Molecular Properties of Bare and Microhydrated Vitamin B5–Calcium Complexes

 , ,
, ,
Abstract
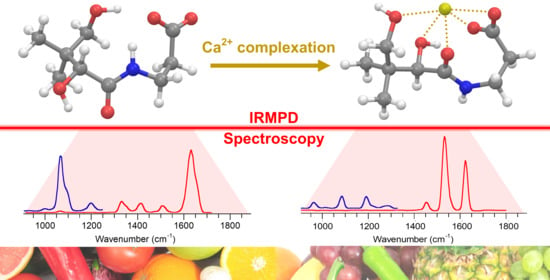
:
1. Introduction
2. Results and Discussion
2.1. Mass Spectra and Fragmentation Channels
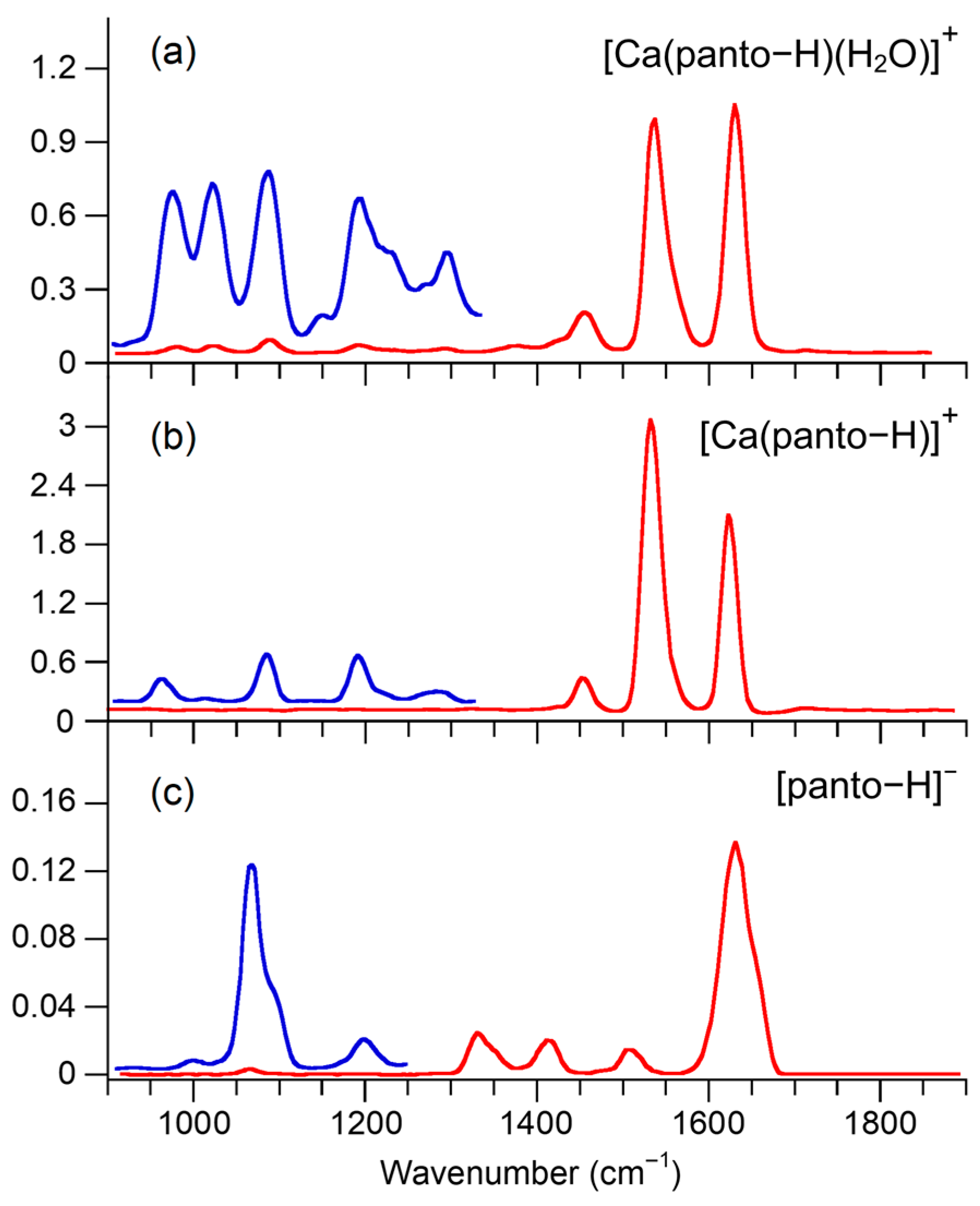
2.2. IRMPD Spectra
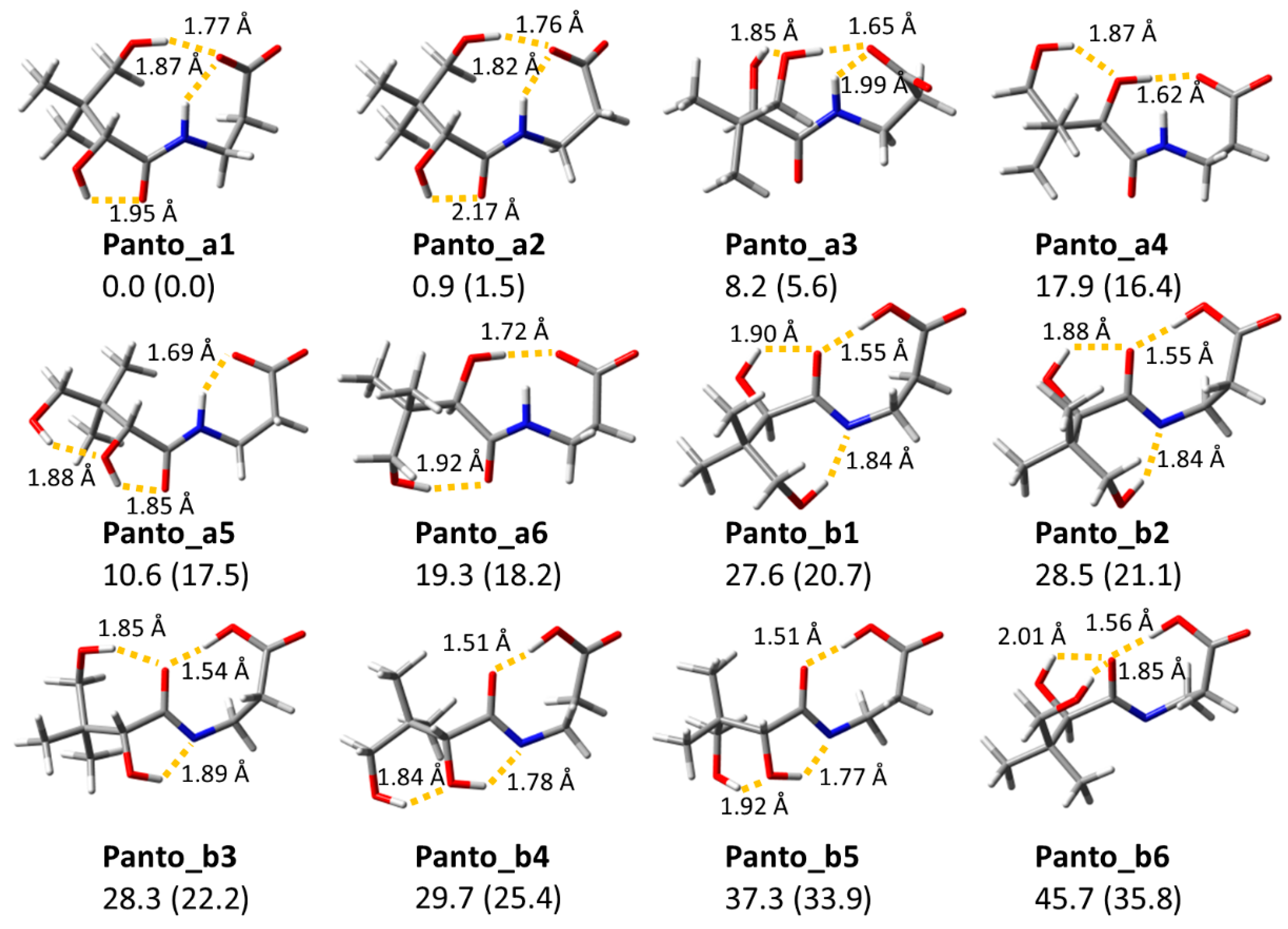
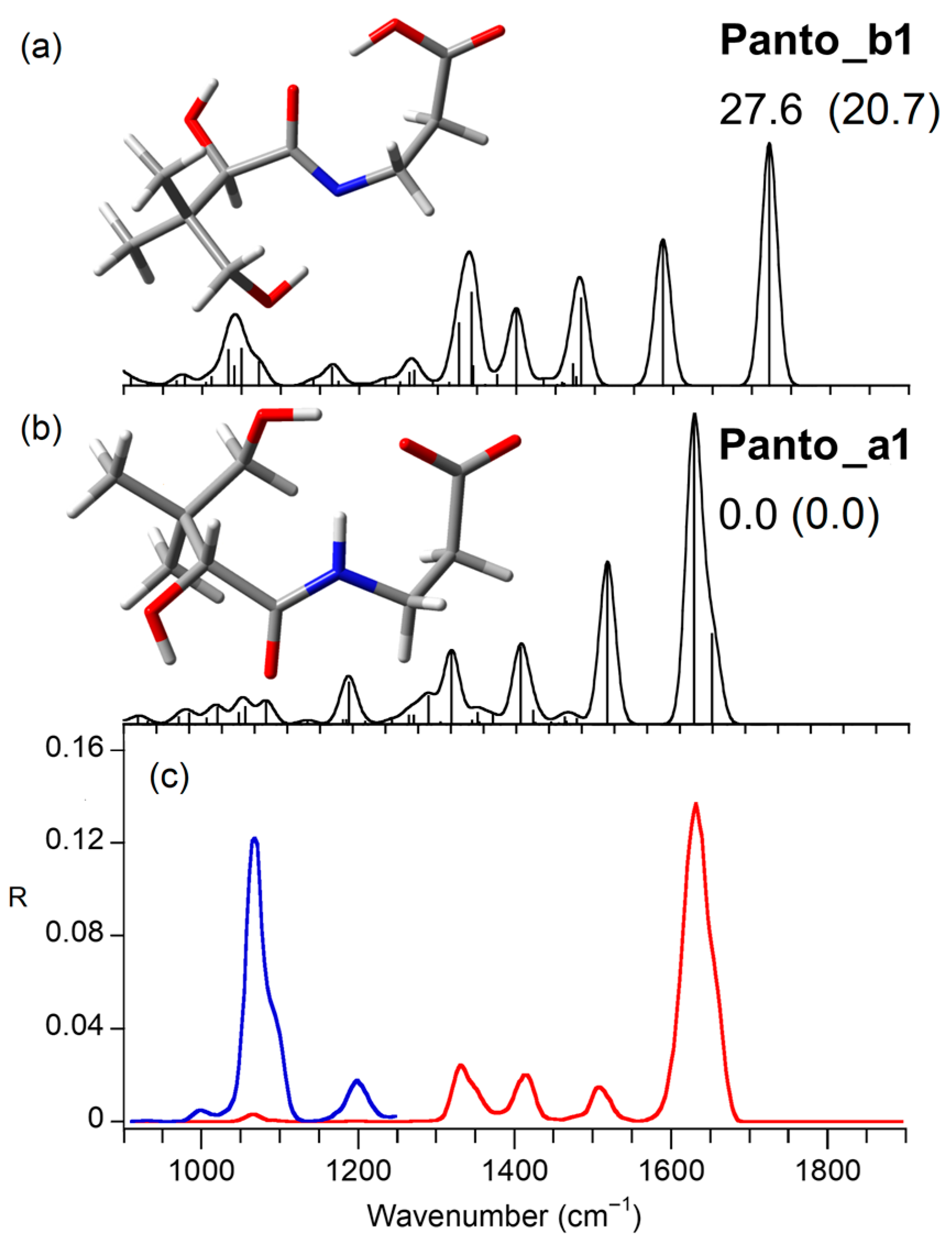
2.3. Computed Structures and Spectral Assignments of [Panto-H]−
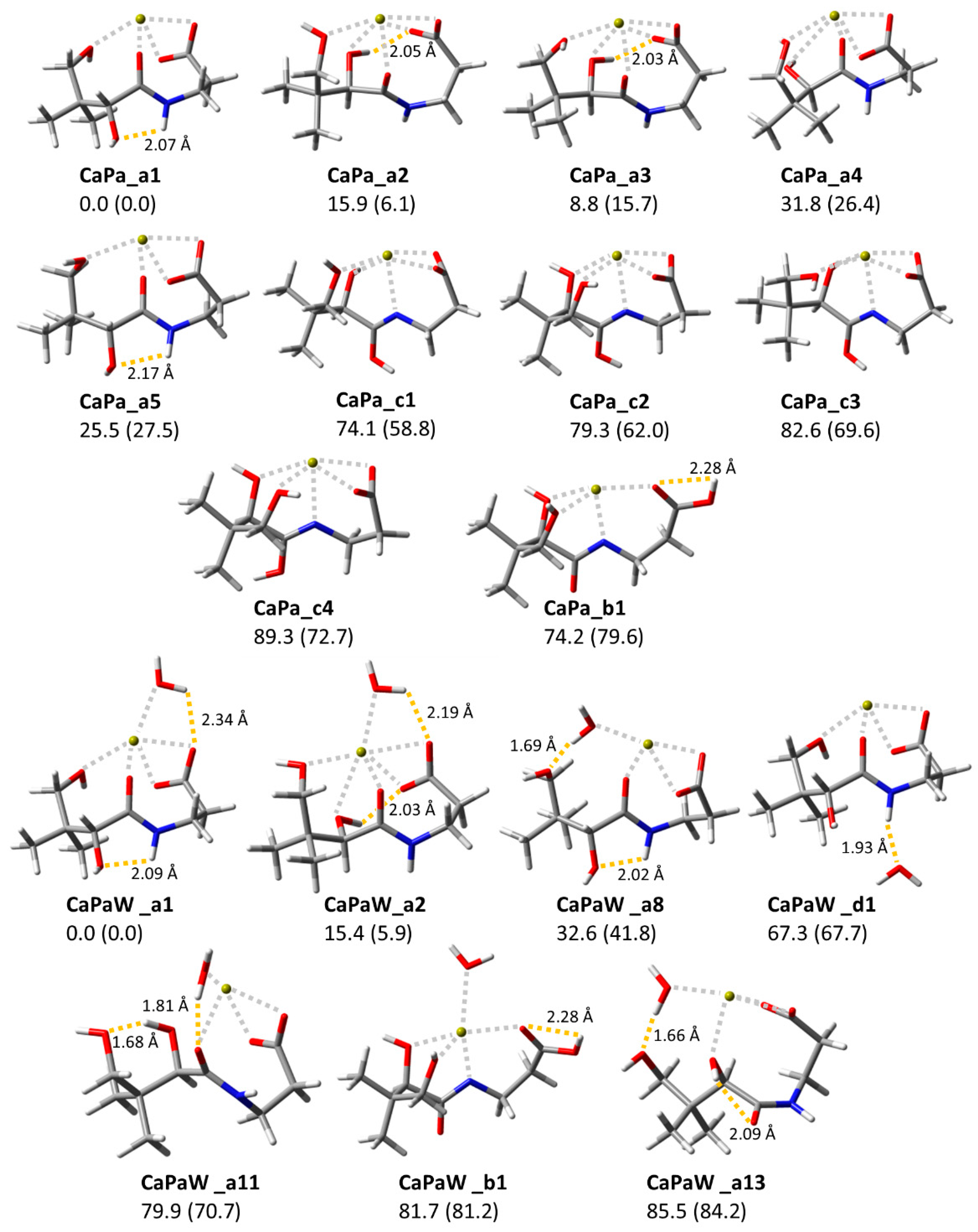
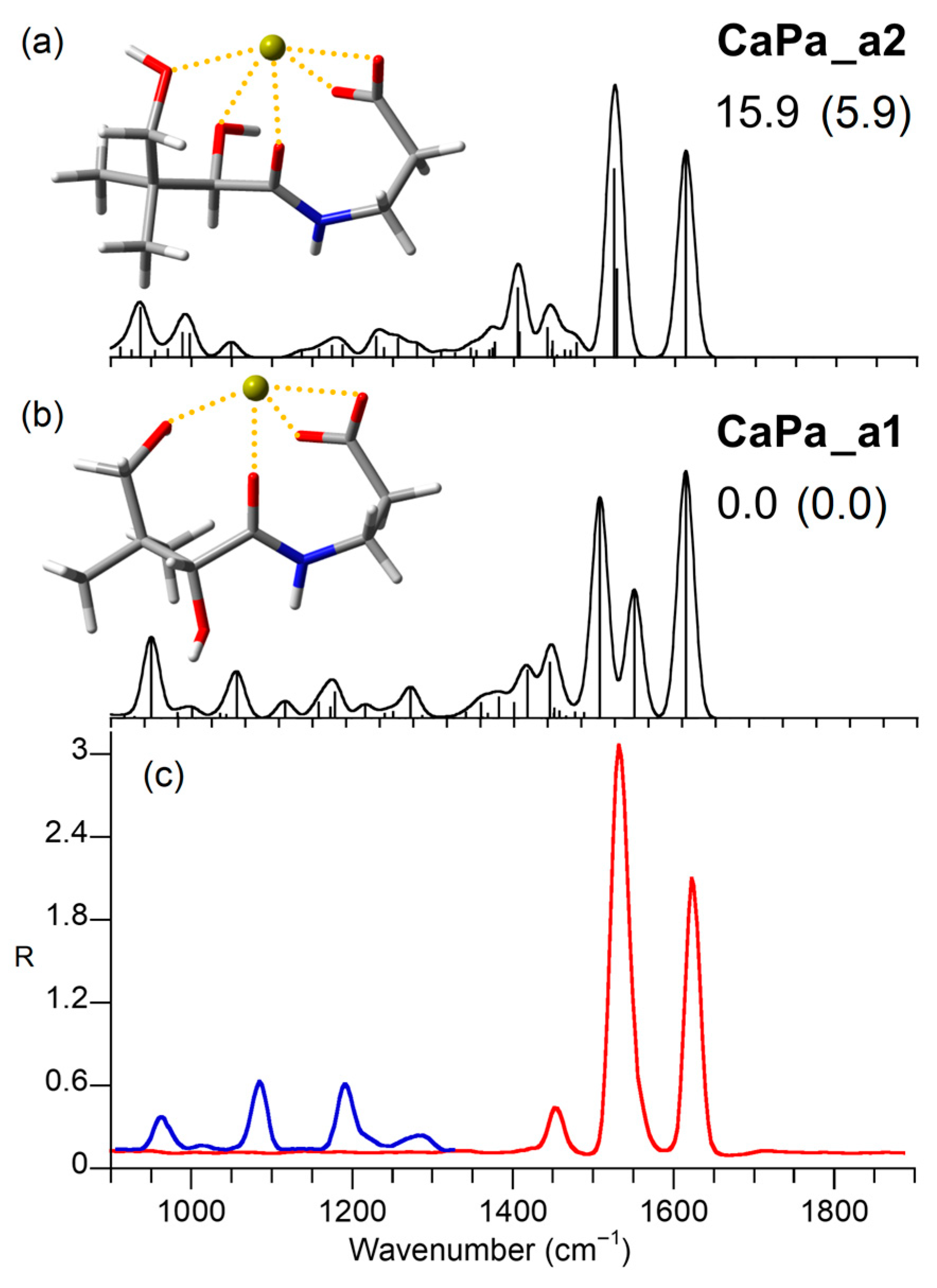
2.4. Computed Structures and Spectral Assignments of [Ca(panto-H)]+
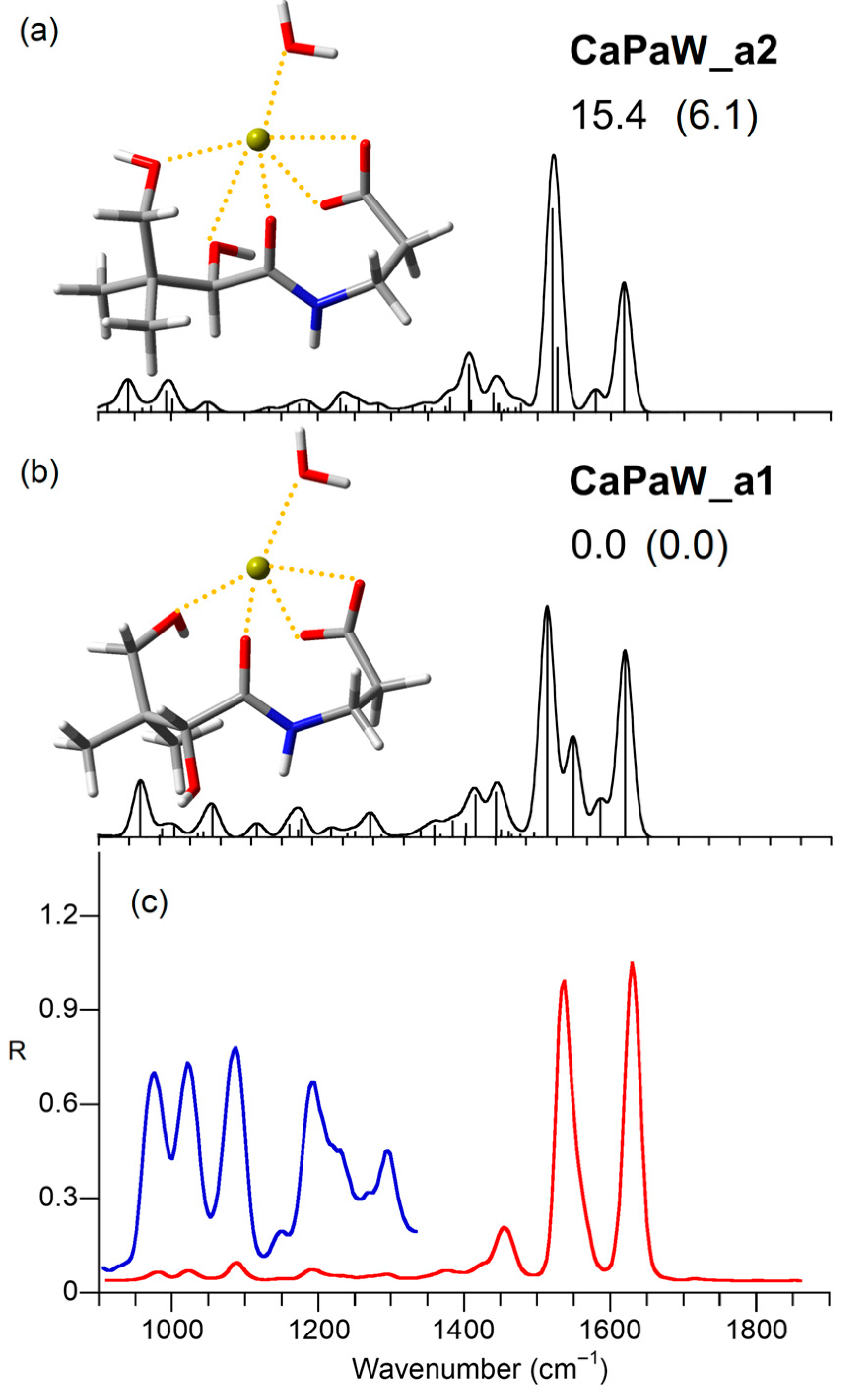
2.5. Computed Structures and Spectral Assignments of [Ca(panto-H)(H2O)]+
3. Materials and Methods
3.1. Sample Preparation
3.2. MS and IRMPD Spectroscopy
3.3. Computational Details/Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gheita, A.A.; Gheita, T.A.; Kenawy, S.A. The potential role of B5: A stitch in time and switch in cytokine. Phyther. Res. 2020, 34, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, M. How Nutrients are Affected by Genetics. In Nutrigenetics; Elsevier: Amsterdam, The Netherlands, 2013; pp. 103–221. [Google Scholar]
- Spry, C.; Macuamule, C.; Lin, Z.; Virga, K.G.; Lee, R.E.; Strauss, E.; Saliba, K.J. Pantothenamides Are Potent, On-Target Inhibitors of Plasmodium falciparum Growth When Serum Pantetheinase Is Inactivated. PLoS ONE 2013, 8, e54974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, M.E.; Smith, A.G.; Abell, C. Biosynthesis of pantothenate. Nat. Prod. Rep. 2004, 21, 695. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, R.; Salvati, A.; Olinga, P.; Boersma, Y.L. Vanin 1: Its Physiological Function and Role in Diseases. Int. J. Mol. Sci. 2019, 20, 3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosohata, K.; Ando, H.; Fujiwara, Y.; Fujimura, A. Vanin-1; A potential biomarker for nephrotoxicant-induced renal injury. Toxicology 2011, 290, 82–88. [Google Scholar] [CrossRef]
- Ellinger, S.; Stehle, P. Efficacy of vitamin supplementation in situations with wound healing disorders: Results from clinical intervention studies. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 588–595. [Google Scholar] [CrossRef]
- Dell’Acqua, G.; Schweikert, K. Panthenyl triacetate transformation, stimulation of metabolic pathways, and wound-healing properties in the human skin. J. Cosmet. Sci. 2012, 63, 1–13. [Google Scholar]
- Kobayashi, D.; Kusama, M.; Onda, M.; Nakahata, N. The Effect of Pantothenic Acid Deficiency on Keratinocyte Proliferation and the Synthesis of Keratinocyte Growth Factor and Collagen in Fibroblasts. J. Pharmacol. Sci. 2011, 115, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Gutzeit, D.; Klaubert, B.; Rychlik, M.; Winterhalter, P.; Jerz, G. Effects of processing and of storage on the stability of pantothenic acid in sea buckthorn products (Hippophaë rhamnoides L. ssp. rhamnoides) assessed by stable isotope dilution assay. J. Agric. Food Chem. 2007, 55, 3978–3984. [Google Scholar] [CrossRef]
- Fatima, Z.; Jin, X.; Zou, Y.; Kaw, H.Y.; Quinto, M.; Li, D. Recent trends in analytical methods for water-soluble vitamins. J. Chromatogr. A 2019, 1606, 360245. [Google Scholar] [CrossRef]
- Mittermayr, R.; Kalman, A.; Trisconi, M.-J.; Heudi, O. Determination of Vitamin B5 in a range of fortified food products by reversed-phase liquid chromatography–mass spectrometry with electrospray ionisation. J. Chromatogr. A 2004, 1032, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.A.; Young, M.F.; Taneja, S.; Rangiah, K. Quantification of B-vitamins from different fresh milk samples using ultra-high performance liquid chromatography mass spectrometry/selected reaction monitoring methods. J. Chromatogr. A 2020, 1609, 460452. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Huhman, D.V.; Sumner, L.W. Mass spectrometry strategies in metabolomics. J. Biol. Chem. 2011, 286, 25435–25442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oomens, J.; Sartakov, B.G.; Meijer, G.; Von Helden, G. Gas-phase infrared multiple photon dissociation spectroscopy of mass-selected molecular ions. Int. J. Mass Spectrom. 2006, 254, 1–19. [Google Scholar] [CrossRef]
- Fridgen, T.D. Infrared consequence spectroscopy of gaseous protonated and metal ion cationized complexes. Mass Spectrom. Rev. 2009, 28, 586–607. [Google Scholar] [CrossRef]
- Eyler, J.R. Infrared multiple photon dissociation spectroscopy of ions in Penning traps. Mass Spectrom. Rev. 2009, 28, 448–467. [Google Scholar] [CrossRef]
- Polfer, N.C.; Oomens, J. Vibrational spectroscopy of bare and solvated ionic complexes of biological relevance. Mass Spectrom. Rev. 2009, 28, 468–494. [Google Scholar] [CrossRef]
- Roithová, J. Characterization of reaction intermediates by ion spectroscopy. Chem. Soc. Rev. 2012, 41, 547–559. [Google Scholar] [CrossRef]
- Polfer, N.C.; Dugourd, P. (Eds.) Laser Photodissociation and Spectroscopy of Mass-Separated Biomolecular Ions; Lecture Notes in Chemistry; Springer International Publishing: Cham, Switzerland, 2013; Volume 83, ISBN 978-3-319-01251-3. [Google Scholar]
- Maitre, P.; Scuderi, D.; Corinti, D.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Applications of Infrared Multiple Photon Dissociation (IRMPD) to the Detection of Posttranslational Modifications. Chem. Rev. 2020, 120, 3261–3295. [Google Scholar] [CrossRef]
- Lanucara, F.; Scuderi, D.; Chiavarino, B.; Fornarini, S.; Maitre, P.; Crestoni, M.E. IR Signature of NO Binding to a Ferrous Heme Center. J. Phys. Chem. Lett. 2013, 4, 2414–2417. [Google Scholar] [CrossRef]
- Chiavarino, B.; Crestoni, M.E.; Schütz, M.; Bouchet, A.; Piccirillo, S.; Steinmetz, V.; Dopfer, O.; Fornarini, S. Cation-Interactions in Protonated Phenylalkylamines. J. Phys. Chem. A 2014, 118, 7130–7138. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Maccelli, A.; Crestoni, M.E.; Cesa, S.; Quaglio, D.; Botta, B.; Ingallina, C.; Mannina, L.; Tintaru, A.; Chiavarino, B.; et al. IR ion spectroscopy in a combined approach with MS/MS and IM-MS to discriminate epimeric anthocyanin glycosides (cyanidin 3-O-glucoside and -galactoside). Int. J. Mass Spectrom. 2019, 444, 116179. [Google Scholar] [CrossRef]
- Corinti, D.; Coletti, C.; Re, N.; Paciotti, R.; Maître, P.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Short-lived intermediates (encounter complexes) in cisplatin ligand exchange elucidated by infrared ion spectroscopy. Int. J. Mass Spectrom. 2019, 435, 7–17. [Google Scholar] [CrossRef]
- Akinyemi, T.E.; Wu, R.R.; Nei, Y.-W.; Cunningham, N.A.; Roy, H.A.; Steill, J.D.; Berden, G.; Oomens, J.; Rodgers, M.T. Influence of Transition Metal Cationization versus Sodium Cationization and Protonation on the Gas-Phase Tautomeric Conformations and Stability of Uracil: Application to [Ura+Cu]+ and [Ura+Ag]+. J. Am. Soc. Mass Spectrom. 2017, 28, 2438–2453. [Google Scholar] [CrossRef] [PubMed]
- Andris, E.; Navrátil, R.; Jašík, J.; Puri, M.; Costas, M.; Que, L.; Roithová, J. Trapping Iron(III)–Oxo Species at the Boundary of the “Oxo Wall”: Insights into the Nature of the Fe(III)–O Bond. J. Am. Chem. Soc. 2018, 140, 14391–14400. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Maccelli, A.; Chiavarino, B.; Maitre, P.; Scuderi, D.; Bodo, E.; Fornarini, S.; Crestoni, M.E. Vibrational signatures of curcumin’s chelation in copper(II) complexes: An appraisal by IRMPD spectroscopy. J. Chem. Phys. 2019, 150, 165101. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Crestoni, M.E.; Chiavarino, B.; Fornarini, S.; Scuderi, D.; Salpin, J.-Y. Insights into Cisplatin Binding to Uracil and Thiouracils from IRMPD Spectroscopy and Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Frison, G.; Chiavarino, B.; Gabano, E.; Osella, D.; Crestoni, M.E.; Fornarini, S. Can an Elusive Platinum (III) Oxidation State be Exposed in an Isolated Complex? Angew. Chem. Int. Ed. 2020, 59, 15595–15598. [Google Scholar] [CrossRef]
- Gholami, A.; Fridgen, T.D. Structures and Unimolecular Reactivity of Gas-Phase [Zn(Proline-H)]+ and [Zn(Proline-H)(H2O)]+. J. Phys. Chem. B 2013, 117, 8447–8456. [Google Scholar] [CrossRef]
- Dunbar, R.C.; Berden, G.; Martens, J.K.; Oomens, J. Divalent Metal-Ion Complexes with Dipeptide Ligands Having Phe and His Side-Chain Anchors: Effects of Sequence, Metal Ion, and Anchor. J. Phys. Chem. A 2015, 119, 9901–9909. [Google Scholar] [CrossRef]
- Nieto, P.; Günther, A.; Berden, G.; Oomens, J.; Dopfer, O. IRMPD Spectroscopy of Metalated Flavins: Structure and Bonding of Lumiflavin Complexes with Alkali and Coinage Metal Ions. J. Phys. Chem. A 2016, 120, 8297–8308. [Google Scholar] [CrossRef]
- Boles, G.C.; Hightower, R.L.; Berden, G.; Oomens, J.; Armentrout, P.B. Zinc and Cadmium Complexation of L -Threonine: An Infrared Multiple Photon Dissociation Spectroscopy and Theoretical Study. J. Phys. Chem. B 2019, 123, 9343–9354. [Google Scholar] [CrossRef] [PubMed]
- Lamsabhi, A.M.; Mó, O.; Yáñez, M.; Salpin, J.-Y. Combined Experimental and Theoretical Survey of the Gas-Phase Reactions of Serine–Ca2+ Adducts. J. Phys. Chem. A 2019, 123, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Gregori, B.; Guidoni, L.; Scuderi, D.; McMahon, T.B.; Chiavarino, B.; Fornarini, S.; Crestoni, M.E. Complexation of halide ions to tyrosine: Role of non-covalent interactions evidenced by IRMPD spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 4429–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciotti, R.; Coletti, C.; Re, N.; Scuderi, D.; Chiavarino, B.; Fornarini, S.; Crestoni, M.E. Serine O-sulfation probed by IRMPD spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 25891–25904. [Google Scholar] [CrossRef] [PubMed]
- Patrick, A.L.; Stedwell, C.N.; Polfer, N.C. Differentiating Sulfopeptide and Phosphopeptide Ions via Resonant Infrared Photodissociation. Anal. Chem. 2014, 86, 5547–5552. [Google Scholar] [CrossRef]
- Fraschetti, C.; Filippi, A.; Guarcini, L.; Steinmetz, V.; Speranza, M. Structure and conformation of protonated d-(+)-biotin in the unsolvated state. J. Phys. Chem. B 2015, 119, 6198–6203. [Google Scholar] [CrossRef]
- Lanucara, F.; Chiavarino, B.; Scuderi, D.; Maitre, P.; Fornarini, S.; Crestoni, M.E. Kinetic control in the CID-induced elimination of H3PO4 from phosphorylated serine probed using IRMPD spectroscopy. Chem. Commun. 2014, 50, 3845–3848. [Google Scholar] [CrossRef]
- Crestoni, M.E.; Chiavarino, B.; Scuderi, D.; Di Marzio, A.; Fornarini, S. Discrimination of 4-hydroxyproline diastereomers by vibrational spectroscopy of the gaseous protonated species. J. Phys. Chem. B 2012, 116, 8771–8779. [Google Scholar] [CrossRef]
- Acharya, B.; Kaushalya, W.K.D.N.; Martens, J.; Berden, G.; Oomens, J.; Patrick, A.L. A Combined Infrared Ion Spectroscopy and Computational Chemistry Study of Hydroxyproline Isomers. J. Am. Soc. Mass Spectrom. 2020, 31, 1205–1211. [Google Scholar] [CrossRef]
- Martens, J.; Koppen, V.; Berden, G.; Cuyckens, F.; Oomens, J. Combined Liquid Chromatography-Infrared Ion Spectroscopy for Identification of Regioisomeric Drug Metabolites. Anal. Chem. 2017, 89, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Lepere, V.; Le Barbu-Debus, K.; Clavaguéra, C.; Scuderi, D.; Piani, G.; Simon, A.-L.; Chirot, F.; MacAleese, L.; Dugourd, P.; Zehnacker, A. Chirality-dependent structuration of protonated or sodiated polyphenylalanines: IRMPD and ion mobility studies. Phys. Chem. Chem. Phys. 2016, 18, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Cismesia, A.P.; Bell, M.R.; Tesler, L.F.; Alves, M.; Polfer, N.C. Infrared ion spectroscopy: An analytical tool for the study of metabolites. Analyst 2018, 143, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Molano-Arevalo, J.C.; Gonzalez, W.; Jeanne Dit Fouque, K.; Miksovska, J.; Maitre, P.; Fernandez-Lima, F. Insights from ion mobility-mass spectrometry, infrared spectroscopy, and molecular dynamics simulations on nicotinamide adenine dinucleotide structural dynamics: NAD+ vs. NADH. Phys. Chem. Chem. Phys. 2018, 20, 7043–7052. [Google Scholar] [CrossRef]
- Martens, J.; Van Outersterp, R.E.; Vreeken, R.J.; Cuyckens, F.; Coene, K.L.M.; Engelke, U.F.; Kluijtmans, L.A.J.; Wevers, R.A.; Buydens, L.M.C.; Redlich, B.; et al. Infrared ion spectroscopy: New opportunities for small-molecule identification in mass spectrometry—A tutorial perspective. Anal. Chim. Acta 2020, 1093, 1–15. [Google Scholar] [CrossRef]
- Martens, J.; Berden, G.; Van Outersterp, R.E.; Kluijtmans, L.A.J.; Engelke, U.F.; Van Karnebeek, C.D.M.; Wevers, R.A.; Oomens, J. Molecular identification in metabolomics using infrared ion spectroscopy. Sci. Rep. 2017, 7, 3363. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xue, S.; Yang, J.J. Calciomics: Integrative studies of Ca2+-binding proteins and their interactomes in biological systems. Metallomics 2013, 5, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Coates, R.A.; McNary, C.P.; Boles, G.C.; Berden, G.; Oomens, J.; Armentrout, P.B. Structural characterization of gas-phase cysteine and cysteine methyl ester complexes with zinc and cadmium dications by infrared multiple photon dissociation spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 25799–25808. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Dunbar, R.C.; Polfer, N.C.; Berden, G.; Oomens, J. Metal ion binding to peptides: Oxygen or nitrogen sites? Int. J. Mass Spectrom. 2012, 330–332, 71–77. [Google Scholar] [CrossRef]
- Dunbar, R.C.; Steill, J.D.; Polfer, N.C.; Berden, G.; Oomens, J. Peptide bond tautomerization induced by divalent metal ions: Characterization of the iminol configuration. Angew. Chem. Int. Ed. 2012, 51, 4591–4593. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, R.C.; Berden, G.; Oomens, J. How does a small peptide choose how to bind a metal ion? IRMPD and computational survey of CS versus Iminol binding preferences. Int. J. Mass Spectrom. 2013, 354–355, 356–364. [Google Scholar] [CrossRef]
- Guerrieri, F.; Minicozzi, V.; Morante, S.; Rossi, G.; Furlan, S.; La Penna, G. Modeling the interplay of glycine protonation and multiple histidine binding of copper in the prion protein octarepeat subdomains. JBIC J. Biol. Inorg. Chem. 2009, 14, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Sóvágó, I.; Ősz, K. Metal ion selectivity of oligopeptides. Dalton Trans. 2006, 3841–3854. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Mannina, L.; Chiavarino, B.; Steinmetz, V.; Fornarini, S.; Crestoni, M.E. IRMPD signature of protonated pantothenic acid, an ubiquitous nutrient. Chem. Phys. Lett. 2016, 646, 162–167. [Google Scholar] [CrossRef]
- Rijs, A.M.; Oomens, J. (Eds.) Gas-Phase IR Spectroscopy and Structure of Biological Molecules; Topics in Current Chemistry; Springer International Publishing: Cham, Switzerland, 2015; Volume 364, ISBN 978-3-319-19203-1. [Google Scholar]
- Carnegie, P.D.; Bandyopadhyay, B.; Duncan, M.A. Infrared Spectroscopy of Mn+(H2O) and Mn2+(H2O) via Argon Complex Predissociation. J. Phys. Chem. A 2011, 115, 7602–7609. [Google Scholar] [CrossRef]
- Straka, M.; Andris, E.; Vícha, J.; Růžička, A.; Roithová, J.; Rulíšek, L. Spectroscopic and Computational Evidence of Intramolecular AuI⋅⋅⋅H+−N Hydrogen Bonding. Angew. Chem. Int. Ed. 2019, 58, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Peckelsen, K.; Martens, J.; Berden, G.; Oomens, J.; Dunbar, R.C.; Meijer, A.J.H.M.; Schäfer, M. Gas-phase complexes of Ni2+ and Ca2+ with deprotonated histidylhistidine (HisHis): A model case for polyhistidyl-metal binding motifs. J. Mol. Spectrosc. 2017, 332, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, R.C.; Steill, J.D.; Oomens, J. Conformations and vibrational spectroscopy of metal-ion/polylalanine complexes. Int. J. Mass Spectrom. 2010, 297, 107–115. [Google Scholar] [CrossRef]
- DeLucas, L.; Einspahr, H.; Bugg, C.E. Calcium Binding to Amide Carbonyl Groups: Structure of a Calcium Bromide Salt of D-Pantothenic Acid. Acta Cryst. 1979, 35, 2724–2726. [Google Scholar] [CrossRef]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, USA, 1994. [Google Scholar]
- Burt, M.B.; Decker, S.G.A.; Atkins, C.G.; Rowsell, M.; Peremans, A.; Fridgen, T.D. Structures of Bare and Hydrated [Pb(AminoAcid-H)]+ Complexes Using Infrared Multiple Photon Dissociation Spectroscopy. J. Phys. Chem. B 2011, 115, 11506–11518. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, R.C.; Martens, J.; Berden, G.; Oomens, J. Water Microsolvation Can Switch the Binding Mode of Ni(II) with Small Peptides. J. Phys. Chem. Lett. 2017, 8, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Valadbeigi, Y.; Farrokhpour, H.; Tabrizchi, M. DFT study on the isomerization and tautomerism in vitamins B3 (niacin), B5 (pantothenic acid) and B7 (biotin). Chem. Phys. Lett. 2014, 601, 155–162. [Google Scholar] [CrossRef]
- Corinti, D.; De Petris, A.; Coletti, C.; Re, N.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Cisplatin Primary Complex with l-Histidine Target Revealed by IR Multiple Photon Dissociation (IRMPD) Spectroscopy. ChemPhysChem 2017, 18, 318–325. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L.; Jackson, G.S. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom. Rev. 1998, 17, 1–35. [Google Scholar] [CrossRef]
- Lemaire, J.; Boissel, P.; Heninger, M.; Mauclaire, G.; Bellec, G.; Mestdagh, H.; Simon, A.; Le Caer, S.; Ortega, J.M.; Glotin, F. Gas Phase Infrared Spectroscopy of Selectively Prepared Ions. Phys. Rev. Lett. 2002, 89, 273001–273002. [Google Scholar] [CrossRef] [PubMed]
- Mac Aleese, L.; Simon, A.; McMahon, T.B.; Ortega, J.-M.; Scuderi, D.; Lemaire, J.; Maître, P. Mid-IR spectroscopy of protonated leucine methyl ester performed with an FTICR or a Paul type ion-trap. Int. J. Mass Spectrom. 2006, 249–250, 14–20. [Google Scholar] [CrossRef]
- Bakker, J.M.; Besson, T.; Lemaire, J.; Scuderi, D.; Maître, P. Gas-Phase Structure of a π-Allyl−Palladium Complex: Efficient Infrared Spectroscopy in a 7 T Fourier Transform Mass Spectrometer. J. Phys. Chem. A 2007, 111, 13415–13424. [Google Scholar] [CrossRef]
- Prell, J.S.; O’Brien, J.T.; Williams, E.R. IRPD spectroscopy and ensemble measurements: Effects of different data acquisition and analysis methods. J. Am. Soc. Mass Spectrom. 2010, 21, 800–809. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculated a,b | |||
|---|---|---|---|
| IRMPD a | CaPa_a1 | CaPa_a2 | Vibrational Mode |
| 965 | 950 (126) | 937 (77) | C4′O4′ stretch + C2′O2′ stretch + CH3 bend |
| 989 (146) | C4′O4′ stretch + CH3 bend | ||
| 998 (35) | C1′C2′ stretch + C3N stretch | ||
| 1010 | |||
| 1085 | 1056 (70) | C2′O2′ stretch + CH2 (all) rock | |
| 1192 | 1178 (40) | O2′H bend + C2H2 twist | |
| 1285 | 1271 (47) | NH bend + C2′H bend + C3H bend | |
| 1379 | 1381 (32) | O4′H bend + C4′H bend + CH3 umbrella | |
| 1450 | 1417 (74) | 1405 (107) | C2H2 scissor + C1C2 stretch + sym carboxylate stretch + CH3 umbrella |
| 1407 (38) | C2H2 scissor + C3H2 scissor + CH3 umbrella | ||
| 1445 (86) | O2′H bend + C4′H2 wagging | ||
| 1441 (45) | C2H2 scissor + C3H2 scissor | ||
| 1533 | 1507 (342) | 1524 (291) | antisym carboxylate stretch + C2H2 twist |
| 1549 (200) | 1528 (136) | NH bend (Amide II) + CN stretch | |
| 1625 | 1613 (384) | 1614 (321) | C1′O1′ stretch (Amide I) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corinti, D.; Chiavarino, B.; Scuderi, D.; Fraschetti, C.; Filippi, A.; Fornarini, S.; Crestoni, M.E. Molecular Properties of Bare and Microhydrated Vitamin B5–Calcium Complexes. Int. J. Mol. Sci. 2021, 22, 692. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020692
Corinti D, Chiavarino B, Scuderi D, Fraschetti C, Filippi A, Fornarini S, Crestoni ME. Molecular Properties of Bare and Microhydrated Vitamin B5–Calcium Complexes. International Journal of Molecular Sciences. 2021; 22(2):692. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020692
Chicago/Turabian StyleCorinti, Davide, Barbara Chiavarino, Debora Scuderi, Caterina Fraschetti, Antonello Filippi, Simonetta Fornarini, and Maria Elisa Crestoni. 2021. "Molecular Properties of Bare and Microhydrated Vitamin B5–Calcium Complexes" International Journal of Molecular Sciences 22, no. 2: 692. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020692