiga Moreira, J.; Fais, S. Out of Warburg effect: An effective cancer treatment targeting the tumor specific metabolism and dysregulated pH. Semin. Cancer Biol. 2017, 43, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-W.; Guo, Z.-J.; Chu, A.-L.; Zhang, Y.; Liang, B.; Guo, X.; Chai, T.; Song, R.; Hou, G.; Yuan, J.-J. High incidence of coding gene mutations in mitochondrial DNA in esophageal cancer. Mol. Med. Rep. 2017, 16, 8537–8541. [Google Scholar] [CrossRef] [Green Version]
- Keles, M.; Sahin, I.; Kurt, A.; Bozoglu, C.; Simsek, G.; Kabalar, E.; Tatar, A. Mitochondrial DNA deletions in patients with esophagitis, Barrett’s esophagus, esophageal adenocarcinoma and squamous cell carcinoma. Afr. Health Sci. 2019, 19, 1671. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, N.J.; Feighery, R.; Picardo, S.L.; Lynam-Lennon, N.; Biniecka, M.; McGarrigle, S.A.; Phelan, J.J.; MacCarthy, F.; O’Toole, D.; Fox, E.J.; et al. Changes in mitochondrial stability during the progression of the Barrett’s esophagus disease sequence. BMC Cancer 2016, 16, 497. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Guan, R.; Liao, X.; Ouyang, C.; Rees, T.W.; Liu, J.; Chen, Y.; Ji, L.; Chao, H. A mitochondria-targeting dinuclear Ir–Ru complex as a synergistic photoactivated chemotherapy and photodynamic therapy agent against cisplatin-resistant tumour cells. Chem. Commun. 2019, 55, 12547–12550. [Google Scholar] [CrossRef]
- Galluzzi, L.; Larochette, N.; Zamzami, N.; Kroemer, G. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene 2006, 25, 4812–4830. [Google Scholar] [CrossRef] [Green Version]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc. Chem. Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Ayyalusamy, R. Studies on anticancer activities of antimicrobial peptides. Biochim. Et Biophys. Acta (Bba) Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.B.; Kelley, S.O. Peptide-chlorambucil conjugates combat pgp-dependent drug efflux. ACS Med. Chem. Lett. 2011, 2, 419–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.B.; Pereira, M.P.; Mourtada, R.; Gronda, M.; Horton, K.L.; Hurren, R.; Minden, M.D.; Schimmer, A.D.; Kelley, S.O. Rerouting chlorambucil to mitochondria combats drug deactivation and resistance in cancer cells. Chem. Biol. 2011, 18, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Jean, S.R.; Pereira, M.P.; Kelley, S.O. Structural modifications of mitochondria-targeted chlorambucil alter cell death mechanism but preserve MDR evasion. Mol. Pharm. 2014, 11, 2675–2682. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, T.; Tao, J.; Wan, G.; Zhao, H. Co-delivery of paclitaxel and indocyanine green by PEGylated graphene oxide: A potential integrated nanoplatform for tumor theranostics. RSC Adv. 2016, 6, 15460–15468. [Google Scholar] [CrossRef]
- Xia, L.-J.; Wu, Y.-L.; Ma, J.; Zhang, F.-C. Therapeutic effects of antimicrobial peptide on malignant ascites in a mouse model. Mol. Med. Rep. 2018, 17, 6245–6252. [Google Scholar] [CrossRef] [Green Version]
- Brady, D.; Grapputo, A.; Romoli, O.; Sandrelli, F. Insect Cecropins, Antimicrobial Peptides with Potential Therapeutic Applications. Int. J. Mol. Sci. 2019, 20, 5862. [Google Scholar] [CrossRef] [Green Version]
- Lei, E.K.; Pereira, M.P.; Kelley, S.O. Tuning the intracellular bacterial targeting of peptidic vectors. Angew. Chem. Int. Ed Engl. 2013, 52, 9660–9663. [Google Scholar] [CrossRef]
- Pereira, M.P.; Shi, J.; Kelley, S.O. Peptide Targeting of an Antibiotic Prodrug toward Phagosome-Entrapped Mycobacteria. ACS Infect. Dis. 2015, 1, 586–592. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Doherty, T.; Waring, A.J.; Ruchala, P.; Hong, M. Roles of Arginine and Lysine Residues in the Translocation of a Cell-Penetrating Peptide from13C,31P, and19F Solid-State NMR. Biochemistry 2009, 48, 4587–4595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Martín, F.; Annaval, T.; Buchoux, S.; Sarazin, C.; D’Amelio, N. ADAPTABLE: A comprehensive web platform of antimicrobial peptides tailored to the user’s research. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Xu, D.; Yang, J.; Walker, S.; Zhang, Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci. Rep. 2013, 3, 2619. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5–8. [Google Scholar] [CrossRef]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P.S. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef] [Green Version]
- Mól, A.R.; Castro, M.S.; Fontes, W. NetWheels: A web application to create high quality peptide helical wheel and net projections. BioRxiv 2018, 416347. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, S. Calorimetric and molecular mechanics studies of the thermotropic phase behavior of membrane phospholipids. Biochim. Biophys. Acta 1999, 1422, 273–307. [Google Scholar] [CrossRef]
- Lamy-Freund, M.T.; Riske, K.A. The peculiar thermo-structural behavior of the anionic lipid DMPG. Chem. Phys. Lipids 2003, 122, 19–32. [Google Scholar] [CrossRef]
- Tincho, M.B.; Morris, T.; Meyer, M.; Pretorius, A. Antibacterial Activity of Rationally Designed Antimicrobial Peptides. Int. J. Microbiol. 2020, 2020, 2131535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachowski, A. Phospholipids in animal eukaryotic membranes: Transverse asymmetry and movement. Biochem. J. 1993, 294, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, A.J.; Zwaal, R.F.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; van Deenen, L.L. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta 1973, 323, 178–193. [Google Scholar] [CrossRef]
- Sok, M.; Sentjurc, M.; Schara, M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999, 139, 215–220. [Google Scholar] [CrossRef]
- Zalba, S.; Ten Hagen, T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Arashiki, N.; Saito, M.; Koshino, I.; Kamata, K.; Hale, J.; Mohandas, N.; Manno, S.; Takakuwa, Y. An Unrecognized Function of Cholesterol: Regulating the Mechanism Controlling Membrane Phospholipid Asymmetry. Biochemistry 2016, 55, 3504–3513. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, A.B.; Michalak, K. Lipids as a target for drugs modulating multidrug resistance of cancer cells. Curr. Drug Targets 2003, 4, 23–30. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharm. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, Y.; Tu, Y.; Freter, C.E. Breast Cancer and Lipid Metabolism. In Lipidomics in Health & Disease; Springer: Singapore, 2018; pp. 113–135. [Google Scholar]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Romoli, O.; Mukherjee, S.; Mohid, S.A.; Dutta, A.; Montali, A.; Franzolin, E.; Brady, D.; Zito, F.; Bergantino, E.; Rampazzo, C.; et al. Enhanced Silkworm Cecropin B Antimicrobial Activity against from Single Amino Acid Variation. ACS Infect. Dis. 2019, 5, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Suttmann, H.; Retz, M.; Paulsen, F.; Harder, J.; Zwergel, U.; Kamradt, J.; Wullich, B.; Unteregger, G.; Stöckle, M.; Lehmann, J. Antimicrobial peptides of the Cecropin-family show potent antitumor activity against bladder cancer cells. BMC Urol. 2008, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Kosuge, M.; Tadokoro, A.; Sugiura, Y.; Nishi, M.; Kawata, M.; Sakai, N.; Matile, S.; Futaki, S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, G.; Bang, E.-K.; Montenegro, J.; Matile, S. Cellular uptake: Lessons from supramolecular organic chemistry. Chem. Commun. 2015, 51, 10389–10402. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Wereszczynski, J. Probing the disparate effects of arginine and lysine residues on antimicrobial peptide/bilayer association. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Fuselier, T.; Wimley, W.C. Spontaneous Membrane Translocating Peptides: The Role of Leucine-Arginine Consensus Motifs. Biophys. J. 2017, 113, 835–846. [Google Scholar] [CrossRef]
- LeBarron, J.; London, E. Effect of lipid composition and amino acid sequence upon transmembrane peptide-accelerated lipid transleaflet diffusion (flip-flop). Biochim. Biophys. Acta 2016, 1858, 1812–1820. [Google Scholar] [CrossRef]
- Rydberg, H.A.; Matson, M.; Amand, H.L.; Esbjörner, E.K.; Nordén, B. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry 2012, 51, 5531–5539. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [Green Version]
- Stafford, J.H.; Thorpe, P.E. Increased exposure of phosphatidylethanolamine on the surface of tumor vascular endothelium. Neoplasia 2011, 13, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Emoto, K.; Toyama-Sorimachi, N.; Karasuyama, H.; Inoue, K.; Umeda, M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res. 1997, 232, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; de Graaf, P.W.; Minsky, B.D.; Obertop, H.; Glonek, T. Esophageal cancer phospholipid characterization by 31P NMR. NMR Biomed. 1993, 6, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, D.A.; Harris, F.; Mura, M.; Dennison, S.R. The increasing role of phosphatidylethanolamine as a lipid receptor in the action of host defence peptides. Prog. Lipid Res. 2015, 59, 26–37. [Google Scholar] [CrossRef]
- Marconescu, A.; Thorpe, P.E. Coincident exposure of phosphatidylethanolamine and anionic phospholipids on the surface of irradiated cells. Biochim. Biophys. Acta 2008, 1778, 2217–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggs, J.M. Lipid intermolecular hydrogen bonding: Influence on structural organization and membrane function. Biochim. Biophys. Acta 1987, 906, 353–404. [Google Scholar] [CrossRef]
- Hübner, W.; Blume, A. Interactions at the lipid–water interface. Chem. Phys. Lipids 1998, 96, 99–123. [Google Scholar] [CrossRef]
- Liu, F.; Ruthven, N.A.; Hodges, R.S.; McElhaney, R.N. Effect of Variations in the Structure of a Polyleucine-Based α-Helical Transmembrane Peptide on Its Interaction with Phosphatidylethanolamine Bilayers. Biophys. J. 2004, 87, 2470–2482. [Google Scholar] [CrossRef] [Green Version]
- Galanth, C.; Abbassi, F.; Lequin, O.; Ayala-Sanmartin, J.; Ladram, A.; Nicolas, P.; Amiche, M. Mechanism of antibacterial action of dermaseptin B2: Interplay between helix-hinge-helix structure and membrane curvature strain. Biochemistry 2009, 48, 313–327. [Google Scholar] [CrossRef]
- Komaratat, P.; Kates, M. The lipid composition of a halotolerant species of Staphylococcus epidermidis. Biochim. Biophys. Acta 1975, 398, 464–484. [Google Scholar] [CrossRef]
- Benamara, H.; Rihouey, C.; Jouenne, T.; Alexandre, S. Impact of the biofilm mode of growth on the inner membrane phospholipid composition and lipid domains in Pseudomonas aeruginosa. Biochim. Biophys. Acta 2011, 1808, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopalco, P.; Stahl, J.; Annese, C.; Averhoff, B.; Corcelli, A. Identification of unique cardiolipin and monolysocardiolipin species in Acinetobacter baumannii. Sci. Rep. 2017, 7, 2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, G.F. Lipids of Salmonella typhimurium and Escherichia coli: Structure and Metabolism. J. Bacteriol. 1968, 95, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobby, C.R.; Herndon, J.L.; Morrow, C.A.; Peters, R.E.; Symes, S.J.K.; Giles, D.K. Exogenous fatty acids alter phospholipid composition, membrane permeability, capacity for biofilm formation, and antimicrobial peptide susceptibility in Klebsiella pneumoniae. Microbiologyopen 2019, 8, e00635. [Google Scholar] [CrossRef]
- Randle, C.L.; Albro, P.W.; Dittmer, J.C. The phosphoglyceride composition of gram-negative bacteria and the changes in composition during growth. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1969, 187, 214–220. [Google Scholar] [CrossRef]
- Bishop, D.G.; Op den Kamp, J.A.; van Deenen, L.L. The distribution of lipids in the protoplast membranes of Bacillus subtilis. A study with phospholipase C and trinitrobenzenesulphonic acid. Eur. J. Biochem. 1977, 80, 381–391. [Google Scholar] [CrossRef]
- Shokri, A.; Larsson, G. Characterisation of the Escherichia coli membrane structure and function during fedbatch cultivation. Microb. Cell Fact. 2004, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Eder, A.E.; Munir, S.A.; Hobby, C.R.; Anderson, D.M.; Herndon, J.L.; Siv, A.W.; Symes, S.J.K.; Giles, D.K. Exogenous polyunsaturated fatty acids (PUFAs) alter phospholipid composition, membrane permeability, biofilm formation and motility in Acinetobacter baumannii. Microbiology 2017, 163, 1626–1636. [Google Scholar] [CrossRef]
- Dicko, A.; Bourque, H.; Pézolet, M. Study by infrared spectroscopy of the conformation of dipalmitoylphosphatidylglycerol monolayers at the air–water interface and transferred on solid substrates. Chem. Phys. Lipids 1998, 96, 125–139. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Lewis, R.N.; McElhaney, R.N. Calorimetric and spectroscopic studies of the thermotropic phase behavior of the n-saturated 1,2-diacylphosphatidylglycerols. Biophys. J. 1997, 72, 779–793. [Google Scholar] [CrossRef] [Green Version]
- Kaznessis, Y.N.; Kim, S.; Larson, R.G. Simulations of zwitterionic and anionic phospholipid monolayers. Biophys. J. 2002, 82, 1731–1742. [Google Scholar] [CrossRef] [Green Version]
- Bartels, E.J.H.; Dekker, D.; Amiche, M. Dermaseptins, Multifunctional Antimicrobial Peptides: A Review of Their Pharmacology, Effectivity, Mechanism of Action, and Possible Future Directions. Front. Pharmacol. 2019, 10, 1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luévano-Martínez, L.A.; Forni, M.F.; dos Santos, V.T.; Souza-Pinto, N.C.; Kowaltowski, A.J. Cardiolipin is a key determinant for mtDNA stability and segregation during mitochondrial stress. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 587–598. [Google Scholar]
- Schlame, M.; Brody, S.; Hostetler, K.Y. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 1993, 212, 727–733. [Google Scholar] [CrossRef]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef]
- Lewis, R.N.A.H.; Ruthven, N.A.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- Hasim, S.; Vaughn, E.N.; Donohoe, D.; Gordon, D.M.; Pfiffner, S.; Reynolds, T.B. Influence of phosphatidylserine and phosphatidylethanolamine on farnesol tolerance in Candida albicans. Yeast 2018, 35, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, N.K.; Sarkar, P.; Gaur, N.A.; Chattopadhyay, A.; Prasad, R. Phosphatidylserine decarboxylase governs plasma membrane fluidity and impacts drug susceptibilities of Candida albicans cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2308–2319. [Google Scholar] [CrossRef]
- Perczyk, P.; Wójcik, A.; Wydro, P.; Broniatowski, M. The role of phospholipid composition and ergosterol presence in the adaptation of fungal membranes to harsh environmental conditions–membrane modeling study. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183136. [Google Scholar] [CrossRef]
- Thevissen, K.; Ferket, K.K.A.; François, I.E.J.A.; Cammue, B.P.A. Interactions of antifungal plant defensins with fungal membrane components. Peptides 2003, 24, 1705–1712. [Google Scholar] [CrossRef]
- Lösel, D.M. Lipids in the Structure and Function of Fungal Membranes. Biochem. Cell Walls Membr. Fungi 1990, 119–133. [Google Scholar]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weete, J.D. Fungal Lipids. In Lipid Biochemistry of Fungi and Other Organisms; Springer: Boston, MA, USA, 1980; pp. 9–48. [Google Scholar]
- Griffiths, R.G.; Dancer, J.; O’Neill, E.; Harwood, J.L. Lipid composition of Botrytis cinerea and inhibition of its radiolabelling by the fungicide iprodione. New Phytol. 2003, 160, 199–207. [Google Scholar] [CrossRef]
- Hammond, K.; Ryadnov, M.G.; Hoogenboom, B.W. Atomic force microscopy to elucidate how peptides disrupt membranes. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183447. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Aisenbrey, C.; Marquette, A.; Bechinger, B. The Mechanisms of Action of Cationic Antimicrobial Peptides Refined by Novel Concepts from Biophysical Investigations. Adv. Exp. Med. Biol. 2019, 1117, 33–64. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B. The SMART model: Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Malanovic, N.; Marx, L.; Blondelle, S.E.; Pabst, G.; Semeraro, E.F. Experimental concepts for linking the biological activities of antimicrobial peptides to their molecular modes of action. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183275. [Google Scholar] [CrossRef]
- Binder, H.; Lindblom, G. Charge-dependent translocation of the Trojan peptide penetratin across lipid membranes. Biophys. J. 2003, 85, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.W. DAPTOMYCIN, its membrane-active mechanism vs. that of other antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183395. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Martinez, G.V.; Brown, M.F.; Ramamoorthy, A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry 2004, 43, 8459–8469. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J.; Smith, I.C.; Dufourcq, J. Molecular details of melittin-induced lysis of phospholipid membranes as revealed by deuterium and phosphorus NMR. Biochemistry 1986, 25, 6448–6455. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Rivel, T.; Ramseyer, C. The influence of curvature on the properties of the plasma membrane. Insights from atomistic molecular dynamics simulations. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Brown, M.F. Curvature forces in membrane lipid-protein interactions. Biochemistry 2012, 51, 9782–9795. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Kim, J.-K.; Jeon, D.; Jeong, K.-W.; Shin, A.; Kim, Y. Functional Roles of Aromatic Residues and Helices of Papiliocin in its Antimicrobial and Anti-inflammatory Activities. Sci. Rep. 2015, 5, 12048. [Google Scholar] [CrossRef] [Green Version]
- Guha, S.; Ghimire, J.; Wu, E.; Wimley, W.C. Mechanistic Landscape of Membrane-Permeabilizing Peptides. Chem. Rev. 2019, 119, 6040–6085. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K. The Mechanism of Membrane Permeabilization by Peptides: Still an Enigma. Aust. J. Chem. 2020, 73, 96. [Google Scholar] [CrossRef]
- Ulmschneider, J.P. Charged Antimicrobial Peptides Can Translocate across Membranes without Forming Channel-like Pores. Biophys. J. 2017, 113, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Sani, M.-A.; Separovic, F. Interaction of cationic antimicrobial peptides from Australian frogs with lipid membranes. Pept. Sci. 2018, 110, e24061. [Google Scholar] [CrossRef]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, D.G. Antimicrobial Peptides (AMPs) with Dual Mechanisms: Membrane Disruption and Apoptosis. J. Microbiol. Biotechnol. 2015, 25, 759–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, A.; Pirri, G.; Bozzi, A.; Di Giulio, A.; Aschi, M.; Rinaldi, A.C. Antimicrobial peptides: Natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 2008, 65, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Choi, H.; Weisshaar, J.C. Melittin-Induced Permeabilization, Re-sealing, and Re-permeabilization of E. coli Membranes. Biophys. J. 2018, 114, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Orsi, M.; Noro, M.G.; Essex, J.W. Dual-resolution molecular dynamics simulation of antimicrobials in biomembranes. J. R. Soc. Interface 2011, 8, 826–841. [Google Scholar] [CrossRef] [Green Version]
- Isralewitz, B.; Baudry, J.; Gullingsrud, J.; Kosztin, D.; Schulten, K. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 2001, 19, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J. Umbrella sampling. Wires Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Domański, J.; Hedger, G.; Best, R.B.; Stansfeld, P.J.; Sansom, M.S.P. Convergence and Sampling in Determining Free Energy Landscapes for Membrane Protein Association. J. Phys. Chem. B 2017, 121, 3364–3375. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wires Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200–212. [Google Scholar] [CrossRef]
- Worch, R.; Dudek, A.; Krupa, J.; Szymaniec, A.; Setny, P. Charged N-terminus of Influenza Fusion Peptide Facilitates Membrane Fusion. Int. J. Mol. Sci. 2018, 19, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta 2016, 1858, 1635–1651. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Jo, S.; Lee, H.S.; Klauda, J.B.; Im, W. CHARMM-GUI micelle builder for pure/mixed micelle and protein/micelle complex systems. J. Chem. Inf. Model. 2013, 53, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.J.; Klauda, J.B.; Sodt, A.J. Simulation Best Practices for Lipid Membranes [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5966. [Google Scholar] [CrossRef]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Buchoux, S. FATSLiM: A fast and robust software to analyze MD simulations of membranes. Bioinformatics 2017, 33, 133–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Janert, P.K. Gnuplot in Action: Understanding Data with Graphs; Manning Publications: Stamford, CT, USA, 2010; ISBN 9781933988399. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. Ccp4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Cassilly, C.; Reynolds, T. PS, It’s Complicated: The Roles of Phosphatidylserine and Phosphatidylethanolamine in the Pathogenesis of Candida albicans and Other Microbial Pathogens. J. Fungi 2018, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konarzewska, P.; Wang, Y.; Han, G.-S.; Goh, K.J.; Gao, Y.-G.; Carman, G.M.; Xue, C. Phosphatidylserine synthesis is essential for viability of the human fungal pathogenCryptococcus neoformans. J. Biol. Chem. 2019, 294, 2329–2339. [Google Scholar] [CrossRef] [Green Version]
- Bukata, L.; Altabe, S.; de Mendoza, D.; Ugalde, R.A.; Comerci, D.J. Phosphatidylethanolamine synthesis is required for optimal virulence of Brucella abortus. J. Bacteriol. 2008, 190, 8197–8203. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Martín, F.; D’Amelio, N. Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study. Int. J. Mol. Sci. 2021, 22, 691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020691
Ramos-Martín F, D’Amelio N. Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study. International Journal of Molecular Sciences. 2021; 22(2):691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020691
Chicago/Turabian StyleRamos-Martín, Francisco, and Nicola D’Amelio. 2021. "Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study" International Journal of Molecular Sciences 22, no. 2: 691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020691