Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice

 , and
, and 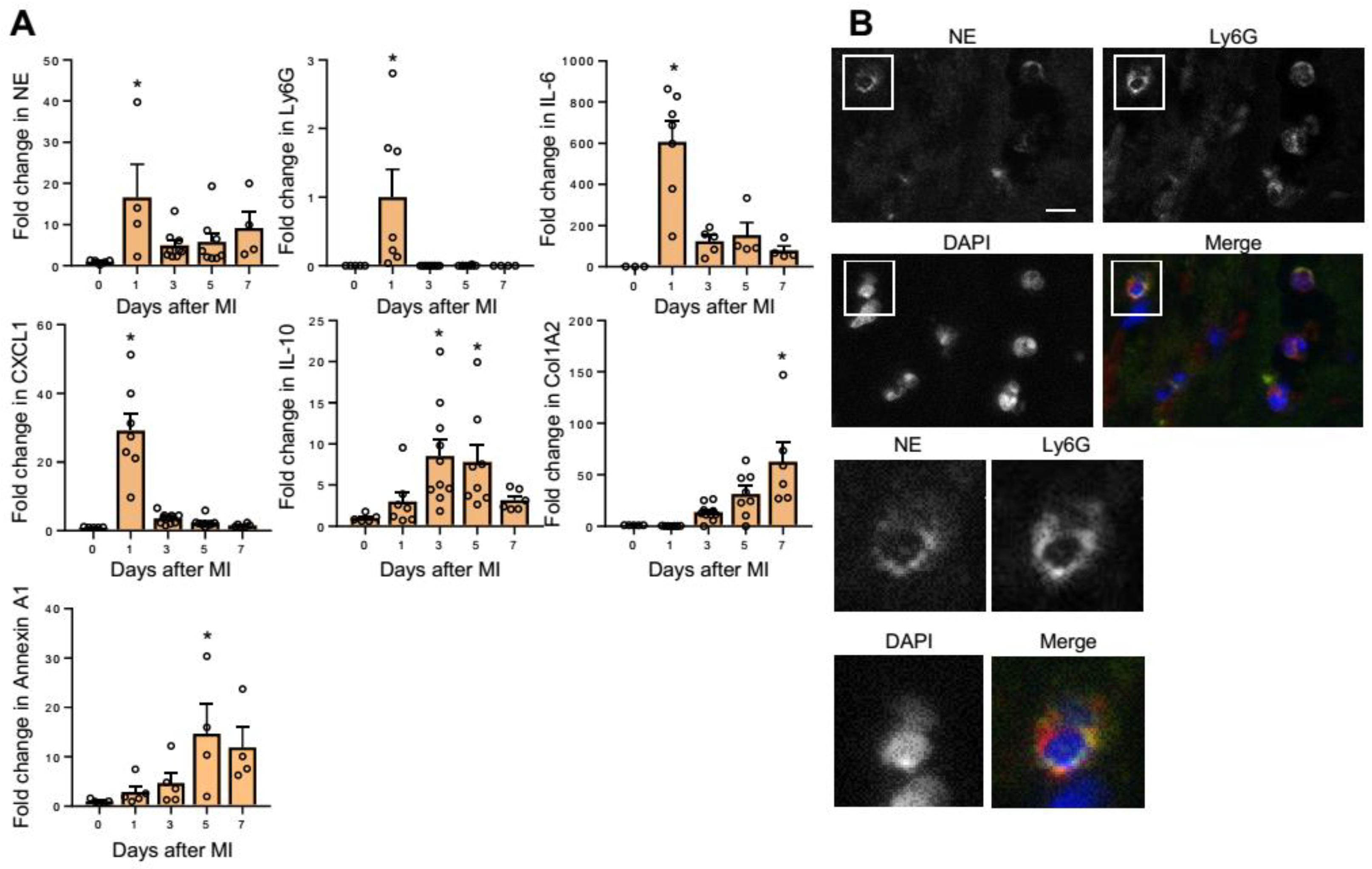{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. NE Expression in MI Hearts
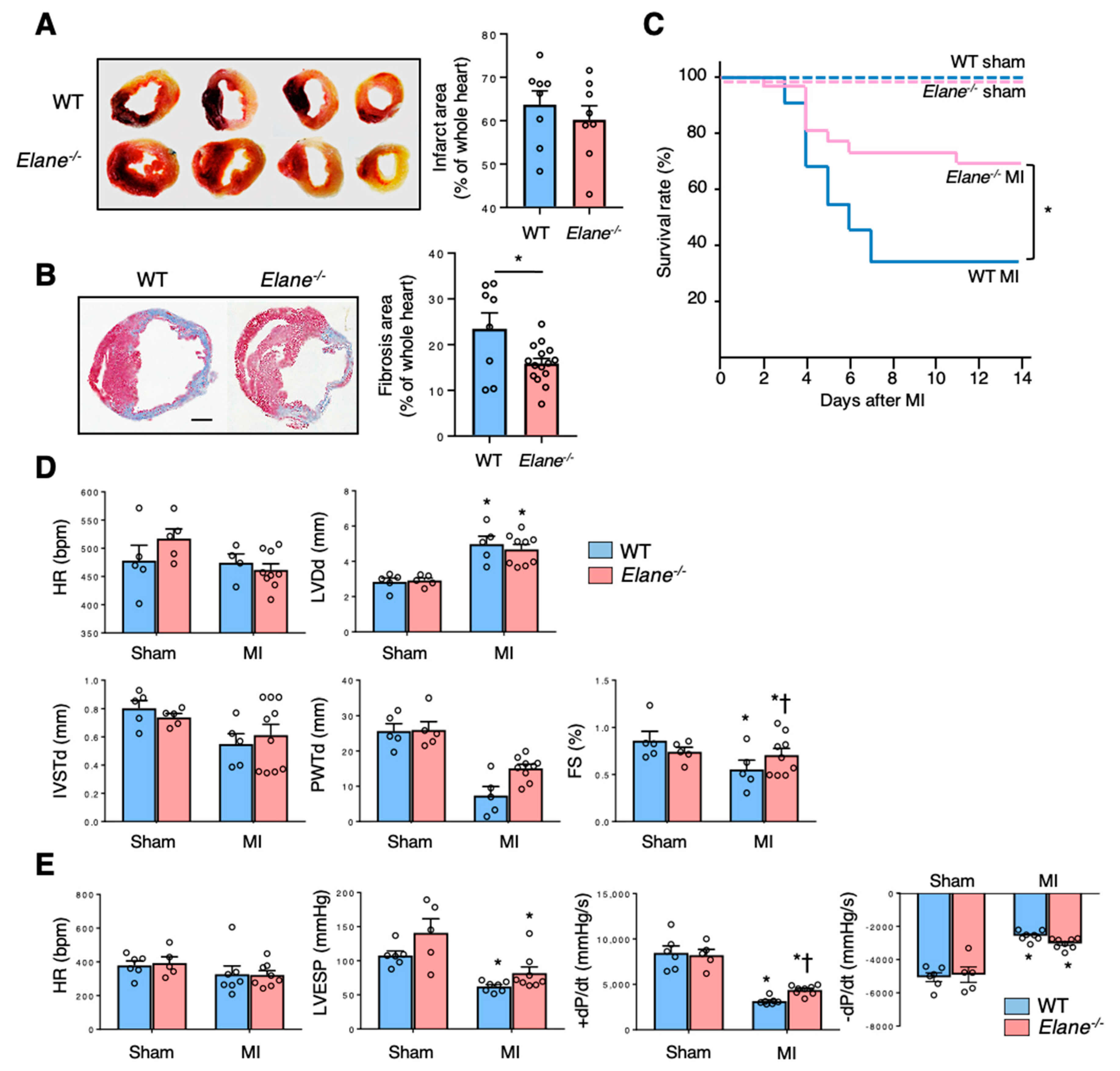
2.2. NE Deficiency Improves Cardiac Function Post-MI
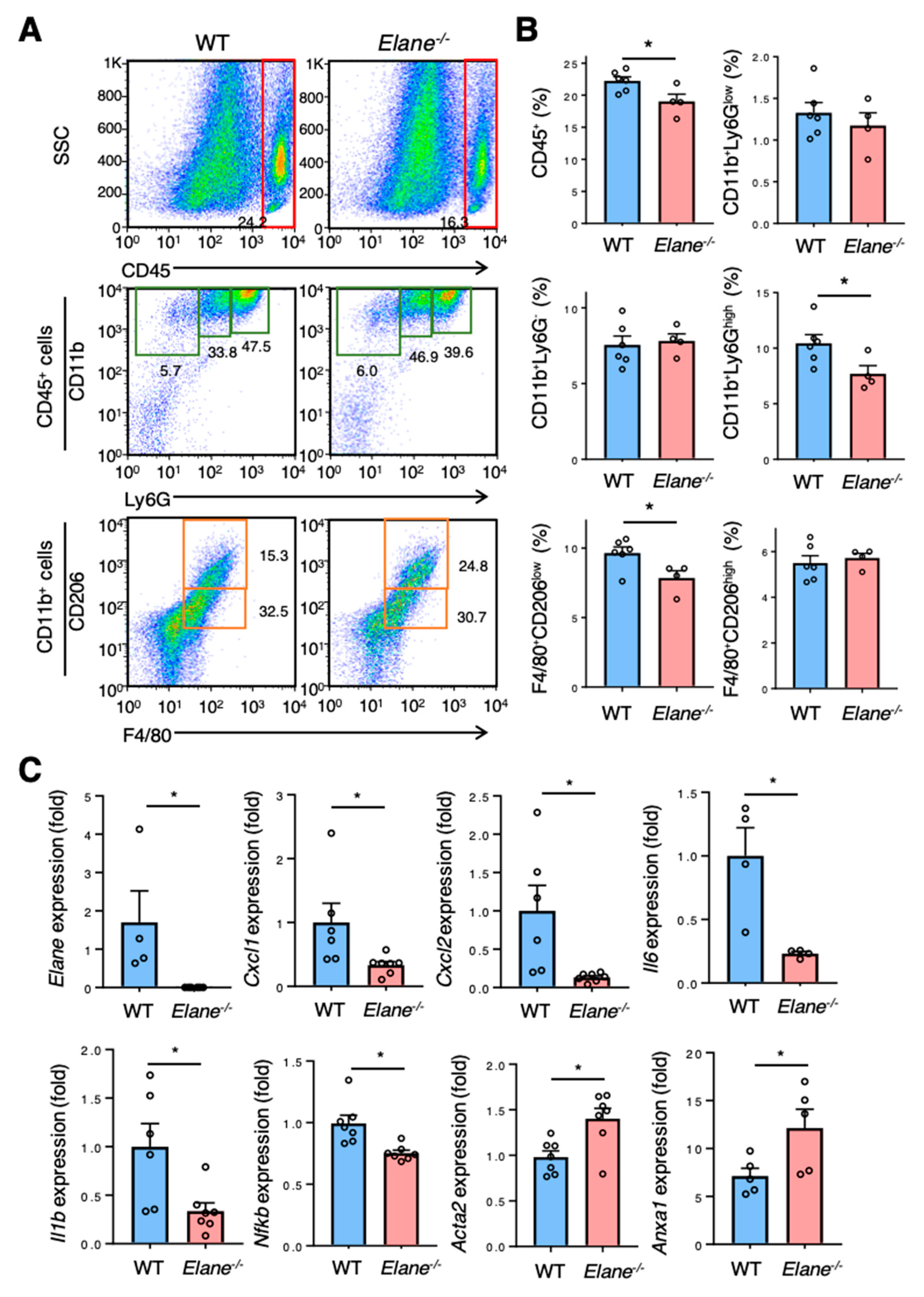
2.3. NE Deficiency Suppresses Excessive Inflammation Post-MI
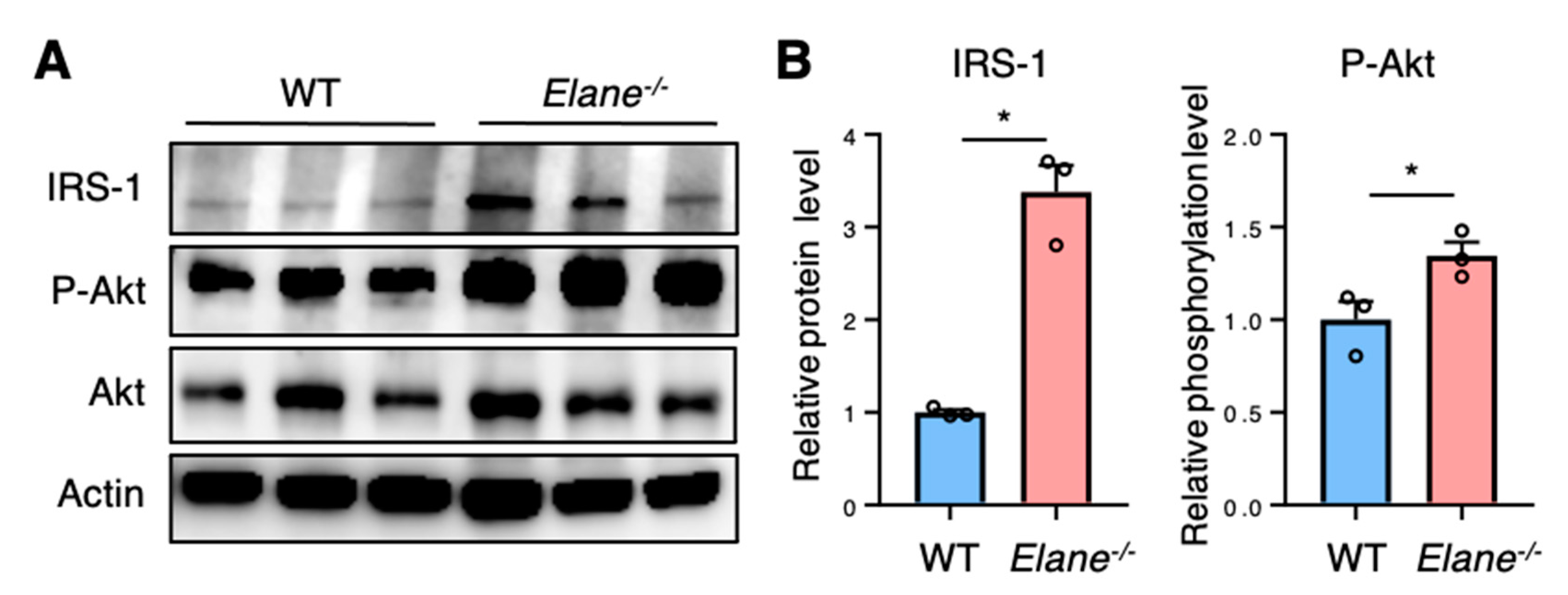
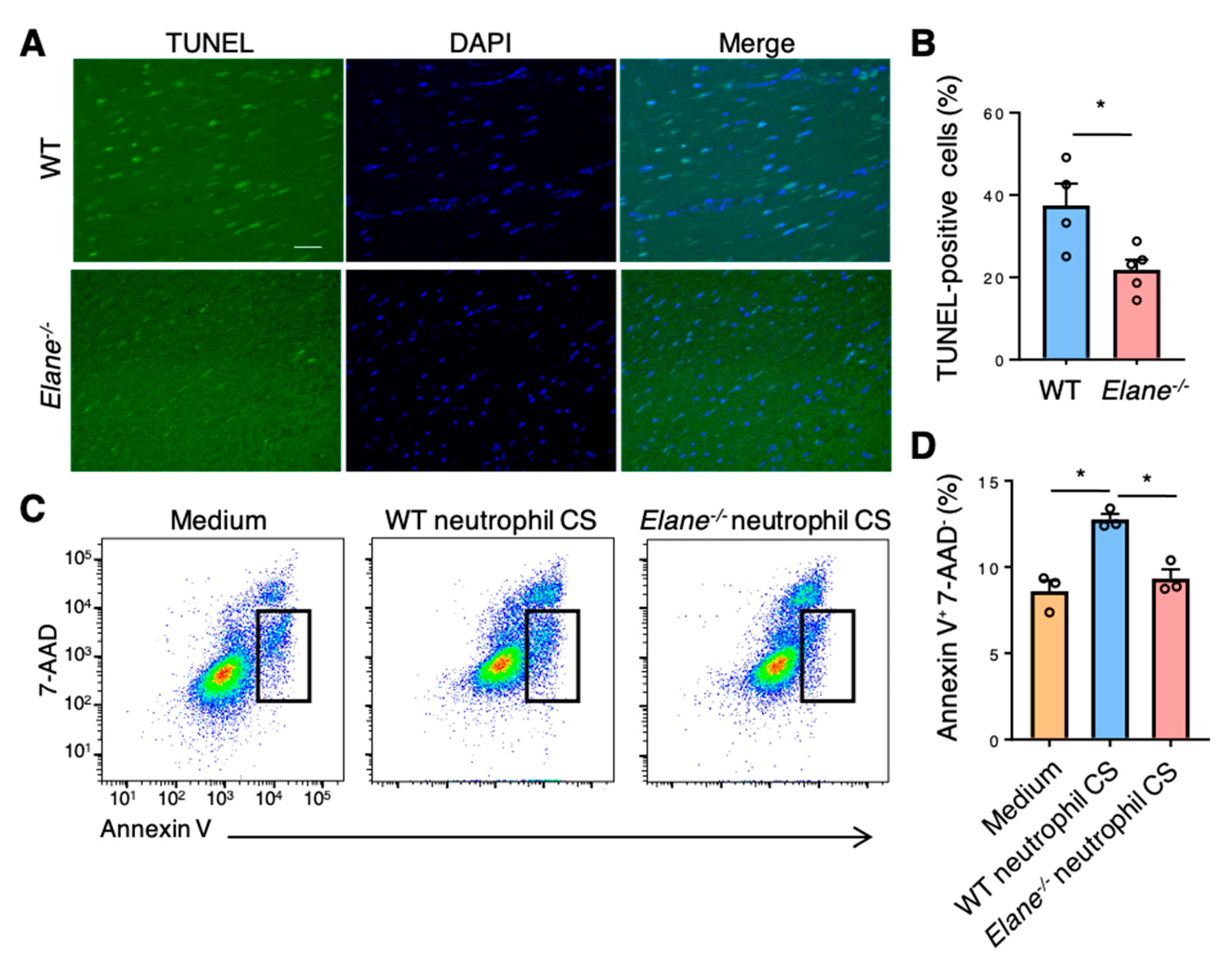
2.4. NE Deficiency Reduces Cardiomyocyte Apoptosis by Activating Insulin/Akt Signaling
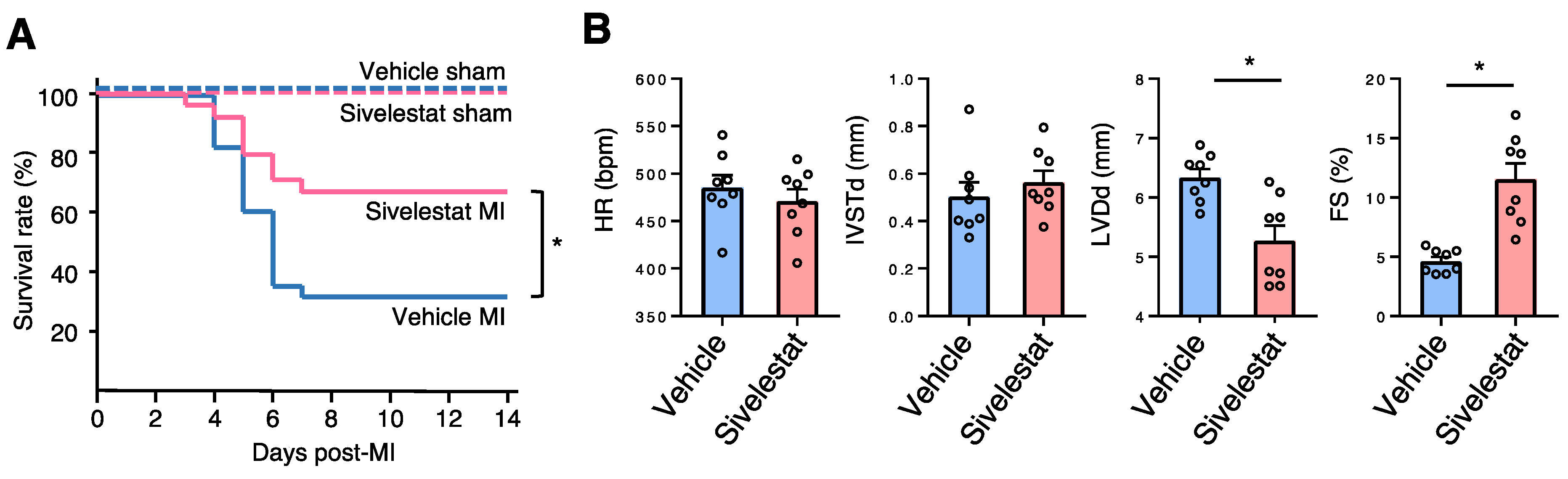
2.5. NE Inhibitor Improves Survival and Cardiac Function Post-MI
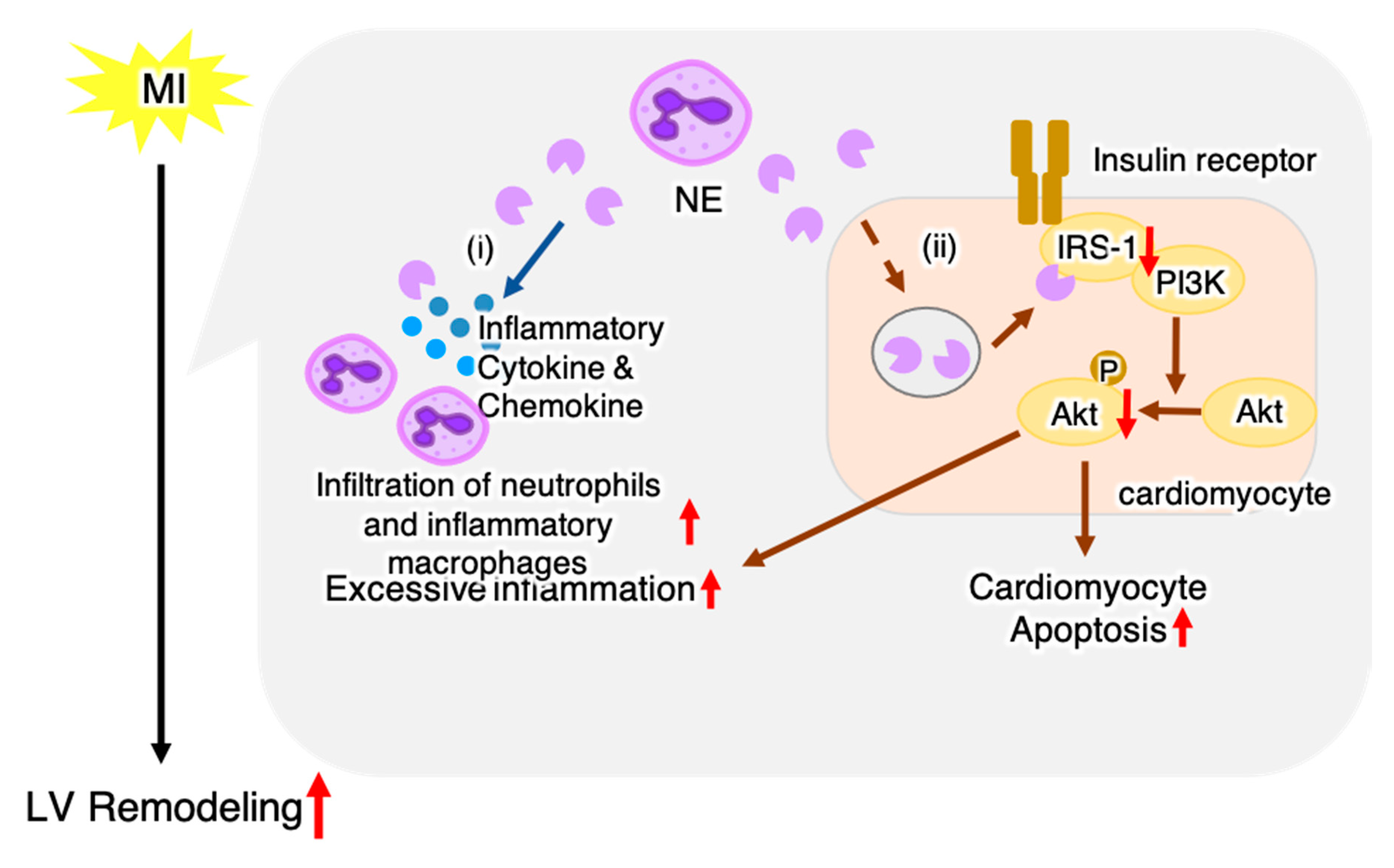
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. MI Model
4.3. Survival Analysis
4.4. Echocardiography
4.5. Histopathological and Immunohistochemical Examination
4.6. Infarct Size Evaluation
4.7. TUNEL Assay
4.8. Flow Cytometry
4.9. Cell Culture of HL-1 Cells
4.10. Flow Cytometric Analysis of Cardiomyocyte Apoptosis
4.11. RNA Extraction and Quantitative Reverse-Transcription Polymerase Chain Reaction
4.12. Western Blotting
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | cluster of differentiation |
| Col1a2 | collagen type I alpha 2 chain |
| CS | culture supernatant |
| CXCL | C-X-C motif chemokine ligand |
| DAPI | 4′,6-diamidino-2-phenylindole |
| FS | fractional shortening |
| HR | heart rate |
| IL | interleukin |
| IRS-1 | insulin receptor substrate-1 |
| IVSTd | interventricular septal in end-diastole |
| LV | left ventricular |
| LVDd | left ventricular diameter at end-diastole |
| LVESP | left ventricular end-systolic pressure |
| Ly6G | lymphocyte antigen 6 complex, locus G |
| MI | myocardial infarction |
| mRNA | messenger RNA |
| NE | neutrophil elastase |
| NFκB | nuclear factor kappa B |
| PBS | phosphate-buffered saline |
| PI3K | phosphoinositide 3-kinase |
| PWTd | left ventricular posterior wall in end-diastole |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| WT | wild-type |
References
- Bennett, J.E.; Stevens, G.A.; Mathers, C.D.; Bonita, R.; Rehm, J.; Kruk, M.E.; Riley, L.M.; Dain, K.; Kengne, A.P.; Chalkidou, K.; et al. NCD Countdown 2030: Worldwide trends in non-communicable disease mortality and progress towards Sustainable Development Goal target 3.4. Lancet 2018, 392, 1072–1088. [Google Scholar] [CrossRef] [Green Version]
- Wolk, M.J.; Bailey, S.R.; Doherty, J.U.; Douglas, P.S.; Hendel, R.C.; Kramer, C.M.; Min, J.K.; Patel, M.R.; Rosenbaum, L.; Shaw, L.J.; et al. ACCF/AHA/ASE/ASNC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2013 multimodality appropriate use criteria for the detection and risk assessment of stable ischemic heart disease. J. Am. Coll. Cardiol. 2014, 63, 380–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eghbalzadeh, K.; Georgi, L.; Louis, T.; Zhao, H.; Keser, U.; Weber, C.; Mollenhauer, M.; Conforti, A.; Wahlers, T.; Paunel-Görgülü, A. Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation after Myocardial Infarction. Front. Immunol. 2019, 10, 2313. [Google Scholar] [CrossRef]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- López-Boado, Y.S.; Espinola, M.; Bahr, S.; Belaaouaj, A. Neutrophil Serine Proteinases Cleave Bacterial Flagellin, Abrogating Its Host Response-Inducing Activity. J. Immunol. 2004, 172, 509–515. [Google Scholar] [CrossRef]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.; Jackson, M.; Nicoll, J.J.; Millar, A.; Dawes, J.; Muir, A.L. Inflammatory response, neutrophil activation, and free radical production after acute myocardial infarction: Effect of thrombolytic treatment. Br. Heart J. 1990, 63, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Hajjar, R.J. AKT signalling in the failing heart. Eur. J. Heart Fail. 2011, 13, 825–829. [Google Scholar] [CrossRef]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; Ofrecio, J.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol 2015, 98, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Anzai, T. Post-infarction inflammation and left ventricular remodeling: A double-edged sword. Circ. J. 2013, 77, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzai, T. Inflammatory mechanisms of cardiovascular remodeling. Circ. J. 2018, 82, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, S.; Chotirmall, S.H.; Greene, C.M.; McElvaney, N.G. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/toll-like receptor pathway. J. Biol. Chem. 2011, 286, 7692–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.T.N. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Adkison, A.M.; Raptis, S.Z.; Kelley, D.G.; Pham, C.T.N. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J. Clin. Investig. 2002, 109, 363–371. [Google Scholar] [CrossRef]
- Young, R.E.; Thompson, R.D.; Larbi, K.Y.; La, M.; Roberts, C.E.; Shapiro, S.D.; Perretti, M.; Nourshargh, S. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J. Immunol. 2004, 172, 4493–4502. [Google Scholar] [CrossRef] [Green Version]
- Otaka, N.; Shibata, R.; Ohashi, K.; Uemura, Y.; Kambara, T.; Enomoto, T.; Ogawa, H.; Ito, M.; Kawanishi, H.; Maruyama, S.; et al. Myonectin is an exercise-induced Myokine that protects the heart from ischemia-reperfusion injury. Circ. Res. 2018, 123, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Mackman, N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 2002, 277, 32124–32132. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Geng, J.; Xu, Z.; Chen, Y.; Zhang, X.W. Neutrophil gelatinase-associated lipocalin2 exaggerates cardiomyocyte hypoxia injury by inhibiting integrin β3 signaling. Med. Sci. Monit. 2019, 25, 5426–5434. [Google Scholar] [CrossRef] [PubMed]
- Hojo, Y.; Saito, T.; Kondo, H. Role of apoptosis in left ventricular remodeling after acute myocardial infarction. J. Cardiol. 2012, 60, 91–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbate, A.; Bussani, R.; Amin, M.S.; Vetrovec, G.W.; Baldi, A. Acute myocardial infarction and heart failure: Role of apoptosis. Int. J. Biochem. Cell Biol. 2006, 38, 1834–1840. [Google Scholar] [CrossRef]
- Kandel, E.S.; Hay, N. The Regulation and Activities of the Multifunctional Serine/Threonine Kinase Akt/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Shiojima, I.; Ozasa, Y.; Yoshida, M.; Holzenberger, M.; Kahn, C.R.; Walsh, K.; Igarashi, T.; Abel, E.D.; Komuro, I. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J. Mol. Cell Cardiol. 2009, 47, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaki, K.; Okada, T.; Nakayama, Y.; Nagao, Y.; Kobayashi, K.; Sakai, Y.; Mohri, T.; Amino, T.; Nakai, H.; Kawamura, M. Non-peptidic inhibitors of human neutrophil elastase: The design and synthesis of sulfonanilide-containing inhibitors. Bioorganic. Med. Chem. 1996, 4, 2115–2134. [Google Scholar] [CrossRef]
- Kimura, T.; Tajiri, K.; Sato, A.; Sakai, S.; Wang, Z.; Yoshida, T.; Uede, T.; Hiroe, M.; Aonuma, K.; Ieda, M.; et al. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc. Res. 2019, 115, 614–624. [Google Scholar] [CrossRef]
- Yonebayashi, S.; Tajiri, K.; Murakoshi, N.; Xu, D.; Li, S.; Feng, D.; Okabe, Y.; Yuan, Z.; Song, Z.; Aonuma, K.; et al. MAIR-II deficiency ameliorates cardiac remodelling post-myocardial infarction by suppressing TLR9-mediated macrophage activation. J. Cell Mol. Med. 2020, 24, 14481–14490. [Google Scholar] [CrossRef]
- Xu, D.; Murakoshi, N.; Igarashi, M.; Hirayama, A.; Ito, Y.; Seo, Y.; Tada, H.; Aonuma, K. PPAR-γ activator pioglitazone prevents age-related atrial fibrillation susceptibility by improving antioxidant capacity and reducing apoptosis in a rat model. J. Cardiovasc. Electrophysiol. 2012, 23, 209–217. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogura, Y.; Tajiri, K.; Murakoshi, N.; Xu, D.; Yonebayashi, S.; Li, S.; Okabe, Y.; Feng, D.; Shimoda, Y.; Song, Z.; et al. Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. Int. J. Mol. Sci. 2021, 22, 722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020722
Ogura Y, Tajiri K, Murakoshi N, Xu D, Yonebayashi S, Li S, Okabe Y, Feng D, Shimoda Y, Song Z, et al. Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. International Journal of Molecular Sciences. 2021; 22(2):722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020722
Chicago/Turabian StyleOgura, Yukino, Kazuko Tajiri, Nobuyuki Murakoshi, DongZhu Xu, Saori Yonebayashi, Siqi Li, Yuta Okabe, Duo Feng, Yuzuno Shimoda, Zoughu Song, and et al. 2021. "Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice" International Journal of Molecular Sciences 22, no. 2: 722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020722