Novel Tyrosine Kinase Targets in Urothelial Carcinoma

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Targeting ERB-B: Epidermal Growth Factor Receptor (EGFR) and HER-2
2.1. Molecular Biology of the EGFR
2.2. Molecular Biology of the HER2 Receptor
2.3. Clinical Trials of EGFR
2.4. Clinical Trials of HER-2
3. Targeting Fibroblast Growth Factor Receptor (FGFR)
3.1. Molecular Biology of FGFR
3.2. Clinical Trials in FGFR
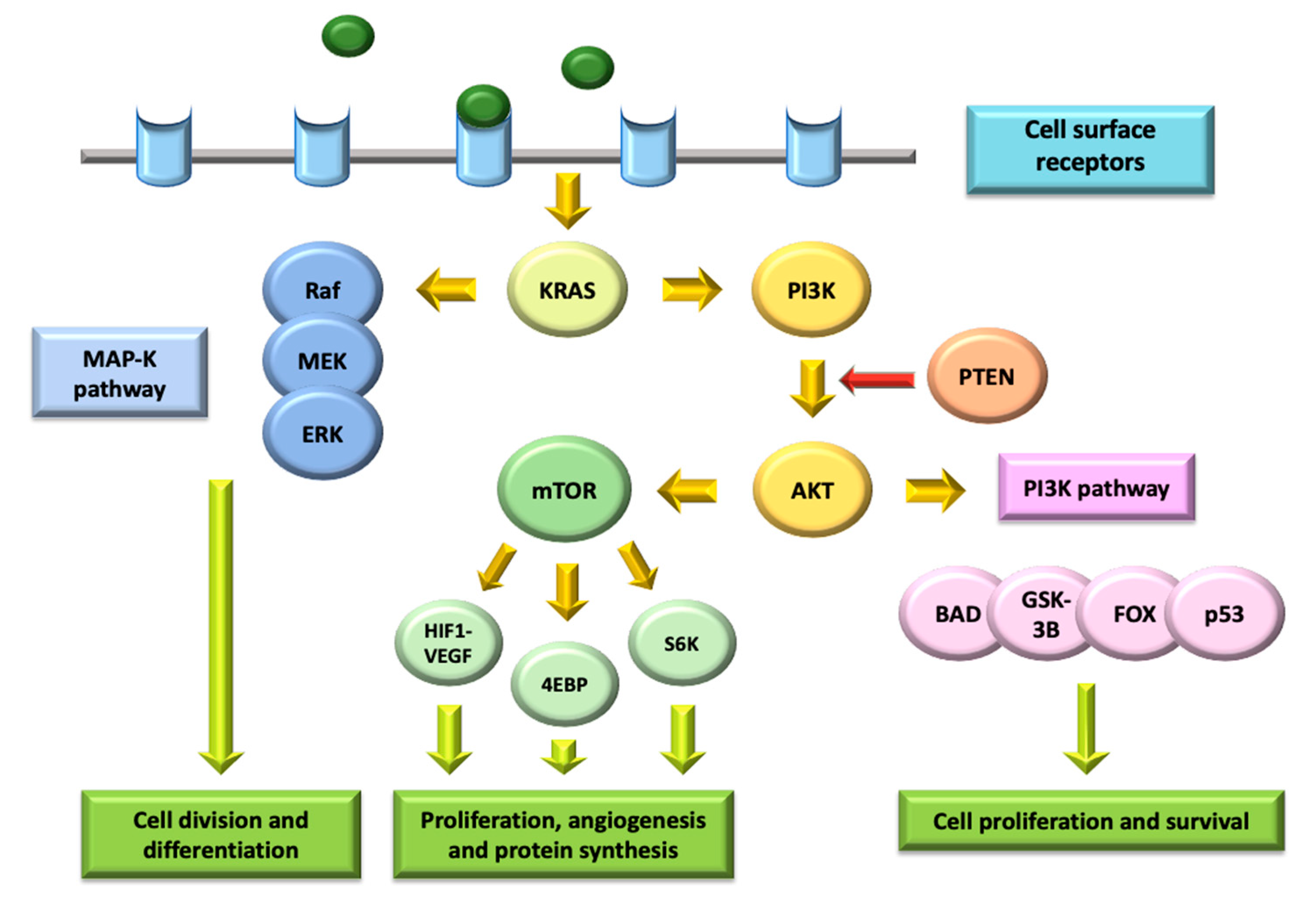
4. Targeting the PI3K/AKT/mTOR Pathway
4.1. Molecular Biology of the PI3K/AKT/mTOR Pathway
4.2. Clinical Trials in PI3K/AKT/mTOR Pathway
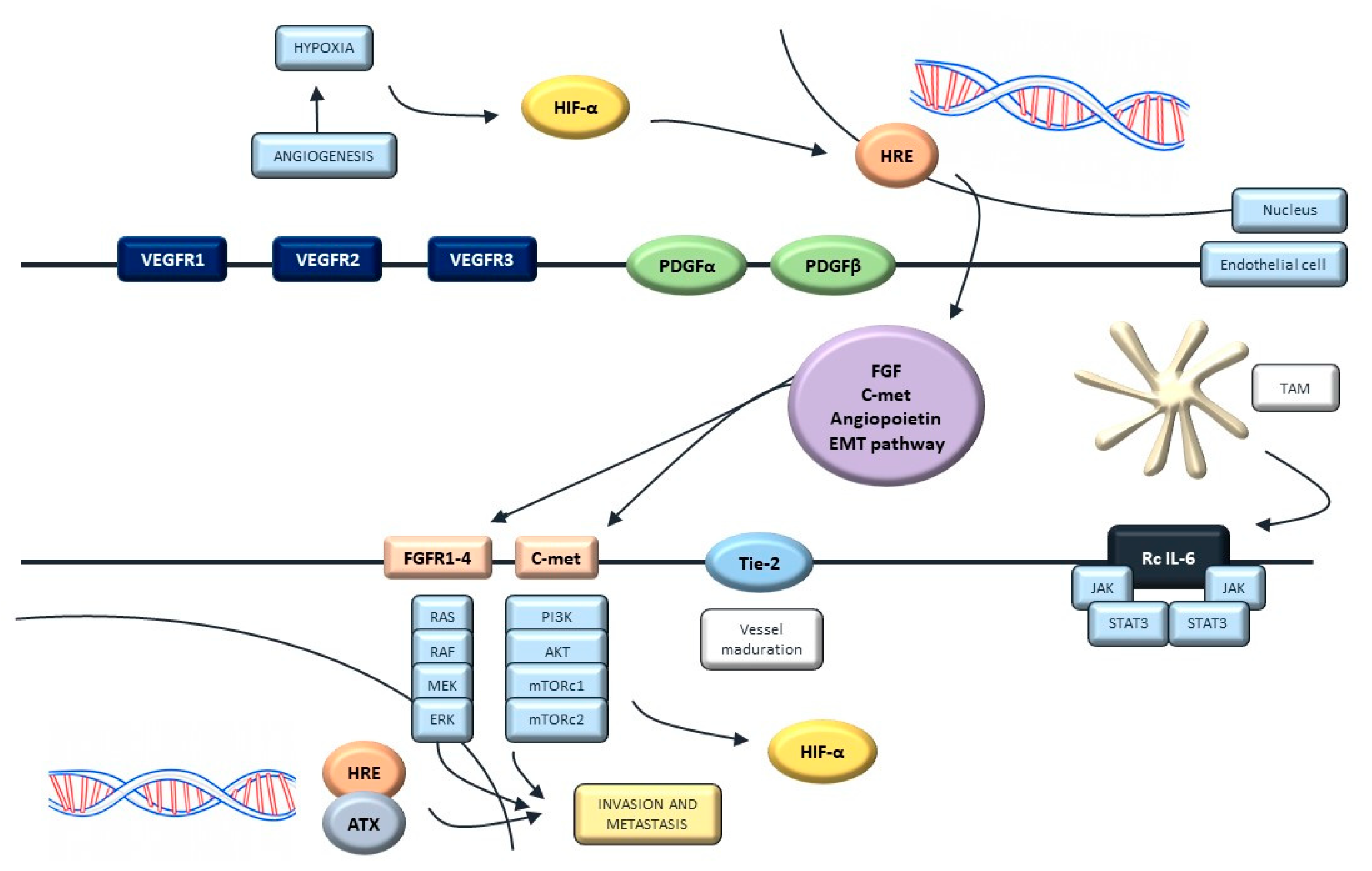
5. Targeting Angiogenesis
5.1. Molecular Biology of Angiogenesis
5.2. Clinical Trials Targeting Angiogenesis
6. Other Targets in Urothelial Carcinoma
6.1. Targeting the Anaplastic Lymphoma Kinase (ALK)
6.2. Targeting NOTCH Pathway
6.3. Targeting c-MET and SRC
6.4. Targeting Bruton’s Tyrosine-Kinase (BTK)
6.5. Targeting AXL
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AE | Adverse Events |
| ALK | Anaplastic Lymphoma Kinase |
| BC | Bladder Cancer |
| BCG | Bacillus Calmette–Guérin |
| BCRP | Bruton’s Tyrosine-Kinase |
| COX-2 | Cyclooxygenase-2 |
| CRS | Central Serous Retinophaty |
| CT | Chemotherapy |
| DFS | Disease-Free Survival |
| DLL | Delta-like canonical NOTCH ligand |
| DLT | Dose-limiting toxicity |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| FDA | Food and Drug Administration |
| FGF | Fibroblast Growth Factor |
| FGFR | Fibroblast Growth Factor Receptor |
| FISH | Fluorescence In Situ Hybridization |
| HDI | Human Development Index |
| HGF | Hepatocyte Growth Factor |
| HIP | Hypoxia Inducible Factor |
| HR | Hazard Ratio |
| IHC | Immunohistochemistry |
| iNOS | Inducible Nitric Oxide Synthase |
| IT | Immunotherapy |
| ICI | Immune Checkpoint Inhibitors |
| JAG | Jagged Canonical NOTCH ligand |
| MAPK | Mitogen Activated Protein-Kinase |
| MIBC | Muscle-Invasive Bladder Cancer |
| miRNAs | micro-RNAs |
| mOS | Median Overall Survival |
| mPFS | Median Progression–Free Survival |
| mTOR | Mamalian Target of Rapamycin |
| mUC | Metastatic Urothelial Cancer |
| NGS | Next-Generation Sequencing |
| NMIBC | Non-Muscle-Invasive Bladder Cancer |
| ORR | Overall Response Rate |
| OS | Overall Survival |
| PCNA | Proliferating Cell Nuclear Antigen |
| PDGF | Platelet-Derived Growth Factor |
| PFS | Progression-Free Survival |
| PI3K | Phosphatidylinositol-3-kinase |
| PKC | Protein kinase C |
| PLC | Phosphatidylinositol-Specific Phospholipase C |
| PUMA | p53-Upregulated Modulator of Apoptosis |
| RFS | Recurrence-free survival |
| RON | Recepteur d’Origine Nantais |
| SCC | Squamous Cell Carcinoma |
| TCGA | The Cancer Genome Atlas |
| TSC | Tuberous Sclerosis Complex |
| TGF | Transforming Growth Factor |
| TMB | Tumor Mutation Burden |
| UUT-TCC | Upper urinary tract transitional cell carcinoma |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Bladder. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/30-Bladder-fact-sheet.pdf (accessed on 20 December 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder Cancer. Nat. Rev. Dis. Primer 2017, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-Invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Mar, N.; Dayyani, F. Management of Urothelial Bladder Cancer in Clinical Practice: Real-World Answers to Difficult Questions. J. Oncol. Pract. 2019, 15, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Orsola, A.; Leow, J.J.; Wiegel, T.; De Santis, M.; Horwich, A. Bladder Cancer: ESMO Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, iii40–iii48. [Google Scholar] [CrossRef] [PubMed]
- Bladder Cancer Treatment Recommendations. Available online: https://www.esmo.org/guidelines/genitourinary-cancers/bladder-cancer/eupdate-bladder-cancer-treatment-recommendations2 (accessed on 11 December 2020).
- Montazeri, K.; Bellmunt, J. Erdafitinib for the Treatment of Metastatic Bladder Cancer. Expert Rev. Clin. Pharmacol. 2020, 13, 1–6. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Choudhury, A. Bladder Preservation for Muscle Invasive Bladder Cancer. Bladder Cancer 2016, 2, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Ericson, K.M.; Isinger, A.P.; Isfoss, B.L.; Nilbert, M.C. Low Frequency of Defective Mismatch Repair in a Population-Based Series of Upper Urothelial Carcinoma. BMC Cancer 2005, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Iyer, G.; Rosenberg, J.E. Novel Therapies in Urothelial Carcinoma: A Biomarker-Driven Approach. Ann. Oncol. 2018, 29, 2302–2312. [Google Scholar] [CrossRef]
- Lo, H.-W.; Hung, M.-C. Nuclear EGFR Signalling Network in Cancers: Linking EGFR Pathway to Cell Cycle Progression, Nitric Oxide Pathway and Patient Survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanabata, Y.; Nakajima, Y.; Morita, K.; Kayamori, K.; Omura, K. Coexpression of SGLT1 and EGFR Is Associated with Tumor Differentiation in Oral Squamous Cell Carcinoma. Odontology 2012, 100, 156–163. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, X.; Ali-Osman, F.; Keir, S.; Lo, H.-W. EGFR and EGFRvIII Interact with PUMA to Inhibit Mitochondrial Translocalization of PUMA and PUMA-Mediated Apoptosis Independent of EGFR Kinase Activity. Cancer Lett. 2010, 294, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jahng, W.J.; Di Vizio, D.; Lee, J.S.; Jhaveri, R.; Rubin, M.A.; Shisheva, A.; Freeman, M.R. The Phosphoinositide Kinase PIKfyve Mediates Epidermal Growth Factor Receptor Trafficking to the Nucleus. Cancer Res. 2007, 67, 9229–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, L.-Y.; Tseng, J.T.; Lee, Y.-C.; Xia, W.; Wang, Y.-N.; Wu, M.-L.; Chuang, Y.-H.; Lai, C.-H.; Chang, W.-C. Nuclear Epidermal Growth Factor Receptor (EGFR) Interacts with Signal Transducer and Activator of Transcription 5 (STAT5) in Activating Aurora-A Gene Expression. Nucleic Acids Res. 2008, 36, 4337–4351. [Google Scholar] [CrossRef]
- Wang, S.-C.; Lien, H.-C.; Xia, W.; Chen, I.-F.; Lo, H.-W.; Wang, Z.; Ali-Seyed, M.; Lee, D.-F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and Transactivation of the COX-2 Promoter by Nuclear Tyrosine Kinase Receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Wang, Y.-N.; Xia, W.; Hsu, S.-C.; Lai, C.-C.; Li, L.-Y.; Chang, W.-C.; Wang, Y.; Hsu, M.-C.; Yu, Y.-L.; et al. RNA Helicase A Is a DNA-Binding Partner for EGFR-Mediated Transcriptional Activation in the Nucleus. Proc. Natl. Acad. Sci. USA 2010, 107, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-C.; Nakajima, Y.; Yu, Y.-L.; Xia, W.; Chen, C.-T.; Yang, C.-C.; McIntush, E.W.; Li, L.-Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine Phosphorylation Controls PCNA Function through Protein Stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Yu, S.; Zhuang, L.; Xia, S.; Zhao, Z.; Rong, L. Induction of ERBB2 Nuclear Transport after Radiation in Breast Cancer Cells. J. Huazhong Univ. Sci. Technol. [Med. Sci.] 2009, 29, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Sjödahl, G.; Chebil, G.; Liedberg, F.; Höglund, M. HER2 and EGFR Amplification and Expression in Urothelial Carcinoma Occurs in Distinct Biological and Molecular Contexts. Oncotarget 2017, 8, 48905–48914. [Google Scholar] [CrossRef] [Green Version]
- Mooso, B.A.; Vinall, R.L.; Mudryj, M.; Yap, S.A.; deVere White, R.W.; Ghosh, P.M. The Role of EGFR Family Inhibitors in Muscle Invasive Bladder Cancer: A Review of Clinical Data and Molecular Evidence. J. Urol. 2015, 193, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Stern, D.F. The Biology of ErbB-2/Neu/HER-2 and Its Role in Cancer. Biochim. Biophys. Acta 1994, 1198, 165–184. [Google Scholar] [CrossRef]
- Van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor Protein-Tyrosine Kinases and Their Signal Transduction Pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Alroy, I.; Yarden, Y. The ErbB Signaling Network in Embryogenesis and Oncogenesis: Signal Diversification through Combinatorial Ligand-Receptor Interactions. FEBS Lett. 1997, 410, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 Oncoprotein of Human Carcinomas May Function Solely as a Shared Coreceptor for Multiple Stroma-Derived Growth Factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkas-Kramarski, R.; Shelly, M.; Guarino, B.C.; Wang, L.M.; Lyass, L.; Alroy, I.; Alimandi, M.; Kuo, A.; Moyer, J.D.; Lavi, S.; et al. ErbB Tyrosine Kinases and the Two Neuregulin Families Constitute a Ligand-Receptor Network. Mol. Cell. Biol. 1998, 18, 6090–6101. [Google Scholar] [CrossRef] [Green Version]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Lavi, S.; Iwai, K.; Reiss, Y.; Ciechanover, A.; et al. Ubiquitin Ligase Activity and Tyrosine Phosphorylation Underlie Suppression of Growth Factor Signaling by C-Cbl/Sli-1. Mol. Cell 1999, 4, 1029–1040. [Google Scholar] [CrossRef]
- Kasprzyk, P.G.; Song, S.U.; Di Fiore, P.P.; King, C.R. Therapy of an Animal Model of Human Gastric Cancer Using a Combination of Anti-ErbB-2 Monoclonal Antibodies. Cancer Res. 1992, 52, 2771–2776. [Google Scholar] [PubMed]
- The cancer genome atlas pan-cancer analysis project. Nature 2013, 45, 1113.
- Ross, J.S.; Wang, K.; Gay, L.M.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Jennings, T.A.; Otto, G.A.; Donahue, A.; He, J.; et al. A High Frequency of Activating Extracellular Domain ERBB2 (HER2) Mutation in Micropapillary Urothelial Carcinoma. Clin. Cancer Res. 2014, 20, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, R.E.; Hussain, M.; Bianco, F.J.; Vaishampayan, U.; Tabazcka, P.; Sakr, W.A.; Pontes, J.E.; Wood, D.P.; Grignon, D.J. Her-2/Neu Overexpression in Muscle-Invasive Urothelial Carcinoma of the Bladder: Prognostic Significance and Comparative Analysis in Primary and Metastatic Tumors. Clin. Cancer Res. 2001, 7, 2440–2447. [Google Scholar] [PubMed]
- Arnould, L.; Roger, P.; Macgrogan, G.; Chenard, M.-P.; Balaton, A.; Beauclair, S.; Penault-Llorca, F. Accuracy of HER2 Status Determination on Breast Core-Needle Biopsies (Immunohistochemistry, FISH, CISH and SISH vs FISH). Mod. Pathol. 2012, 25, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.P.; Tangen, C.M.; Van Veldhuizen, P.J.; Goodwin, J.W.; Twardowski, P.W.; Atkins, J.N.; Kakhil, S.R.; Lange, M.K.; Mansukhani, M.; Crawford, E.D. Results of the Southwest Oncology Group Phase II Evaluation (Study S0031) of ZD1839 for Advanced Transitional Cell Carcinoma of the Urothelium. BJU Int. 2010, 105, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Philips, G.K.; Halabi, S.; Sanford, B.L.; Bajorin, D.; Small, E.J. Cancer and Leukemia Group B A Phase II Trial of Cisplatin (C), Gemcitabine (G) and Gefitinib for Advanced Urothelial Tract Carcinoma: Results of Cancer and Leukemia Group B (CALGB) 90102. Ann. Oncol. 2009, 20, 1074–1079. [Google Scholar] [CrossRef]
- Miller, K.; Morant, R.; Stenzl, A.; Zuna, I.; Wirth, M. A Phase II Study of the Central European Society of Anticancer-Drug Research (CESAR) Group: Results of an Open-Label Study of Gemcitabine plus Cisplatin with or without Concomitant or Sequential Gefitinib in Patients with Advanced or Metastatic Transitional Cell Carcinoma of the Urothelium. Urol. Int. 2016, 96, 5–13. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Campanile, A.; Antic, T.; Yap, K.L.; Fitzpatrick, C.A.; Wade, J.L.; Karrison, T.; Stadler, W.M.; Nakamura, Y.; O’Donnell, P.H. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. J. Clin. Oncol. 2016, 34, 2165–2171. [Google Scholar] [CrossRef] [Green Version]
- Font Pous, A.; Puente, J.; Castellano, D.E.; Real, F.X.; Climent, M.A.; Gonzalez del Alba, A.A.; Oudard, S.; Vazquez Mazon, F.J.; Morales Barrera, R.; Virizuela, J.V.; et al. Phase II Trial of Afatinib in Patients with Advanced/Metastatic Urothelial Carcinoma (UC) with Genetic Alterations in ERBB Receptors 1-3 Who Failed on Platinum-Based Chemotherapy (CT). J. Clin. Oncol. 2018, 36, TPS540. [Google Scholar] [CrossRef]
- Wong, Y.-N.; Litwin, S.; Vaughn, D.; Cohen, S.; Plimack, E.R.; Lee, J.; Song, W.; Dabrow, M.; Brody, M.; Tuttle, H.; et al. Phase II Trial of Cetuximab With or Without Paclitaxel in Patients With Advanced Urothelial Tract Carcinoma. J. Clin. Oncol. 2012, 30, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Daignault, S.; Agarwal, N.; Grivas, P.D.; Siefker-Radtke, A.O.; Puzanov, I.; MacVicar, G.R.; Levine, E.G.; Srinivas, S.; Twardowski, P.; et al. A Randomized Phase 2 Trial of Gemcitabine/Cisplatin With or Without Cetuximab in Patients With Advanced Urothelial Carcinoma. Cancer 2014, 120, 2684–2693. [Google Scholar] [CrossRef] [Green Version]
- Jack, S.; Madhivanan, K.; Ramadesikan, S.; Subramanian, S.; Edwards, D.F.; Elzey, B.D.; Dhawan, D.; McCluskey, A.; Kischuk, E.M.; Loftis, A.R.; et al. A Novel, Safe, Fast and Efficient Treatment for Her2-Positive and Negative Bladder Cancer Utilizing an EGF-Anthrax Toxin Chimera. Int. J. Cancer 2020, 146, 449–460. [Google Scholar] [CrossRef]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 Expression Status in Diverse Cancers: Review of Results from 37,992 Patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.H.A.; MacVicar, G.R.; Petrylak, D.P.; Dunn, R.L.; Vaishampayan, U.; Lara, P.N.; Chatta, G.S.; Nanus, D.M.; Glode, L.M.; Trump, D.L.; et al. Trastuzumab, Paclitaxel, Carboplatin, and Gemcitabine in Advanced Human Epidermal Growth Factor Receptor-2/Neu-Positive Urothelial Carcinoma: Results of a Multicenter Phase II National Cancer Institute Trial. J. Clin. Oncol. 2007, 25, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Culine, S.; Vano, Y.; Goldwasser, F.; Théodore, C.; Nguyen, T.; Voog, E.; Banu, E.; Vieillefond, A.; Priou, F.; et al. Multicentre Randomised Phase II Trial of Gemcitabine+platinum, with or without Trastuzumab, in Advanced or Metastatic Urothelial Carcinoma Overexpressing Her2. Eur. J. Cancer 2015, 51, 45–54. [Google Scholar] [CrossRef]
- Wülfing, C.; Machiels, J.-P.H.; Richel, D.J.; Grimm, M.-O.; Treiber, U.; De Groot, M.R.; Beuzeboc, P.; Parikh, R.; Pétavy, F.; El-Hariry, I.A. A Single-Arm, Multicenter, Open-Label Phase 2 Study of Lapatinib as the Second-Line Treatment of Patients with Locally Advanced or Metastatic Transitional Cell Carcinoma. Cancer 2009, 115, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Mamtani, R.; Keefe, S.; Guzzo, T.; Malkowicz, S.B.; Vaughn, D.J. Cisplatin, Gemcitabine, and Lapatinib as Neoadjuvant Therapy for Muscle-Invasive Bladder Cancer. Cancer Res. Treat. 2016, 48, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Huddart, R.A.; Elliott, T.; Sarker, S.-J.; Ackerman, C.; Jones, R.; Hussain, S.; Crabb, S.; Jagdev, S.; Chester, J.; et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib Versus Placebo After First-Line Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 1/2-Positive Metastatic Bladder Cancer. J. Clin. Oncol. 2017, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, B.; Breeze, A.L. Structure, Activation and Dysregulation of Fibroblast Growth Factor Receptor Kinases: Perspectives for Clinical Targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor Signaling Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Vida, A.; Saggese, M.; Hughes, S.; Rudman, S.; Chowdhury, S.; Smith, N.R.; Lawrence, P.; Rooney, C.; Dougherty, B.; Landers, D.; et al. Complexity of FGFR Signalling in Metastatic Urothelial Cancer. J. Hematol. Oncol. J. Hematol. Oncol. 2015, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast Growth Factor Receptor Signaling in Hereditary and Neoplastic Disease: Biologic and Clinical Implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cihoric, N.; Savic, S.; Schneider, S.; Ackermann, I.; Bichsel-Naef, M.; Schmid, R.A.; Lardinois, D.; Gugger, M.; Bubendorf, L.; Zlobec, I.; et al. Prognostic Role of FGFR1 Amplification in Early-Stage Non-Small Cell Lung Cancer. Br. J. Cancer 2014, 110, 2914–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Fibroblast Growth Factor Receptors as Treatment Targets in Clinical Oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef]
- Di Martino, E.; Tomlinson, D.C.; Williams, S.V.; Knowles, M.A. A Place for Precision Medicine in Bladder Cancer: Targeting the FGFRs. Future Oncol. 2016, 12, 2243–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Šuštić, T.; Leite de Oliveira, R.; Lieftink, C.; Halonen, P.; van de Ven, M.; Beijersbergen, R.L.; van den Heuvel, M.M.; Bernards, R.; van der Heijden, M.S. A Functional Genetic Screen Identifies the Phosphoinositide 3-Kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur. Urol. 2017, 71, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour Cell Responses to New Fibroblast Growth Factor Receptor Tyrosine Kinase Inhibitors and Identification of a Gatekeeper Mutation in FGFR3 as a Mechanism of Acquired Resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration FDA Grants Accelerated Approval to Erdafitinib for Metastatic Urothelial Carcinoma. 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-erdafitinib-metastatic-urothelial-carcinoma (accessed on 9 December 2020).
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. ERDAFITINIB in Locally Advanced or Metastatic Urothelial Carcinoma (MUC): Long-Term Outcomes in BLC2001. J. Clin. Oncol. 2020, 38, 5015. [Google Scholar] [CrossRef]
- Janssen Research & Development, LLC. A Phase 3 Study of Erdafitinib Compared With Vinflunine or Docetaxel or Pembrolizumab in Subjects with Advanced Urothelial Cancer and Selected FGFR Gene Aberrations. Available online: clinicaltrials.gov (accessed on 9 December 2020).
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non–T-Cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, V.; Loriot, Y.; Rutkowski, P.; Beato, C.; Felip, E.; Duran, I.; Kowalski, D.; Siena, S.; Cortinovis, D.; Geoffrois, L.; et al. Evolving Development of PD-1 Therapy: Cetrelimab (JNJ-63723283) from Monotherapy to Combination with Erdafitinib. J. Clin. Oncol. 2020, 38, 3055. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizman, N.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.N.; De Giorgi, U.; Gupta, S.; Burris, H.A.; et al. Infigratinib in Upper Tract Urothelial Carcinoma vs Urothelial Carcinoma of the Bladder and Association with Comprehensive Genomic Profiling/Cell-Free DNA Results. J. Clin. Oncol. 2019, 37, 4510. [Google Scholar] [CrossRef]
- Pal, S.K.; Bajorin, D.; Dizman, N.; Hoffman-Censits, J.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.; De Giorgi, U.; Gupta, S.; et al. Infigratinib in Upper Tract Urothelial Carcinoma versus Urothelial Carcinoma of the Bladder and Its Association with Comprehensive Genomic Profiling and/or Cell-Free DNA Results. Cancer 2020, 126, 2597–2606. [Google Scholar] [CrossRef]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in Patients with Advanced Cancers Selected by FGFR MRNA Expression: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncol. 2019, 20, 1454–1466. [Google Scholar] [CrossRef]
- Quinn, D.I.; Petrylak, D.P.; Bellmunt, J.; Necchi, A.; Gurney, H.; Lee, J.-L.; Van Der Heijden, M.S.; Rosenbaum, E.; Penel, N.; Pang, S.-T.; et al. FORT-1: Phase II/III Study of Rogaratinib versus Chemotherapy (CT) in Patients (Pts) with Locally Advanced or Metastatic Urothelial Carcinoma (UC) Selected Based on FGFR1/3 MRNA Expression. J. Clin. Oncol. 2020, 38, 489. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Gajate, P.; Morales-Barrera, R.; Lee, J.-L.; Necchi, A.; Penel, N.; Zagonel, V.; Sierecki, M.R.; Piciu, A.-M.; Ellinghaus, P.; et al. Safety and Preliminary Efficacy of Rogaratinib in Combination with Atezolizumab in a Phase Ib/II Study (FORT-2) of First-Line Treatment in Cisplatin-Ineligible Patients (Pts) with Locally Advanced or Metastatic Urothelial Cancer (UC) and FGFR MRNA Overexpression. J. Clin. Oncol. 2020, 38, 5014. [Google Scholar] [CrossRef]
- Necchi, A.P.D. Interim Results of Fight-201, a Phase 2, Open-Label, Multicenter Study of INCB054828 in Patients (Pts) with Metastatic or Surgically Unresectable Urothelial Carcinoma (UC) Harboring Fibroblast Growth Factor (FGF)/FGF Receptor (FGFR) Genetic Alterations (GA). Ann. Oncol. 2018, 29, Viii303–Viii331. [Google Scholar]
- Meric-Bernstam, F.; Goyal, L.; Tran, B.; Matos, I.; Arkenau, H.-T.; He, H.; Huang, J.; Bahleda, R. Abstract CT238: TAS-120 in Patients with Advanced Solid Tumors Bearing FGF/FGFR Aberrations: A Phase I Study. In Proceedings of the Clinical Trials, AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; p. CT238. [Google Scholar]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-Escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milowsky, M.I.; Dittrich, C.; Durán, I.; Jagdev, S.; Millard, F.E.; Sweeney, C.J.; Bajorin, D.; Cerbone, L.; Quinn, D.I.; Stadler, W.M.; et al. Phase 2 Trial of Dovitinib in Patients with Progressive FGFR3-Mutated or FGFR3 Wild-Type Advanced Urothelial Carcinoma. Eur. J. Cancer 2014, 50, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.M.; Bivalacqua, T.J.; Ross, A.E.; Netto, G.J.; Baras, A.; Park, J.C.; Chapman, C.; Masterson, T.A.; Koch, M.O.; Bihrle, R.; et al. A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12-157. Clin. Cancer Res. 2017, 23, 3003–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Balar, A.; Gravis, G.; Jones, R.; Ravaud, A.; Florence, J.; Grivas, P.; Petrylak, D.P.; Galsky, M.; Carles, J.; et al. An Adaptive, Biomarker Directed Platform Study in Metastatic Urothelial Cancer (BISCAY) with Durvalumab in Combination with Targeted Therapies. Ann. Oncol. 2019, 30, v356–v357. [Google Scholar] [CrossRef]
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Yu, J.; Kim, R. Prevention and Treatment of FGFR Inhibitor-Associated Toxicities. Crit. Rev. Oncol. Hematol. 2020, 155, 103091. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-MTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt Signalling Pathway and Cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Testa, J.R.; Bellacosa, A. AKT Plays a Central Role in Tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Slingerland, J.M. Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression. Cell Cycle Georget. Tex 2003, 2, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.; Parsons, R. PTEN: Life as a Tumor Suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and Downstream of MTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palicharla, V.R.; Maddika, S. HACE1 Mediated K27 Ubiquitin Linkage Leads to YB-1 Protein Secretion. Cell. Signal. 2015, 27, 2355–2362. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the MTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Li, X.; Zhang, J. MTOR Signaling in Cancer and MTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R. MTOR and Lysosome Regulation. F1000prime Rep. 2014, 6, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kim, K.; Sung, H.H.; Jeon, H.G.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Lee, H.M.; Choi, H.-Y.; Kwon, G.-Y.; et al. Molecular Characterization of Urothelial Carcinoma of the Bladder and Upper Urinary Tract. Transl. Oncol. 2018, 11, 37–42. [Google Scholar] [CrossRef]
- Osei-Amponsa, V.; Buckwalter, J.M.; Shuman, L.; Zheng, Z.; Yamashita, H.; Walter, V.; Wildermuth, T.; Ellis-Mohl, J.; Liu, C.; Warrick, J.I.; et al. Hypermethylation of FOXA1 and Allelic Loss of PTEN Drive Squamous Differentiation and Promote Heterogeneity in Bladder Cancer. Oncogene 2020, 39, 1302–1317. [Google Scholar] [CrossRef]
- Necchi, A.; Madison, R.; Raggi, D.; Jacob, J.M.; Bratslavsky, G.; Shapiro, O.; Elvin, J.A.; Vergilio, J.-A.; Killian, J.K.; Ngo, N.; et al. Comprehensive Assessment of Immuno-Oncology Biomarkers in Adenocarcinoma, Urothelial Carcinoma, and Squamous-Cell Carcinoma of the Bladder. Eur. Urol. 2020, 77, 548–556. [Google Scholar] [CrossRef]
- Seront, E.; Rottey, S.; Sautois, B.; Kerger, J.; D’Hondt, L.A.; Verschaeve, V.; Canon, J.-L.; Dopchie, C.; Vandenbulcke, J.M.; Whenham, N.; et al. Phase II Study of Everolimus in Patients with Locally Advanced or Metastatic Transitional Cell Carcinoma of the Urothelial Tract: Clinical Activity, Molecular Response, and Biomarkers. Ann. Oncol. 2012, 23, 2663–2670. [Google Scholar] [CrossRef]
- Milowsky, M.I.; Iyer, G.; Regazzi, A.M.; Al-Ahmadie, H.; Gerst, S.R.; Ostrovnaya, I.; Gellert, L.L.; Kaplan, R.; Garcia-Grossman, I.R.; Pendse, D.; et al. Phase II Study of Everolimus in Metastatic Urothelial Cancer. BJU Int. 2013, 112, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Niegisch, G.; Retz, M.; Thalgott, M.; Balabanov, S.; Honecker, F.; Ohlmann, C.H.; Stöckle, M.; Bögemann, M.; Vom Dorp, F.; Gschwend, J.; et al. Second-Line Treatment of Advanced Urothelial Cancer with Paclitaxel and Everolimus in a German Phase II Trial (AUO Trial AB 35/09). Oncology 2015, 89, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome Sequencing Identifies a Basis for Everolimus Sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seront, E.; Rottey, S.; Filleul, B.; Glorieux, P.; Goeminne, J.-C.; Verschaeve, V.; Vandenbulcke, J.-M.; Sautois, B.; Boegner, P.; Gillain, A.; et al. Phase II Study of Dual Phosphoinositol-3-Kinase (PI3K) and Mammalian Target of Rapamycin (MTOR) Inhibitor BEZ235 in Patients with Locally Advanced or Metastatic Transitional Cell Carcinoma. BJU Int. 2016, 118, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Iyer, G.; Tully, C.M.; Garcia-Grossman, I.R.; Scott, S.N.; Boyd, M.E.; McCoy, A.S.; Berger, M.F.; Al-Ahmadie, H.; Solit, D.B.; Rosenberg, J.E.; et al. Phase 2 Study of the Pan-Isoform PI3 Kinase Inhibitor BKM120 in Metastatic Urothelial Carcinoma Patients. J. Clin. Oncol. 2015, 33, 324. [Google Scholar] [CrossRef]
- Maishi, N.; Hida, K. Tumor Endothelial Cells Accelerate Tumor Metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senger, D.R.; Connolly, D.T.; Van de Water, L.; Feder, J.; Dvorak, H.F. Purification and NH2-Terminal Amino Acid Sequence of Guinea Pig Tumor-Secreted Vascular Permeability Factor. Cancer Res. 1990, 50, 1774–1778. [Google Scholar] [PubMed]
- Semenza, G.L. HIF-1: Using Two Hands to Flip the Angiogenic Switch. Cancer Metastasis Rev. 2000, 19, 59–65. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y.R. Molecular Control of Endothelial Cell Behaviour during Blood Vessel Morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Theodoropoulos, V.E.; Lazaris, A.C.; Sofras, F.; Gerzelis, I.; Tsoukala, V.; Ghikonti, I.; Manikas, K.; Kastriotis, I. Hypoxia-Inducible Factor 1 Alpha Expression Correlates with Angiogenesis and Unfavorable Prognosis in Bladder Cancer. Eur. Urol. 2004, 46, 200–208. [Google Scholar] [CrossRef]
- Pignot, G.; Bieche, I.; Vacher, S.; Güet, C.; Vieillefond, A.; Debré, B.; Lidereau, R.; Amsellem-Ouazana, D. Large-Scale Real-Time Reverse Transcription-PCR Approach of Angiogenic Pathways in Human Transitional Cell Carcinoma of the Bladder: Identification of VEGFA as a Major Independent Prognostic Marker. Eur. Urol. 2009, 56, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, S.; Fauconnet, S.; Chabannes, E.; Henry, P.C.; Adessi, G.; Bittard, H. Serum Levels of Vascular Endothelial Growth Factor as a Prognostic Factor in Bladder Cancer. J. Urol. 2001, 166, 1275–1279. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Amaro, T.; Lobo, F.; Longatto-Filho, A. The Aggressiveness of Urothelial Carcinoma Depends to a Large Extent on Lymphovascular Invasion--the Prognostic Contribution of Related Molecular Markers. Histopathology 2009, 55, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Siefker-Radtke, A.O.; Kamat, A.M.; Corn, P.G.; Matin, S.F.; Grossman, H.B.; Millikan, R.E.; Dinney, C.P.N. Neoadjuvant Chemotherapy with DD-MVAC and Bevacizumab in High-Risk Urothelial Cancer: Results from a Phase II Trial at the M. D. Anderson Cancer Center. J. Clin. Oncol. 2012, 30, 261. [Google Scholar] [CrossRef]
- Balar, A.V.; Apolo, A.B.; Ostrovnaya, I.; Mironov, S.; Iasonos, A.; Trout, A.; Regazzi, A.M.; Garcia-Grossman, I.R.; Gallagher, D.J.; Milowsky, M.I.; et al. Phase II Study of Gemcitabine, Carboplatin, and Bevacizumab in Patients with Advanced Unresectable or Metastatic Urothelial Cancer. J. Clin. Oncol. 2013, 31, 724–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.P.; de Wit, R.; Chi, K.N.; Drakaki, A.; Sternberg, C.N.; Nishiyama, H.; Castellano, D.; Hussain, S.; Fléchon, A.; Bamias, A.; et al. Ramucirumab plus Docetaxel versus Placebo plus Docetaxel in Patients with Locally Advanced or Metastatic Urothelial Carcinoma after Platinum-Based Therapy (RANGE): A Randomised, Double-Blind, Phase 3 Trial. Lancet Lond. 2017, 390, 2266–2277. [Google Scholar] [CrossRef] [Green Version]
- Twardowski, P.; Stadler, W.M.; Frankel, P.; Lara, P.N.; Ruel, C.; Chatta, G.; Heath, E.; Quinn, D.I.; Gandara, D.R. Phase II Study of Aflibercept (VEGF-Trap) in Patients with Recurrent or Metastatic Urothelial Cancer, a California Cancer Consortium Trial. Urology 2010, 76, 923–926. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.J.; Milowsky, M.I.; Gerst, S.R.; Ishill, N.; Riches, J.; Regazzi, A.; Boyle, M.G.; Trout, A.; Flaherty, A.-M.; Bajorin, D.F. Phase II Study of Sunitinib in Patients with Metastatic Urothelial Cancer. J. Clin. Oncol. 2010, 28, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; González-Larriba, J.L.; Prior, C.; Maroto, P.; Carles, J.; Castellano, D.; Mellado, B.; Gallardo, E.; Perez-Gracia, J.L.; Aguilar, G.; et al. Phase II Study of Sunitinib as First-Line Treatment of Urothelial Cancer Patients Ineligible to Receive Cisplatin-Based Chemotherapy: Baseline Interleukin-8 and Tumor Contrast Enhancement as Potential Predictive Factors of Activity. Ann. Oncol. 2011, 22, 2646–2653. [Google Scholar] [CrossRef]
- Geldart, T.; Chester, J.; Casbard, A.; Crabb, S.; Elliott, T.; Protheroe, A.; Huddart, R.A.; Mead, G.; Barber, J.; Jones, R.J.; et al. SUCCINCT: An Open-Label, Single-Arm, Non-Randomised, Phase 2 Trial of Gemcitabine and Cisplatin Chemotherapy in Combination with Sunitinib as First-Line Treatment for Patients with Advanced Urothelial Carcinoma. Eur. Urol. 2015, 67, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Grivas, P.D.; Daignault, S.; Tagawa, S.T.; Nanus, D.M.; Stadler, W.M.; Dreicer, R.; Kohli, M.; Petrylak, D.P.; Vaughn, D.J.; Bylow, K.A.; et al. Double-Blind, Randomized, Phase 2 Trial of Maintenance Sunitinib versus Placebo after Response to Chemotherapy in Patients with Advanced Urothelial Carcinoma. Cancer 2014, 120, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Iyer, G.; Apolo, A.B.; Regazzi, A.M.; Garcia-Grossman, I.R.; Pendse, D.; Ostrovnaya, I.; Chou, J.F.; Bochner, B.; Dalbagni, G.; et al. Phase II Trial of Neoadjuvant Gemcitabine (G) and Cisplatin (C) with Sunitinib in Patients (Pts) with Muscle-Invasive Bladder Cancer (MIBC). J. Clin. Oncol. 2012, 30, 4581. [Google Scholar] [CrossRef]
- Necchi, A.; Mariani, L.; Zaffaroni, N.; Schwartz, L.H.; Giannatempo, P.; Crippa, F.; Morosi, C.; Lanocita, R.; Sava, T.; Ortega, C.; et al. Pazopanib in Advanced and Platinum-Resistant Urothelial Cancer: An Open-Label, Single Group, Phase 2 Trial. Lancet Oncol. 2012, 13, 810–816. [Google Scholar] [CrossRef]
- Pili, R.; Qin, R.; Flynn, P.J.; Picus, J.; Millward, M.; Ho, W.M.; Pitot, H.; Tan, W.; Miles, K.M.; Erlichman, C.; et al. A Phase II Safety and Efficacy Study of the Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor Pazopanib in Patients with Metastatic Urothelial Cancer. Clin. Genitourin. Cancer 2013, 11, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Gerullis, H.; Eimer, C.; Ecke, T.H.; Georgas, E.; Arndt, C.; Otto, T. Combined Treatment with Pazopanib and Vinflunine in Patients with Advanced Urothelial Carcinoma Refractory after First-Line Therapy. Anticancer. Drugs 2013, 24, 422–425. [Google Scholar] [CrossRef]
- Narayanan, S.; Lam, A.; Vaishampayan, U.; Harshman, L.; Fan, A.; Pachynski, R.; Poushnejad, S.; Haas, D.; Li, S.; Srinivas, S. Phase II Study of Pazopanib and Paclitaxel in Patients With Refractory Urothelial Cancer. Clin. Genitourin. Cancer 2016, 14, 432–437. [Google Scholar] [CrossRef]
- Jones, R.J.; Hussain, S.A.; Protheroe, A.S.; Birtle, A.; Chakraborti, P.; Huddart, R.A.; Jagdev, S.; Bahl, A.; Stockdale, A.; Sundar, S.; et al. Randomized Phase II Study Investigating Pazopanib Versus Weekly Paclitaxel in Relapsed or Progressive Urothelial Cancer. J. Clin. Oncol. 2017, 35, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Apolo, A.B.; Parnes, H.L.; Francis, D.C.; Cordes, L.M.; Berninger, M.; Lamping, E.; Costello, R.; Trepel, J.B.; Merino, M.J.; Folio, L.; et al. A Phase II Study of Cabozantinib in Patients (Pts) with Relapsed or Refractory Metastatic Urothelial Carcinoma (MUC). J. Clin. Oncol. 2016, 34. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.4534 (accessed on 19 November 2020).
- Apolo, A.B.; Nadal, R.; Tomita, Y.; Davarpanah, N.N.; Cordes, L.M.; Steinberg, S.M.; Cao, L.; Parnes, H.L.; Costello, R.; Merino, M.J.; et al. Cabozantinib in Patients with Platinum-Refractory Metastatic Urothelial Carcinoma: An Open-Label, Single-Centre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1099–1109. [Google Scholar] [CrossRef]
- Bergerot, P.; Lamb, P.; Wang, E.; Pal, S.K. Cabozantinib in Combination with Immunotherapy for Advanced Renal Cell Carcinoma and Urothelial Carcinoma: Rationale and Clinical Evidence. Mol. Cancer Ther. 2019, 18, 2185–2193. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Tabata, K.; Hori, Y.; Tachino, S.; Okamoto, K.; Matsui, J. Abstract A92: Effects of Lenvatinib on Tumor-Associated Macrophages Enhance Antitumor Activity of PD-1 Signal Inhibitors. In Proceedings of the Immune Modulators; American Association for Cancer Research, Boston, MA, USA, 5–9 December 2015; p. A92. [Google Scholar]
- Lacal, P.M.; Graziani, G. Therapeutic Implication of Vascular Endothelial Growth Factor Receptor-1 (VEGFR-1) Targeting in Cancer Cells and Tumor Microenvironment by Competitive and Non-Competitive Inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Taylor, M.H.; Lee, C.-H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- De Pontual, L.; Kettaneh, D.; Gordon, C.T.; Oufadem, M.; Boddaert, N.; Lees, M.; Balu, L.; Lachassinne, E.; Petros, A.; Mollet, J.; et al. Germline Gain-of-Function Mutations of ALK Disrupt Central Nervous System Development. Hum. Mutat. 2011, 32, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabbó, F.; Barreca, A.; Piva, R.; Inghirami, G. European T-Cell Lymphoma Study Group ALK Signaling and Target Therapy in Anaplastic Large Cell Lymphoma. Front. Oncol. 2012, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical Features and Outcome of Patients with Non-Small-Cell Lung Cancer Who Harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [Green Version]
- Mano, H. ALKoma: A Cancer Subtype with a Shared Target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellmunt, J.; Selvarajah, S.; Rodig, S.; Salido, M.; de Muga, S.; Costa, I.; Bellosillo, B.; Werner, L.; Mullane, S.; Fay, A.P.; et al. Identification of ALK Gene Alterations in Urothelial Carcinoma. PLoS ONE 2014, 9, e103325. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israël, A. A Novel Proteolytic Cleavage Involved in Notch Signaling: The Role of the Disintegrin-Metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Bray, S.J. Notch Signalling: A Simple Pathway Becomes Complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Lin, C.; Zheng, H.; Wang, C.; Yang, L.; Chen, S.; Li, B.; Zhou, Y.; Tan, H.; Li, Y. Mutations Increased Overexpression of Notch1 in T-Cell Acute Lymphoblastic Leukemia. Cancer Cell Int. 2012, 12, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraver, A.; Fernandez-Marcos, P.J.; Cash, T.P.; Mendez-Pertuz, M.; Dueñas, M.; Maietta, P.; Martinelli, P.; Muñoz-Martin, M.; Martínez-Fernández, M.; Cañamero, M.; et al. NOTCH Pathway Inactivation Promotes Bladder Cancer Progression. J. Clin. Investig. 2015, 125, 824–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Gust, K.M.; Wyatt, A.W.; Goriki, A.; Jäger, W.; Awrey, S.; Li, N.; Oo, H.Z.; Altamirano-Dimas, M.; Buttyan, R.; et al. Not All NOTCH Is Created Equal: The Oncogenic Role of NOTCH2 in Bladder Cancer and Its Implications for Targeted Therapy. Clin. Cancer Res. 2016, 22, 2981–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Wang, D.-S.; Li, Q.-J.; Sun, W.; Zhang, Y.; Dou, K.-F. The Down-Regulation of Notch1 Inhibits the Invasion and Migration of Hepatocellular Carcinoma Cells by Inactivating the Cyclooxygenase-2/Snail/E-Cadherin Pathway in Vitro. Dig. Dis. Sci. 2013, 58, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. NOTCH Decoys That Selectively Block DLL/NOTCH or JAG/NOTCH Disrupt Angiogenesis by Unique Mechanisms to Inhibit Tumor Growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Y.; Kong, D.; Ahmad, A.; Banerjee, S.; Sarkar, F.H. Cross-Talk between MiRNA and Notch Signaling Pathways in Tumor Development and Progression. Cancer Lett. 2010, 292, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and Novel Roles of the MET Oncogene in Cancer: A Coherent Approach to Targeted Therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Miyata, Y.; Sagara, Y.; Kanda, S.; Hayashi, T.; Kanetake, H. Phosphorylated Hepatocyte Growth Factor Receptor/c-Met Is Associated with Tumor Growth and Prognosis in Patients with Bladder Cancer: Correlation with Matrix Metalloproteinase–2 and –7 and E-Cadherin. Hum. Pathol. 2009, 40, 496–504. [Google Scholar] [CrossRef]
- Yamasaki, K.; Mukai, S.; Nagai, T.; Nakahara, K.; Fujii, M.; Terada, N.; Ohno, A.; Sato, Y.; Toda, Y.; Kataoka, H.; et al. Matriptase-Induced Phosphorylation of MET Is Significantly Associated with Poor Prognosis in Invasive Bladder Cancer; an Immunohistochemical Analysis. Int. J. Mol. Sci. 2018, 19, 3708. [Google Scholar] [CrossRef] [Green Version]
- Kluth, M.; Reynolds, K.; Rink, M.; Chun, F.; Dahlem, R.; Fisch, M.; Höppner, W.; Wagner, W.; Doh, O.; Terracciano, L.; et al. Reduced Membranous MET Expression Is Linked to Bladder Cancer Progression. Cancer Genet. 2014, 207, 147–152. [Google Scholar] [CrossRef]
- Frame, M.C. Src in Cancer: Deregulation and Consequences for Cell Behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Yeatman, T.J. A Renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kwarcinski, F.E.; Brandvold, K.R.; Phadke, S.; Beleh, O.M.; Johnson, T.K.; Meagher, J.L.; Seeliger, M.A.; Stuckey, J.A.; Soellner, M.B. Conformation-Selective Analogues of Dasatinib Reveal Insight into Kinase Inhibitor Binding and Selectivity. ACS Chem. Biol. 2016, 11, 1296–1304. [Google Scholar] [CrossRef]
- Buggy, J.J.; Elias, L. Bruton Tyrosine Kinase (BTK) and Its Role in B-Cell Malignancy. Int. Rev. Immunol. 2012, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s Tyrosine Kinase (BTK) as a Promising Target in Solid Tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.R.; Liss, M.A.; Muldong, M.T.; Palazzi, K.; Strasner, A.; Ammirante, M.; Varki, N.; Shabaik, A.; Howell, S.; Kane, C.J.; et al. Tumor Infiltrating B-Cells Are Increased in Prostate Cancer Tissue. J. Transl. Med. 2014, 12, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, F.; Yu, L.; Shi, Q.; Tang, A.; Cheng, J. Ibrutinib in CLL/SLL: From Bench to Bedside (Review). Oncol. Rep. 2019, 42, 2213–2227. [Google Scholar] [CrossRef]
- Deeks, E.D. Ibrutinib: A Review in Chronic Lymphocytic Leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef] [PubMed]
- ASCO GU 2019: Interim Analysis of Ibrutinib Plus Paclitaxel for Patients with Metastatic Urothelial Carcinoma Previously Treated with Platinum-Based Chemotherapy. Available online: https://www.urotoday.com/conference-highlights/asco-gu-2019/asco-gu-2019-bladder-cancer/110296-asco-gu-2019-interim-analysis-of-ibrutinib-plus-paclitaxel-for-patients-with-metastatic-urothelial-carcinoma-previously-treated-with-platinum-based-chemotherapy.html (accessed on 24 November 2020).
- Lee, W.P.; Liao, Y.; Robinson, D.; Kung, H.J.; Liu, E.T.; Hung, M.C. Axl-Gas6 Interaction Counteracts E1A-Mediated Cell Growth Suppression and Proapoptotic Activity. Mol. Cell. Biol. 1999, 19, 8075–8082. [Google Scholar] [CrossRef] [Green Version]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of Phosphatidylinositol 3-Kinase-Dependent Pathway and Src for Gas6-Axl Mitogenic and Survival Activities in NIH 3T3 Fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple Roles for the Receptor Tyrosine Kinase Axl in Tumor Formation. Cancer Res. 2005, 65, 9294–9303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.-C.; Lay, J.-D.; Huang, J.-S.; Cheng, A.-L.; Tang, J.-L.; Lin, M.-T.; Lai, G.-M.; Chuang, S.-E. Receptor Tyrosine Kinase AXL Is Induced by Chemotherapy Drugs and Overexpression of AXL Confers Drug Resistance in Acute Myeloid Leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.-M.; Gilmer, T.M. Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macleod, K.; Mullen, P.; Sewell, J.; Rabiasz, G.; Lawrie, S.; Miller, E.; Smyth, J.F.; Langdon, S.P. Altered ErbB Receptor Signaling and Gene Expression in Cisplatin-Resistant Ovarian Cancer. Cancer Res. 2005, 65, 6789–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 Controls Motility of Bladder Cancer Cells via Transcriptional Upregulation of the Receptor Tyrosine Kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- British Library EThOS: Evaluating the Role of the Receptor Tyrosine Kinase AXL in Bladder Cancer. Available online: https://ethos.bl.uk/OrderDetails.do?uin=uk.bl.ethos.718696 (accessed on 19 November 2020).
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Compérat, E.M.; Cowan, N.C.; Gakis, G.; Hernández, V.; Linares Espinós, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-Invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020. [Google Scholar] [CrossRef]
- Treatment of Muscle-invasive and Advanced Bladder Cancer in 2020—Patel—2020—CA: A Cancer Journal for Clinicians—Wiley Online Library. Available online: https://0-acsjournals-onlinelibrary-wiley-com.brum.beds.ac.uk/doi/10.3322/caac.21631 (accessed on 9 December 2020).
- Petrylak, D.P.; Balar, A.V.; O’Donnell, P.H.; McGregor, B.A.; Heath, E.; Yu, E.Y.; Galsky, M.D.; Noah, M.H.; Gartner, E.M.; Melhem-Bertrandt, A.; et al. Results of Enfortumab Vedotin Monotherapy for Locally Advanced or Metastatic Urothelial Cancer Previously Treated with Platinum and Immune Checkpoint Inhibitors. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differentiation | MIBC | Oncogenic Mechanisms | Mutations | Possible Targeted Therapies | Median OS (Years) | |
|---|---|---|---|---|---|---|
| Luminal Papillary | Urothelial/ Luminal | 24% | FGFR3–55% | FGFR3–40% | FGFR targeted therapies | 4 |
| CDKN2A–33% | KDM6A–38% | |||||
| PPARG | ||||||
| Luminal non-specified | 8% | PPARG–76% | ELF3–35% | 1.8 | ||
| Luminal unstable | 15% | PPARG–89% | TP53–76% | 2.9 | ||
| Erb-B2–39% | ERCC2–22% | |||||
| E2F3/SOX4–76% | ||||||
| Stroma rich | Urothelial/ | 35% | EGFR | TP53–61% | EGFR targeted therapies | 1.2 |
| Squamous | RB1–25% | ICI | ||||
| Basal/Squamous | Squamous | 35% | EGFR | TP53–61% | EGFR targeted therapies | 1.2 |
| RB1–25% | ICI | |||||
| Neuroendocrine-like | Neuroendocrine | 3% | TP53-, RB1- | TP53–94% | <1 | |
| RB1–39% |
| Study Design (NCT Identifier and Code) | Study Phase | Experimental Treatment | Population | Estimated n | Primary Endpoint | Estimated Study Completion Date |
|---|---|---|---|---|---|---|
| BLC2001 (NCT02365597) | Phase II | Erdafitinib | mUC with FGR3 mutation or FGFR2/3 fusion afterchemotherapy treatment | 236 | ORR | 30 June 2022 (Recruiting) |
| NCT03390504 | Phase III | Erdafitinib Pembrolizumab | mUC with FGFR alterations as second or third line of treatment | 631 | OS | 5 November 2021 (Recruiting) |
| NORSE study (NCT03473743) | Phase I/II | Erdafitinib+cetrelimimab Erdafitinib+(cis/carbo)platin | mUC with selected FGFR alterations | 160 | DLT | 17 March 2023 (Recruiting) |
| NCT04172675 | Phase II | Erdafitinib | NMIBC with FGFR mutations or fusions and recurred after BCG therapy | 280 | RFS | 10 June 2026 (Recruiting) |
| NCT01004224 | Phase I | Infigratinib | Solid tumors with FGFR alterations | 208 | DLP | 8 October 2018 (Completed) |
| NCT04197986 | Phase III | Infigratinib | UC with FGFR3 alterations as adjuvant treatment | 218 | OS | 31 January 2025 (Recruiting) |
| NCT01976741 | Phase I | Rogaratinib | Several solid tumors without/with FGFR alterations | 168 | DLP | 11 March 2019 (Completed) |
| FORT-1 (NCT03410693) | Phase II/III | Rogaratinib | mUC with FGFR1/3 after platinum-based chemotherapy | 172 | ORR | 27 October 2020 (Completed) |
| FORT-2 (NCT03473756) | Phase Ib/II | Rogaratinib+atezolizumab | UC with FGFR alterations as first line of treatment | 210 | DLP | 4 September 2024 (Recruiting) |
| FIGHT-201 (NCT02872714) | Phase II | Pemigatinib | mUC with FGFR alterations | 263 | ORR | 31 March 2021 (Active, no recruiting) |
| FIGHT-205 (NCT04003610) | Phase II | Pemigatinib+atezolizumab Pemigatinib | mUC with FGFR3 alteration and not eligible to cisplatin | 6 | PFS | 31 January 2026 (Recruiting) |
| NCT02052778 | Phase I | TAS 120 | Tumors with FGF/FGFR alterations | 386 | DLT | 29 May 2021 (Active, not recruiting) |
| NCT01948297 | Phase I | Debio 1347-101 | Tumors with FGFR 1, 2, 3 alterations | 77 | DLT | 26 June 2020 (Terminated) |
| BISCAY (NCT02546661) | Phase I | AZD4547 AZD4547+durvalumab | MIBC who progressed prior line of treatment | 156 | DLT | 14 February 2022 (Active, not recruiting) |
| NCT04045613 | Phase I/II | Derazantinib Atezolizumab Derazantinib ± atezolizumab | mUC with FGFR alterations | 306 | ORR | Recruiting (May 2022) |
| NCT00790426 | Phase II | Dovitinib | UC | 48 | OS | April 2012 (Completed) |
| NCT01732107 | Phase II | Dovitinib | NMIUC with FGFR3 alterations | 13 | ORR | 6 March 2017 (Completed) |
| Drug | AEs Any Grade (%) | AEs Grade 3/4 (%) | Reference |
|---|---|---|---|
| Erdafitinib | Hyperphosphatemia (77%) | Hyponatremia (11%) Stomatitis (10%) Asthenia (7%) Nail dystrophy (6%) Hand-foot syndrome (5%) | [61] |
| Stomatitis (58%) | |||
| Diarrhea (51%) | |||
| Dry mouth (46%) | |||
| Central serous retinopathy (27%) | |||
| Onycholysis (18%) | |||
| Infigratinib | Hyperphosphatemia (46.3%) | Hyperlipasemia (10.4%) Fatigue (7.5%) Anemia (7.5%) Hand-foot syndrome (7.5%) Hypophosphatemia (7.5%) | [68] |
| Increase in serum creatinine (41.8%) | |||
| Constipation (37.3%) | |||
| Fatigue (37.3%) | |||
| Anemia (35.8%) | |||
| Rogaratinib | Hyperphosphatemia (60%) | Fatigue (9%) Anemia (6%) Urinary tract infection (8%) Hyperlipasemia (8%) | [71] |
| Diarrhea (49%) | |||
| Decreased appetite (36%) | |||
| Fatigue (24%) | |||
| Nausea (28%) | |||
| Urinary tract infection (11%) | |||
| Pemigatinib | Diarrhea (40%) | Urinary tract infection (7%) Fatigue (6%) | [74] |
| Alopecia (32%) | |||
| Fatigue (29%) | |||
| Constipation (28%) | |||
| Dry mouth (28%) | |||
| Debio-1347 | Hyperphosphatemia (76%) | Hyperphosphatemia (21%) Anemia (12%) Dyspnea (5%) ALT increased (3%) Stomatitis (3%) | [76] |
| Diarrhea (41%) | |||
| Nausea (40%) | |||
| Fatigue (40%) | |||
| Constipation (38%) | |||
| Decreased appetite (33%) | |||
| Nail changes (31%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Jiménez, J.; Albarrán-Fernández, V.; Pozas, J.; Román-Gil, M.S.; Esteban-Villarrubia, J.; Carrato, A.; Rosero, A.; Grande, E.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Novel Tyrosine Kinase Targets in Urothelial Carcinoma. Int. J. Mol. Sci. 2021, 22, 747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020747
Torres-Jiménez J, Albarrán-Fernández V, Pozas J, Román-Gil MS, Esteban-Villarrubia J, Carrato A, Rosero A, Grande E, Alonso-Gordoa T, Molina-Cerrillo J. Novel Tyrosine Kinase Targets in Urothelial Carcinoma. International Journal of Molecular Sciences. 2021; 22(2):747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020747
Chicago/Turabian StyleTorres-Jiménez, Javier, Víctor Albarrán-Fernández, Javier Pozas, María San Román-Gil, Jorge Esteban-Villarrubia, Alfredo Carrato, Adriana Rosero, Enrique Grande, Teresa Alonso-Gordoa, and Javier Molina-Cerrillo. 2021. "Novel Tyrosine Kinase Targets in Urothelial Carcinoma" International Journal of Molecular Sciences 22, no. 2: 747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020747