Cell-Based Tracers as Trojan Horses for Image-Guided Surgery

 ,
,  , ,
, ,  , , and
, , and
Abstract
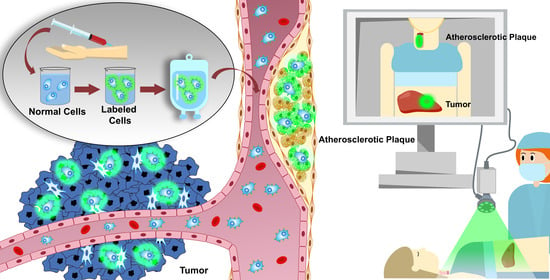
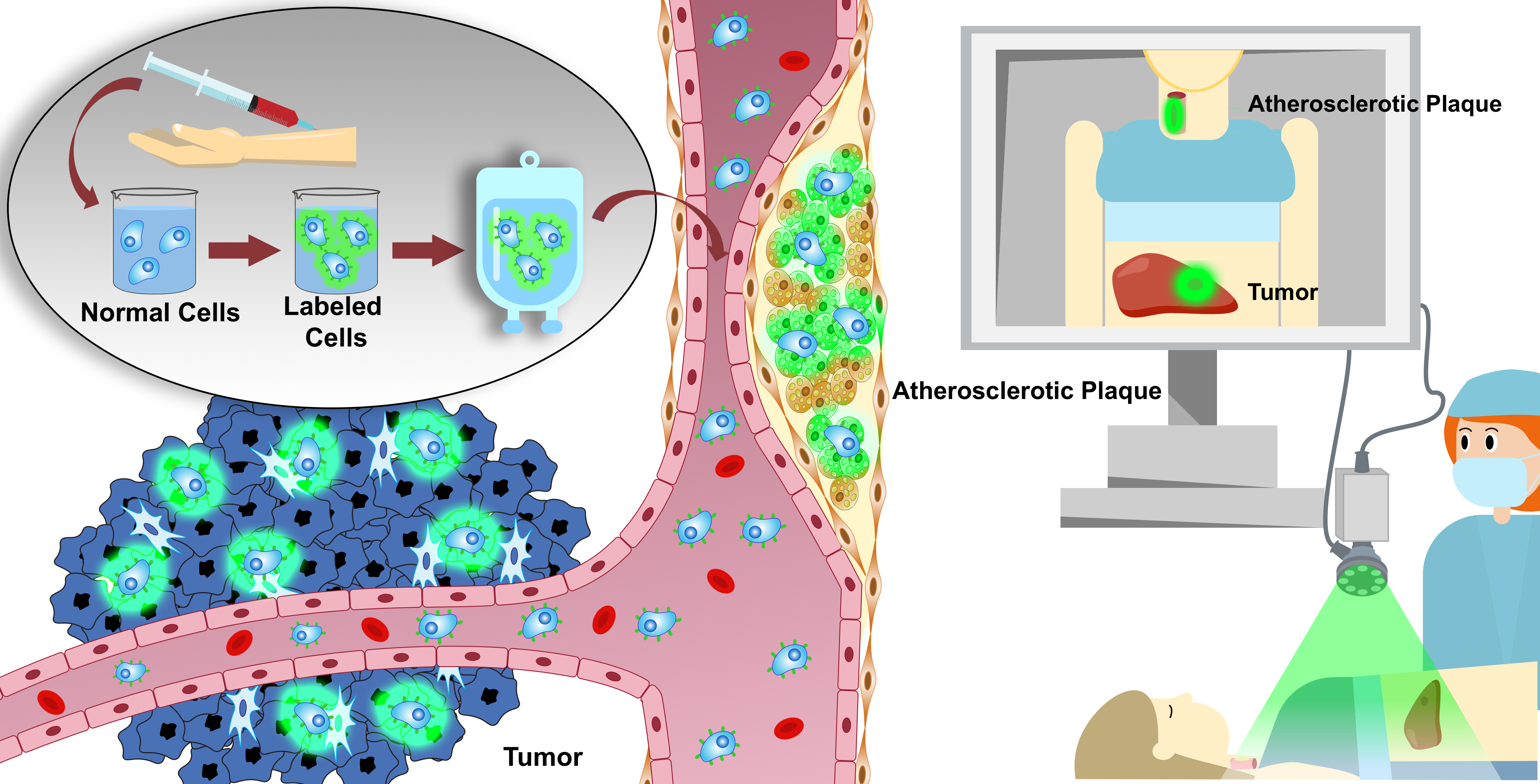
:
1. Introduction to Image-Guided Surgery
2. Cell-Based Carriers for IGS
2.1. Leukocyte-Based IGS Carriers
2.1.1. Monocyte/Macrophage-Based IGS Carriers
2.1.2. Neutrophil-Based IGS Carriers
2.1.3. T Cell-Based IGS Carriers
2.1.4. NK Cells-Based IGS Carriers
2.2. Platelet-Based IGS Carriers
2.3. Mesenchymal Stromal Cell-Based IGS Carriers
3. Non-Cellular and Bacterial Agents for IGS
3.1. Extracellular Vesicle/Exosome-Based IGS Carriers
3.2. Bacterium-Based IGS Carriers
3.3. Virus-Based IGS Carriers
4. Synthetic Nanoparticles for IGS
5. Discussion and Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ApoE | Apolipoprotein E |
| B cell | Bursa of Fabricius cell |
| BCG | bacillus Calmette–Guérin |
| CAR | Chimeric antigen receptor |
| CCL2 | C–C motif chemokine ligand 2 |
| CNT | Carbon nanotube |
| COPD | Chronic obstructive pulmonary disease |
| CT | Computed tomography |
| CTL | Cytotoxic T lymphocyte |
| Cy5 | Cyanine 5 |
| (im)DC | (immature murine) Dendritic cell |
| DiR | 1:1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide |
| DNA | Deoxyribonucleic acid |
| EGF(R) | Epidermal growth factor (receptor) |
| EPR effect | Enhanced permeability and retention effect |
| EV | Extracellular vesicle |
| FDG | Fluorodeoxyglucose |
| FITC | Fluorescein isothiocyanate |
| HER(2/neu) | Human epidermal growth factor receptor (2/neu) |
| HIV | Human immunodeficiency virus |
| HPF | Heparin, protamine, and ferumoxytol |
| HSV | Herpes simplex virus |
| ICG | Indocyanine green |
| IGS | Image-guided surgery |
| LAMP2b | lysosome-associated membrane glycoprotein 2b |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MHC | Major histocompatibility complex |
| MIP | Macrophage inflammatory protein |
| MRI | Magnetic resonance imaging |
| MSC | Mesenchymal stromal cell |
| MV | Microvesicle |
| NIRF | Near-infrared fluorescence |
| NK cell | Natural killer cell |
| PEG-PLA | Polyethylene glycol-Poly lactic acid |
| PET | Positron emission tomography |
| RNA | Ribonucleic acid; mi = micro, si = short interfering, lnc = long non-coding |
| SPECT | Single photon emission computed tomography |
| SWCNT | Single-walled carbon nanotube |
| T cell | Thymus cell |
| Th | T helper |
| TNFα | Tumor necrosis factor α |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF(R) | Vascular endothelial growth factor (receptor) |
References
- Vahrmeijer, A.L.; Hutteman, M.; van der Vorst, J.R.; van de Velde, C.J.; Frangioni, J.V. Image-guided cancer surgery using near-infrared fluorescence. Nat. Rev. Clin. Oncol. 2013, 10, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoven, P.; Ooms, S.; van Manen, L.; van der Bogt, K.E.A.; van Schaik, J.; Hamming, J.F.; Vahrmeijer, A.L.; Van der Vorst, J.R.; Mieog, J.S. A systematic review of the use of near-infrared fluorescence imaging in patients with peripheral artery disease. J. Vasc. Surg. 2019, 70, 286–297.e1. [Google Scholar] [CrossRef]
- Rosenthal, E.L.; Warram, J.M.; de Boer, E.; Basilion, J.P.; Biel, M.A.; Bogyo, M.; Bouvet, M.; Brigman, B.E.; Colson, Y.L.; DeMeester, S.R.; et al. Successful Translation of Fluorescence Navigation During Oncologic Surgery: A Consensus Report. J. Nucl. Med. 2016, 57, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Vahrmeijer, A.L.; Frangioni, J.V. Seeing the invisible during surgery. Br. J. Surg. 2011, 98, 749–750. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Kang, H.; Baek, Y.; El Fakhri, G.; Kuang, A.; Choi, H.S. Real-Time Imaging of Brain Tumor for Image-Guided Surgery. Adv. Healthc. Mater. 2018, 7, e1800066. [Google Scholar] [CrossRef]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R. Peptides as drug delivery vehicles across biological barriers. J. Pharm. Investig. 2018, 48, 89–111. [Google Scholar] [CrossRef]
- Lamberts, L.E.; Koch, M.; de Jong, J.S.; Adams, A.L.L.; Glatz, J.; Kranendonk, M.E.G.; Terwisscha van Scheltinga, A.G.; Jansen, L.; de Vries, J.; Lub-de Hooge, M.N.; et al. Tumor-Specific Uptake of Fluorescent Bevacizumab-IRDye800CW Microdosing in Patients with Primary Breast Cancer: A Phase I Feasibility Study. Clin. Cancer Res. 2017, 23, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Barth, C.W.; Gibbs, S.L. Fluorescence Image-Guided Surgery—A Perspective on Contrast Agent Development. Proc. SPIE Int. Soc. Opt. Eng. 2020, 11222. [Google Scholar] [CrossRef]
- Hernot, S.; van Manen, L.; Debie, P.; Mieog, J.S.D.; Vahrmeijer, A.L. Latest developments in molecular tracers for fluorescence image-guided cancer surgery. Lancet Oncol. 2019, 20, e354–e367. [Google Scholar] [CrossRef]
- Pogue, B.W.; Rosenthal, E.L.; Achilefu, S.; van Dam, G.M. Perspective review of what is needed for molecular-specific fluorescence-guided surgery. J. Biomed. Opt. 2018, 23, 100601. [Google Scholar] [CrossRef] [Green Version]
- Lauri, C.; Iezzi, R.; Rossi, M.; Tinelli, G.; Sica, S.; Signore, A.; Posa, A.; Tanzilli, A.; Panzera, C.; Taurino, M.; et al. Imaging Modalities for the Diagnosis of Vascular Graft Infections: A Consensus Paper amongst Different Specialists. J. Clin. Med. 2020, 9, 1510. [Google Scholar] [CrossRef]
- Van der Vaart, M.G.; Meerwaldt, R.; Slart, R.H.; van Dam, G.M.; Tio, R.A.; Zeebregts, C.J. Application of PET/SPECT imaging in vascular disease. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Annovazzi, A.; Bonanno, E.; Arca, M.; D’Alessandria, C.; Marcoccia, A.; Spagnoli, L.G.; Violi, F.; Scopinaro, F.; De Toma, G.; Signore, A. 99mTc-interleukin-2 scintigraphy for the in vivo imaging of vulnerable atherosclerotic plaques. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 117–126. [Google Scholar] [CrossRef]
- Li, T.; Dong, H.; Zhang, C.; Mo, R. Cell-based drug delivery systems for biomedical applications. Nano Res. 2018, 11, 5240–5257. [Google Scholar] [CrossRef]
- David, B.A.; Kubes, P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol. Rev. 2019, 289, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Nwabo Kamdje, A.H.; Kamga, P.T.; Simo, R.T.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Mesenchymal stromal cells’ role in tumor microenvironment: Involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129–141. [Google Scholar] [CrossRef]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef] [Green Version]
- Muthana, M.; Rodrigues, S.; Chen, Y.Y.; Welford, A.; Hughes, R.; Tazzyman, S.; Essand, M.; Morrow, F.; Lewis, C.E. Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer Res. 2013, 73, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthana, M.; Giannoudis, A.; Scott, S.D.; Fang, H.Y.; Coffelt, S.B.; Morrow, F.J.; Murdoch, C.; Burton, J.; Cross, N.; Burke, B.; et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011, 71, 1805–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippmann, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenzana, A.; Margheri, F.; Chilla, A.; Biagioni, A.; Margheri, G.; Calorini, L.; Fibbi, G.; Del Rosso, M. Endothelial Progenitor Cells as Shuttle of Anticancer Agents. Hum. Gene Ther. 2016, 27, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Oren, R.; Addadi, Y.; Narunsky Haziza, L.; Dafni, H.; Rotkopf, R.; Meir, G.; Fishman, A.; Neeman, M. Fibroblast recruitment as a tool for ovarian cancer detection and targeted therapy. Int. J. Cancer. 2016, 139, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, T.P.; Stuart, M.; Oosting, J.; Tollenaar, R.; Sier, C.F.M.; Mesker, W.E. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer. 2019, 19, 284. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: A foe or ally? Cell Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Back, M.; Hansson, G.K. Anti-inflammatory therapies for atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 199–211. [Google Scholar] [CrossRef]
- Muthana, M.; Kennerley, A.J.; Hughes, R.; Fagnano, E.; Richardson, J.; Paul, M.; Murdoch, C.; Wright, F.; Payne, C.; Lythgoe, M.F.; et al. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat. Commun. 2015, 6, 8009. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, D.; Mei, D.; Zhang, H.; Wang, Z.; He, B.; Dai, W.; Zhang, H.; Wang, X.; Zhang, Q. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J. Control. Release 2015, 204, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Qin, J.; Han, L.; Zhao, W.; Liang, J.; Xie, Z.; Yang, P.; Wang, J. Exploiting macrophages as targeted carrier to guide nanoparticles into glioma. Oncotarget 2016, 7, 37081–37091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almuhaideb, A.; Papathanasiou, N.; Bomanji, J. 18F-FDG PET/CT imaging in oncology. Ann. Saudi Med. 2011, 31, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, B.A.; Hoilund-Carlsen, P.F. [(1)(8)F]-fluorodeoxyglucose PET imaging of atherosclerosis. PET Clin. 2015, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kivimae, S.; Dolor, A.; Szoka, F.C. Macrophage-based cell therapies: The long and winding road. J. Control. Release 2016, 240, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.; Dong, X.; Shi, X.; Zhang, C.; Wang, Z. Neutrophil-Based Drug Delivery Systems. Adv. Mater. 2018, 30, e1706245. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Cho, J.; Malik, A.B. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 2014, 9, 204–210. [Google Scholar] [CrossRef]
- Tregay, N.; Begg, M.; Cahn, A.; Farahi, N.; Povey, K.; Madhavan, S.; Simmonds, R.; Gillett, D.; Solanki, C.; Wong, A.; et al. Use of autologous (99m)Technetium-labelled neutrophils to quantify lung neutrophil clearance in COPD. Thorax 2019, 74, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Vaas, M.; Enzmann, G.; Perinat, T.; Siler, U.; Reichenbach, J.; Licha, K.; Kipar, A.; Rudin, M.; Engelhardt, B.; Klohs, J. Non-invasive near-infrared fluorescence imaging of the neutrophil response in a mouse model of transient cerebral ischaemia. J. Cereb. Blood Flow Metab. 2017, 37, 2833–2847. [Google Scholar] [CrossRef] [Green Version]
- Puncher, M.R.; Blower, P.J. Autoradiography and density gradient separation of technetium-99m-exametazime (HMPAO) labelled leucocytes reveals selectivity for eosinophils. Eur. J. Nucl. Med. 1994, 21, 1175–1182. [Google Scholar] [CrossRef]
- Lukawska, J.J.; Livieratos, L.; Sawyer, B.M.; Lee, T.; O’Doherty, M.; Blower, P.J.; Kofi, M.; Ballinger, J.R.; Corrigan, C.J.; Gnanasegaran, G.; et al. Real-time differential tracking of human neutrophil and eosinophil migration in vivo. J. Allergy Clin. Immunol. 2014, 133, 233–239.e1. [Google Scholar] [CrossRef]
- Hadrup, S.; Donia, M.; Thor Straten, P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013, 6, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanca, T.; Silva-Santos, B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. Oncoimmunology. 2012, 1, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Henze, J.; Tacke, F.; Hardt, O.; Alves, F.; Al Rawashdeh, W. Enhancing the Efficacy of CAR T Cells in the Tumor Microenvironment of Pancreatic Cancer. Cancers 2020, 12, 1389. [Google Scholar] [CrossRef] [PubMed]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897. [Google Scholar] [CrossRef]
- Roddie, C.; O’Reilly, M.; Dias Alves Pinto, J.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340. [Google Scholar] [CrossRef]
- Jones, R.B.; Mueller, S.; Kumari, S.; Vrbanac, V.; Genel, S.; Tager, A.M.; Allen, T.M.; Walker, B.D.; Irvine, D.J. Antigen recognition-triggered drug delivery mediated by nanocapsule-functionalized cytotoxic T-cells. Biomaterials 2017, 117, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Torcellan, T.; Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. In vivo photolabeling of tumor-infiltrating cells reveals highly regulated egress of T-cell subsets from tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 5677–5682. [Google Scholar] [CrossRef] [Green Version]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [Green Version]
- Leong, J.W.; Fehniger, T.A. Human NK cells: SET to kill. Blood 2011, 117, 2297–2298. [Google Scholar] [CrossRef] [Green Version]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Siegler, E.L.; Kim, Y.J.; Chen, X.; Siriwon, N.; Mac, J.; Rohrs, J.A.; Bryson, P.D.; Wang, P. Combination Cancer Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells as Drug Carriers. Mol. Ther. 2017, 25, 2607–2619. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent anti-tumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Papayannakos, C.; Daniel, R. Understanding lentiviral vector chromatin targeting: Working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013, 20, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Romero, H.; Silvia, S.; Beland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, Y.; Lin, Z.; Liu, X.; Li, J.; Yang, H.; Tan, W. Equipping Natural Killer Cells with Specific Targeting and Checkpoint Blocking Aptamers for Enhanced Adoptive Immunotherapy in Solid Tumors. Angew. Chem. Int. Ed. Engl. 2020, 59, 12022–12028. [Google Scholar] [CrossRef]
- Yang, S.; Wen, J.; Li, H.; Xu, L.; Liu, Y.; Zhao, N.; Zeng, Z.; Qi, J.; Jiang, W.; Han, W.; et al. Aptamer-Engineered Natural Killer Cells for Cell-Specific Adaptive Immunotherapy. Small 2019, 15, e1900903. [Google Scholar] [CrossRef]
- Daldrup-Link, H.E.; Meier, R.; Rudelius, M.; Piontek, G.; Piert, M.; Metz, S.; Settles, M.; Uherek, C.; Wels, W.; Schleger, J.; et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radiol. 2005, 15, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Wang, X.; Zheng, L.; Lyu, T.; Figini, M.; Wang, B.; Procissi, D.; Shangguan, J.; Sun, C.; Pan, L.; et al. MRI-guided interventional natural killer cell delivery for liver tumor treatment. Cancer Med. 2018, 7, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Meller, B.; Frohn, C.; Brand, J.M.; Lauer, I.; Schelper, L.F.; von Hof, K.; Kirchner, H.; Richter, E.; Baehre, M. Monitoring of a new approach of immunotherapy with allogenic (111)In-labelled NK cells in patients with renal cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Matera, L.; Galetto, A.; Bello, M.; Baiocco, C.; Chiappino, I.; Castellano, G.; Stacchini, A.; Satolli, M.A.; Mele, M.; Sandrucci, S.; et al. In vivo migration of labeled autologous natural killer cells to liver metastases in patients with colon carcinoma. J. Transl. Med. 2006, 4, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, F.; Rapisarda, A.S.; Stabile, H.; Malviya, G.; Manni, I.; Bonanno, E.; Piaggio, G.; Gismondi, A.; Santoni, A.; Signore, A. In Vivo Imaging of Natural Killer Cell Trafficking in Tumors. J. Nucl. Med. 2015, 56, 1575–1580. [Google Scholar] [CrossRef] [Green Version]
- Meier, R.; Piert, M.; Piontek, G.; Rudelius, M.; Oostendorp, R.A.; Senekowitsch-Schmidtke, R.; Henning, T.D.; Wels, W.S.; Uherek, C.; Rummeny, E.J.; et al. Tracking of [18F]FDG-labeled natural killer cells to HER2/neu-positive tumors. Nucl. Med. Biol. 2008, 35, 579–588. [Google Scholar] [CrossRef]
- Wang, B.; Zheng, J. Platelet generation in vivo and in vitro. Springerplus 2016, 5, 787. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 42632. [Google Scholar] [CrossRef]
- Blumenreich, M.S. The White Blood Cell and Differential Count. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Plantureux, L.; Mege, D.; Crescence, L.; Carminita, E.; Robert, S.; Cointe, S.; Brouilly, N.; Ezzedine, W.; Dignat-George, F.; Dubois, C.; et al. The Interaction of Platelets with Colorectal Cancer Cells Inhibits Tumor Growth but Promotes Metastasis. Cancer Res. 2020, 80, 291–303. [Google Scholar] [CrossRef]
- Saito, H.; Fushida, S.; Miyashita, T.; Oyama, K.; Yamaguchi, T.; Tsukada, T.; Kinoshita, J.; Tajima, H.; Ninomiya, I.; Ohta, T.; et al. Potential of extravasated platelet aggregation as a surrogate marker for overall survival in patients with advanced gastric cancer treated with preoperative docetaxel, cisplatin and S-1: A retrospective observational study. BMC Cancer 2017, 17, 294. [Google Scholar] [CrossRef] [Green Version]
- Mikami, J.; Kurokawa, Y.; Takahashi, T.; Miyazaki, Y.; Yamasaki, M.; Miyata, H.; Nakajima, K.; Takiguchi, S.; Mori, M.; Doki, Y. Anti-tumor effect of antiplatelet agents in gastric cancer cells: An in vivo and in vitro study. Gastric Cancer 2016, 19, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, S.; Miyashita, T.; Inokuchi, M.; Hayashi, H.; Oyama, K.; Tajima, H.; Takamura, H.; Ninomiya, I.; Ahmed, A.K.; Harman, J.W.; et al. Platelets surrounding primary tumor cells are related to chemoresistance. Oncol. Rep. 2016, 36, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, R.; Peyruchaud, O. Metastasis: New functional implications of platelets and megakaryocytes. Blood 2016, 128, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Rinder, H.M.; Smith, B.R. In vitro evaluation of stored platelets: Is there hope for predicting posttransfusion platelet survival and function? Transfusion 2003, 43, 2–6. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, P.F.; Tomson, B.; Brand, A. In vivo tracking of transfused platelets for recovery and survival studies: An appraisal of labeling methods. Transfus. Apher. Sci. 2010, 42, 53–61. [Google Scholar] [CrossRef]
- Strassel, C.; Gachet, C.; Lanza, F. On the Way to in vitro Platelet Production. Front. Med. (Lausanne) 2018, 5, 239. [Google Scholar] [CrossRef] [Green Version]
- Kola, S.M.; Kumar, P.; Choonara, Y.E.; du Toit, L.C.; Pillay, V. Hypothesis: Can drug-loaded platelets be used as delivery vehicles for blood-brain barrier penetration? Med. Hypotheses 2019, 125, 75–78. [Google Scholar] [CrossRef]
- Weller, M.; Le Rhun, E.; Preusser, M.; Tonn, J.C.; Roth, P. How we treat glioblastoma. ESMO Open 2019, 4, e000520. [Google Scholar] [CrossRef] [Green Version]
- Wach, J.; Goetz, C.; Shareghi, K.; Scholz, T.; Hesselmann, V.; Mager, A.K.; Gottschalk, J.; Vatter, H.; Kremer, P. Dual-Use Intraoperative MRI in Glioblastoma Surgery: Results of Resection, Histopathologic Assessment, and Surgical Site Infections. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2019, 80, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.K.; Kean, R.; Bongiorno, E.; Hooper, D.C.; Jin, Y.Y.; Wickstrom, E.; McCue, P.A.; Thakur, M.L. Targeting VPAC1, Receptors for Imaging Glioblastoma. Mol. Imaging Biol. 2019, 22, 293–302. [Google Scholar] [CrossRef]
- Dai, L.; Gu, N.; Chen, B.A.; Marriott, G. Human platelets repurposed as vehicles for in vivo imaging of myeloma xenotransplants. Oncotarget 2016, 7, 21076–21090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Alam, M.A.; Shaw, J.; Dasgupta, A.K. Drug delivery using platelet cancer cell interaction. Pharm. Res. 2013, 30, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.C.; Guilmette, K.M.; Simons, E.R. Fluorescent labeling of human platelets. Blood 1975, 46, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidt, T.; Deininger, F.; Peter, K.; Goldschmidt, J.; Pethe, A.; Hagemeyer, C.E.; Neudorfer, I.; Zirlik, A.; Weber, W.A.; Bode, C.; et al. Activated platelets in carotid artery thrombosis in mice can be selectively targeted with a radiolabeled single-chain antibody. PLoS ONE 2011, 6, e18446. [Google Scholar] [CrossRef]
- Hartwig, J.H.; Bokoch, G.M.; Carpenter, C.L.; Janmey, P.A.; Taylor, L.A.; Toker, A.; Stossel, T.P. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 1995, 82, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, C.H.; DomBourian, M.G.; Millward, P.A. Platelet transfusion for patients with cancer. Cancer Control. 2015, 22, 47–51. [Google Scholar] [CrossRef]
- Stegner, D.; Dutting, S.; Nieswandt, B. Mechanistic explanation for platelet contribution to cancer metastasis. Thromb. Res. 2014, 133, S149–S157. [Google Scholar] [CrossRef]
- Meikle, C.K.; Kelly, C.A.; Garg, P.; Wuescher, L.M.; Ali, R.A.; Worth, R.G. Cancer and Thrombosis: The Platelet Perspective. Front. Cell Dev. Biol. 2016, 4, 147. [Google Scholar] [CrossRef] [Green Version]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Krueger, T.E.G.; Thorek, D.L.J.; Denmeade, S.R.; Isaacs, J.T.; Brennen, W.N. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl. Med. 2018, 7, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Lan, W.; Wang, F.; Zhang, C.; Liu, X.; Chen, Q. AAV-iRFP labelling of human mesenchymal stem cells for near-infrared fluorescence imaging. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G. Functional heterogeneity of mesenchymal stem cells: Implications for cell therapy. J. Cell Biochem. 2012, 113, 2806–2812. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Concise review: Two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 2013, 31, 2042–2046. [Google Scholar] [CrossRef]
- English, K.; Mahon, B.P. Allogeneic mesenchymal stem cells: Agents of immune modulation. J. Cell Biochem. 2011, 112, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frodermann, V.; van Duijn, J.; van Pel, M.; van Santbrink, P.J.; Bot, I.; Kuiper, J.; De Jager, S.C. Mesenchymal Stem Cells Reduce Murine Atherosclerosis Development. Sci. Rep. 2015, 5, 15559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois, S.; Usunier, B.; Douay, L.; Benderitter, M.; Chapel, A. Long-Term Quantitative Biodistribution and Side Effects of Human Mesenchymal Stem Cells (hMSCs) Engraftment in NOD/SCID Mice following Irradiation. Stem. Cells Int. 2014, 2014, 939275. [Google Scholar] [CrossRef] [PubMed]
- Semont, A.; Francois, S.; Mouiseddine, M.; Francois, A.; Sache, A.; Frick, J.; Thierry, D.; Chapel, A. Mesenchymal stem cells increase self-renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Adv. Exp. Med. Biol. 2006, 585, 19–30. [Google Scholar]
- Bax, N.A.; van Oorschot, A.A.; Maas, S.; Braun, J.; van Tuyn, J.; de Vries, A.A.; Gittenberger-de Groot, A.C.; Goumans, M.J. In vitro epithelial-to-mesenchymal transformation in human adult epicardial cells is regulated by TGFbeta-signaling and WT1. Basic Res. Cardiol. 2011, 106, 829–847. [Google Scholar] [CrossRef] [Green Version]
- Voswinkel, J.; Francois, S.; Simon, J.M.; Benderitter, M.; Gorin, N.C.; Mohty, M.; Fouillard, L.; Chaper, A. Use of mesenchymal stem cells (MSC) in chronic inflammatory fistulizing and fibrotic diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 180–192. [Google Scholar] [CrossRef]
- Porada, C.D.; Almeida-Porada, G. Mesenchymal stem cells as therapeutics and vehicles for gene and drug delivery. Adv. Drug Deliv Rev. 2010, 62, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Kraitchman, D.L.; Tatsumi, M.; Gilson, W.D.; Ishimori, T.; Kedziorek, D.; Walczak, P.; Segars, W.P.; Chen, H.H.; Fritzges, D.; Izbudak, I.; et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation 2005, 112, 1451–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Jeong, C.H.; Woo, J.S.; Ryu, C.H.; Lee, J.H.; Jeun, S.S. In vivo near-infrared imaging for the tracking of systemically delivered mesenchymal stem cells: Tropism for brain tumors and biodistribution. Int. J. Nanomed. 2016, 11, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–692. [Google Scholar] [CrossRef] [PubMed]
- de Witte, S.F.H.; Luk, F.; Sierra Parraga, J.M.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells. 2018, 36, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, B.M.; Martin, K.; Ali, M.T.; Kumar, A.H.; Pillai, M.G.; Kumar, S.P.; Pillai, M.G.; Kumar, S.P.; O’Sullivan, J.F.; Whelan, D.; et al. Bone Marrow-Derived Mesenchymal Stem Cells Have Innate Procoagulant Activity and Cause Microvascular Obstruction Following Intracoronary Delivery: Amelioration by Antithrombin Therapy. Stem Cells. 2015, 33, 2726–2737. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Lara, P.; Chan, A.B.; Cruz, L.J.; Quest, A.F.G.; Kogan, M.J. Exploiting the Natural Properties of Extracellular Vesicles in Targeted Delivery towards Specific Cells and Tissues. Pharmaceutics 2020, 12, 1022. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Strobel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Kanada, M.; Kim, B.D.; Hardy, J.W.; Ronald, J.A.; Bachmann, M.H.; Bernard, M.P.; Perez, G.I.; Zarea, A.A.; Ge, T.J.; Withrow, A.; et al. Microvesicle-Mediated Delivery of Minicircle DNA Results in Effective Gene-Directed Enzyme Prodrug Cancer Therapy. Mol. Cancer Ther. 2019, 18, 2331–2342. [Google Scholar] [CrossRef] [Green Version]
- Xitong, D.; Xiaorong, Z. Targeted therapeutic delivery using engineered exosomes and its applications in cardiovascular diseases. Gene 2016, 575, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lu, Y.; Li, X. Biological characteristics of exosomes and genetically engineered exosomes for the targeted delivery of therapeutic agents. J. Drug Target. 2019, 28, 129–141. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver anti-tumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Zhang, J.; Zhao, Q.; Zhuang, W.; Ding, J.; Zhang, C.; Gao, H.; Pang, D.W.; Pu, K.; Xie, H.Y. Molecularly Engineered Macrophage-Derived Exosomes with Inflammation Tropism and Intrinsic Heme Biosynthesis for Atherosclerosis Treatment. Angew. Chem. Int. Ed. Engl. 2020, 59, 4068–4074. [Google Scholar] [CrossRef]
- Simpson, R.J.; Jensen, S.S.; Lim, J.W. Proteomic profiling of exosomes: Current perspectives. Proteomics 2008, 8, 4083–4099. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Gao, J.Q. Exosomes as novel bio-carriers for gene and drug delivery. Int. J. Pharm. 2017, 521, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Takahashi, Y.; Takakura, Y. Possibility of Exosome-Based Therapeutics and Challenges in Production of Exosomes Eligible for Therapeutic Application. Biol. Pharm. Bull. 2018, 41, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhom, K.; Obi, P.O.; Saleem, A. A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef]
- Zhang, M.; Li, M.; Du, L.; Zeng, J.; Yao, T.; Jin, Y. Paclitaxel-in-liposome-in-bacteria for inhalation treatment of primary lung cancer. Int. J. Pharm. 2020, 578, 119177. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, S.; Esmaeili, A.; Barzegari, A.; Rafi, M.A.; Omidi, Y. Bioengineered smart bacterial carriers for combinational targeted therapy of solid tumours. J. Drug Target. 2020, 28, 700-13. [Google Scholar] [CrossRef] [PubMed]
- Zu, C.; Wang, J. Tumor-colonizing bacteria: A potential tumor targeting therapy. Crit. Rev. Microbiol. 2014, 40, 225–235. [Google Scholar] [CrossRef]
- Morrissey, D.; O’Sullivan, G.C.; Tangney, M. Tumour targeting with systemically administered bacteria. Curr. Gene Ther. 2010, 10, 3–14. [Google Scholar] [CrossRef]
- Miyaguchi, J.; Shiga, K.; Ogawa, K.; Suzuki, F.; Katagiri, K.; Saito, D.; Ikeda, A.; Horii, A.; Watanabe, M.; Igimi, S. Treatment with Lactobacillus Retards the Tumor Growth of Head and Neck Squamous Cell Carcinoma Cells Inoculated in Mice. Tohoku J. Exp. Med. 2018, 245, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Redelman-Sidi, G.; Glickman, M.S.; Bochner, B.H. The mechanism of action of BCG therapy for bladder cancer--a current perspective. Nat. Rev. Urol. 2014, 11, 153–162. [Google Scholar] [CrossRef]
- Bilsen, M.P.; van Meijgaarden, K.E.; de Jong, H.K.; Joosten, S.A.; Prins, C.; Kroft, L.J.M.; Jonker, J.T.; Crobach, S.; Pelger, R.C.; Ottenhoff, T.H.; et al. A novel view on the pathogenesis of complications after intravesical BCG for bladder cancer. Int. J. Infect. Dis. 2018, 72, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlec, A.; Zavrsnik, J.; Butinar, M.; Turk, B.; Strukelj, B. In vivo imaging of Lactococcus lactis, Lactobacillus plantarum and Escherichia coli expressing infrared fluorescent protein in mice. Microb. Cell Fact. 2015, 14, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H. Engineering bacteria toward tumor targeting for cancer treatment: Current state and perspectives. Appl Microbiol. Biotechnol. 2012, 93, 517–523. [Google Scholar] [CrossRef]
- Duong, M.T.; Qin, Y.; You, S.H.; Min, J.J. Bacteria-cancer interactions: Bacteria-based cancer therapy. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, S.; Choi, M.H.; Yang, J.E.; Shim, H.E.; Song, L.; Song, H.Y.; Choi, J.K.; Jeon, J. Technetium-99m-based simple and convenient radiolabeling of Escherichia coli for in vivo tracking of microorganisms. J. Radioanal. Nucl. Chem. 2018, 317, 997–1003. [Google Scholar] [CrossRef]
- Lindquist, B.L.; Lebenthal, E.; Lee, P.C.; Stinson, M.W.; Merrick, J.M. Adherence of Salmonella typhimurium to small-intestinal enterocytes of the rat. Infect. Immun. 1987, 55, 3044–3050. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Emerson, C.C.; Yao, J.; Young, C.; Stewart, P.L.; Shayakhmetov, D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020, 12, eabc6659. [Google Scholar] [CrossRef]
- Delwar, Z.M.; Liu, G.; Kuo, Y.; Lee, C.; Bu, L.; Rennie, P.S.; Jia, W.W. Tumour-specific triple-regulated oncolytic herpes virus to target glioma. Oncotarget 2016, 7, 28658–28669. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Xue, J.; Guo, J.; Qian, Z.; Achilefu, S.; Gu, Y. Improved targeting of ligand-modified adenovirus as a new near infrared fluorescence tumor imaging probe. Bioconjug. Chem. 2011, 22, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Spronken, M.I.; Short, K.R.; Herfst, S.; Bestebroer, T.M.; Vaes, V.P.; van der Hoeven, B.; Koster, A.J.; Kremers, G.J.; Scott, D.P.; Gultyaev, A.P.; et al. Optimisations and Challenges Involved in the Creation of Various Bioluminescent and Fluorescent Influenza A Virus Strains for In Vitro and In Vivo Applications. PLoS ONE 2015, 10, e0133888. [Google Scholar] [CrossRef]
- Hofherr, S.E.; Adams, K.E.; Chen, C.Y.; May, S.; Weaver, E.A.; Barry, M.A. Real-time dynamic imaging of virus distribution in vivo. PLoS ONE 2011, 6, e17076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella Man, Y.K.; Foster, J.; Carapuca, E.; Davies, J.A.; Parker, A.L.; Sosabowski, J.; Hallden, G. Systemic delivery and SPECT/CT in vivo imaging of (125)I-labelled oncolytic adenoviral mutants in models of pancreatic cancer. Sci Rep. 2019, 9, 12840. [Google Scholar] [CrossRef]
- Siddique, S.; Chow, J.C.L. Application of Nanomaterials in Biomedical Imaging and Cancer Therapy. Nanomaterials 2020, 10, 1700. [Google Scholar] [CrossRef]
- Mody, N.; Tekade, R.K.; Mehra, N.K.; Chopdey, P.; Jain, N.K. Dendrimer, liposomes, carbon nanotubes and PLGA nanoparticles: One platform assessment of drug delivery potential. AAPS PharmSciTech 2014, 15, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, B.; Hojjat-Farsangi, M.; Mohammadi, H.; Anvari, E.; Ghalamfarsa, G.; Yousefi, M.; Jadidi-Niaragh, F. Nanoparticles and targeted drug delivery in cancer therapy. Immunol. Lett. 2017, 190, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Basel, M.T.; Balivada, S.; Wang, H.; Shrestha, T.B.; Seo, G.M.; Pyle, M.; Abayaweera, G.; Dani, R.; Koper, O.B.; Tamura, M.; et al. Cell-delivered magnetic nanoparticles caused hyperthermia-mediated increased survival in a murine pancreatic cancer model. Int. J. Nanomed. 2012, 7, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.J.; Bu, J.; Kubiatowicz, L.J.; Chen, S.S.; Kim, Y.; Hong, S. Peptide-nanoparticle conjugates: A next generation of diagnostic and therapeutic platforms? Nano Converg. 2018, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Nanoparticles evading the reticuloendothelial system: Role of the supported bilayer. Biochim. Biophys. Acta 2009, 1788, 2259–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wang, J.; Rao, T.; He, X.; Xu, T. Pharmaceutical applications of dendrimers: Promising nanocarriers for drug delivery. Front. Biosci. 2008, 13, 1447–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunoqrot, S.; Bugno, J.; Lantvit, D.; Burdette, J.E.; Hong, S. Prolonged blood circulation and enhanced tumor accumulation of folate-targeted dendrimer-polymer hybrid nanoparticles. J. Control. Release 2014, 191, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.C.; Zhao, X.; Hirahara, K.; Miyamoto, Y.; Ando, Y.; Iijima, S. The smallest carbon nanotube. Nature 2000, 408, 50. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Suenaga, K.; Iijima, S. Smallest carbon nanotube assigned with atomic resolution accuracy. Nano Lett. 2008, 8, 459–462. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y.; Inoue, S.; Suzuki, T.; Jones, R.O.; Ando, Y. Smallest carbon nanotube is 3 a in diameter. Phys. Rev. Lett. 2004, 92, 125502. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Patharkar, A.; Kuche, K.; Maheshwari, R.; Deb, P.K.; Kalia, K.; Tekade, R.K. Functionalized carbon nanotubes as emerging delivery system for the treatment of cancer. Int. J. Pharm. 2018, 548, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhao, N. A Targeted Nanoprobe Based on Carbon Nanotubes-Natural Biopolymer Chitosan Composites. Nanomaterials 2016, 6, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vankayala, R.; Bahena, E.; Guerrero, Y.; Singh, S.P.; Ravoori, M.K.; Kundra, V.; Anvari, B. Virus-Mimicking Nanoparticles for Targeted Near Infrared Fluorescence Imaging of Intraperitoneal Ovarian Tumors in Mice. Ann. Biomed. Eng. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.K.; Medina-Sanchez, M.; Edmondson, R.J.; Schmidt, O.G. Engineering microrobots for targeted cancer therapies from a medical perspective. Nat. Commun. 2020, 11, 5618. [Google Scholar] [CrossRef]
- Qiao, Y.; Yang, F.; Xie, T.; Du, Z.; Zhong, D.; Qi, Y.; Li, Y.; Li, W.; Lu, Z.; Rao, J.; et al. Engineered algae: A novel oxygen-generating system for effective treatment of hypoxic cancer. Sci. Adv. 2020, 6, eaba5996. [Google Scholar] [CrossRef]
- Roddenberry, G. Star Trek: The Next Generation; Paramount Television: Los Angeles, CA, USA, 1987. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
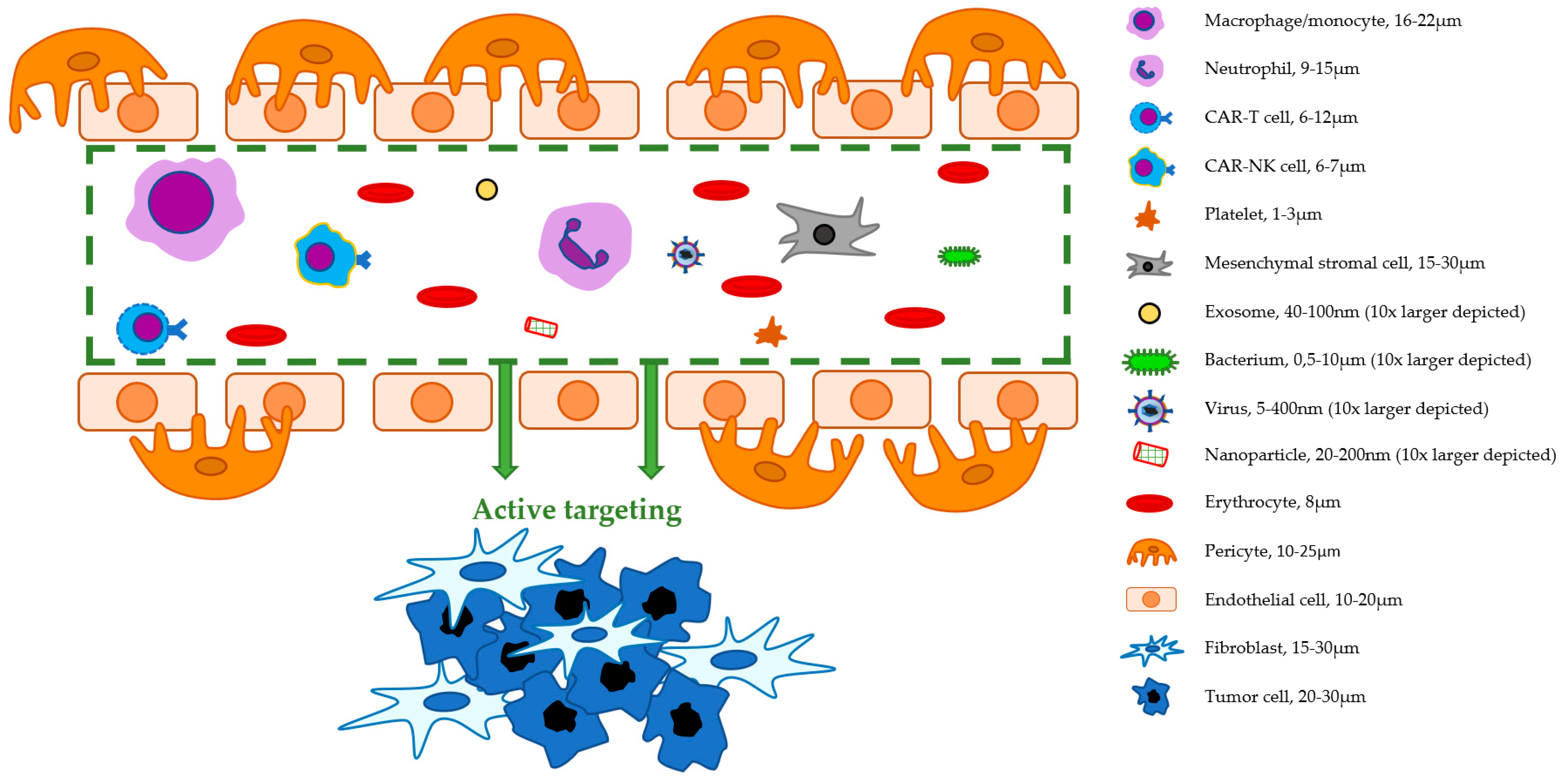
| Macrophages/ Monocytes | Neutrophils | (CAR-)T Cells | (CAR-)NK Cells | Platelets | MSCs | Exosomes | Bacteria | Viruses | Nanoparticles | |
|---|---|---|---|---|---|---|---|---|---|---|
| Diameter | 16–22 μm | 9–15 μm | 6–12 μm | 6–7 μm | 1–3 μm | 15–30 μm | 40–100 nm | 0.5–10 μm | 5–400 nm | 20–200 nm |
| Biocompatibility | + + + | + + + | + +(+) | + + + | + + + | + + + | + + (+) | - - - | - - | +/− |
| Targeting potential | + + | + + | + + + | + + + | + | + | + | + + | + + + | + + + |
| Feasibility & applicability | + | +/− | - | +/− | + | +/− | +/− | +/− | + | + |
| Specific advantages | 1. Active tumor-homing 2. Easily labeled | 1. Active tumor-homing 2. Abundance in blood | 1. CAR specificity | 1. CAR specificity 2. Less associated with off-target effects (compared to T cells) 3. Relatively inexpensive (compared to T cells) | 1. Penetrative capabilities 2. Availability in the circulation | 1. Fast ex-vivo expansion 2. Relatively easy acquisition | 1. Non-immunogenic 2. Penetrative abilities 3. Physiochemical stability | 1. Easily genetically manipulated and cultured 2. Low production costs 3. Tumor-specific replication 4. Variety in suitable species | 1. Easily modified 2. Variety in suitable strains 3. Tumor specificity 4. Ability to replicate | 1. Multitude of design possibilities 2. High specificity potential 3. Humanization to increase biocompatibility 4. High surface binding potential |
| Specific challenges | 1. Avoiding disease promoting subtypes 2. Modification process-induced cell alterations | 1. Acquisition 2. Fragile & easily activated in culture 3. Modification process-induced cell alterations | 1. Costs of cell acquisition and modification 2. Efficiency of cell acquisition and modification 3. Harmful off-target responses 4. Modification process-induced cell alterations | 1. Relative inefficiency of gene transduction methods 2. Relative lack of NK cell-specific CARs 3. Modification process-induced cell alterations | 1. Platelet aggregation 2. Platelet-tumor cell interactions | 1. Trapping in lungs, liver, and spleen 2. Coagulation inducing properties 3. Modification process-induced cell alterations | 1.Purification techniques; yield 2. Costs of large scale production | 1. Patho- and immunogenecity | 1. Memory-induced immune responses (neutralizing antibodies) 2. Patho- and immunogenicity 3. Species-specific non-availability of anti-viral agents | 1. Large pools needed 2. Rapid clearance 3. Expensive manufacturing and processing 4. Limited amount surface targeting agents |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sier, V.Q.; de Vries, M.R.; van der Vorst, J.R.; Vahrmeijer, A.L.; van Kooten, C.; Cruz, L.J.; de Geus-Oei, L.-F.; Ferreira, V.; Sier, C.F.M.; Alves, F.; et al. Cell-Based Tracers as Trojan Horses for Image-Guided Surgery. Int. J. Mol. Sci. 2021, 22, 755. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020755
Sier VQ, de Vries MR, van der Vorst JR, Vahrmeijer AL, van Kooten C, Cruz LJ, de Geus-Oei L-F, Ferreira V, Sier CFM, Alves F, et al. Cell-Based Tracers as Trojan Horses for Image-Guided Surgery. International Journal of Molecular Sciences. 2021; 22(2):755. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020755
Chicago/Turabian StyleSier, Vincent Q., Margreet R. de Vries, Joost R. van der Vorst, Alexander L. Vahrmeijer, Cornelis van Kooten, Luis J. Cruz, Lioe-Fee de Geus-Oei, Valerie Ferreira, Cornelis F. M. Sier, Frauke Alves, and et al. 2021. "Cell-Based Tracers as Trojan Horses for Image-Guided Surgery" International Journal of Molecular Sciences 22, no. 2: 755. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020755