Common Transcriptional Program of Liver Fibrosis in Mouse Genetic Models and Humans

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
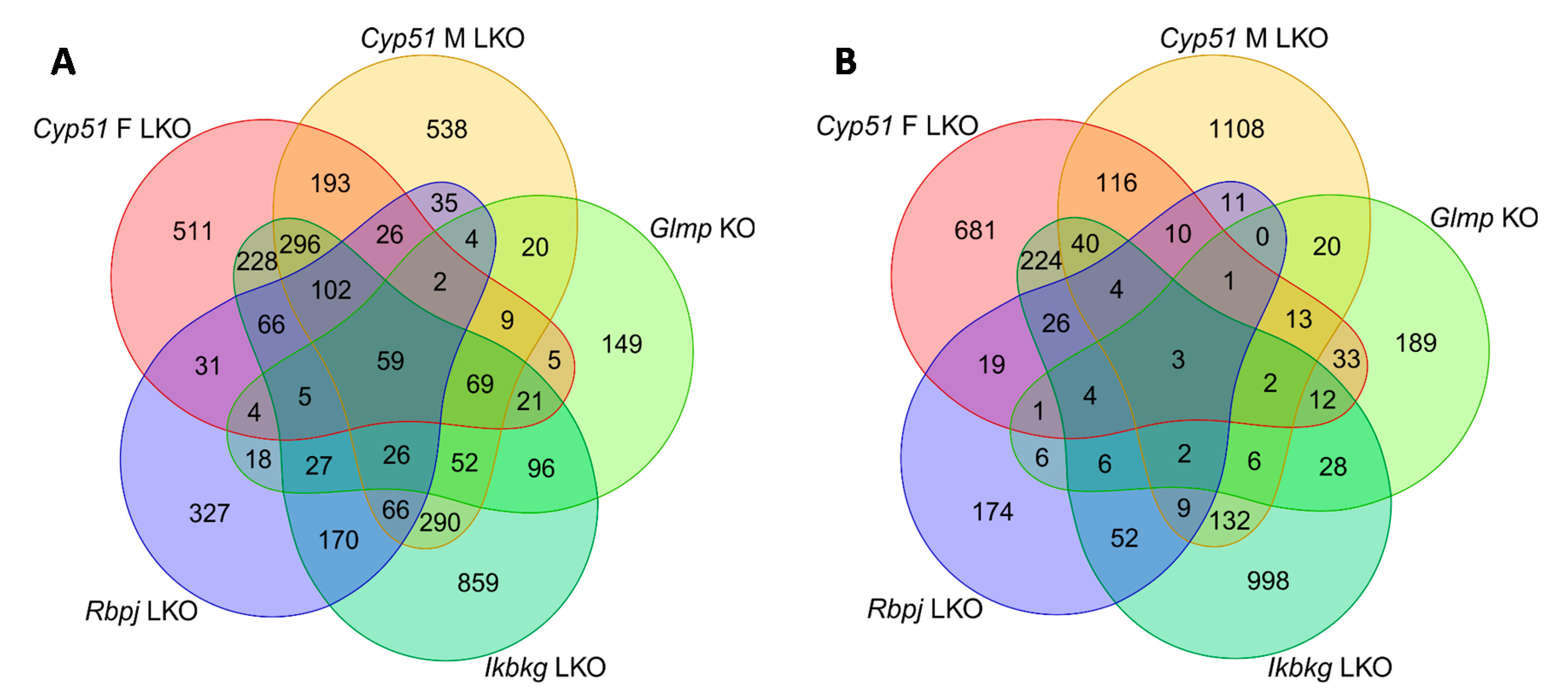
2.1. Similar Transcriptome Alterations Caused by Different Genetic Defects
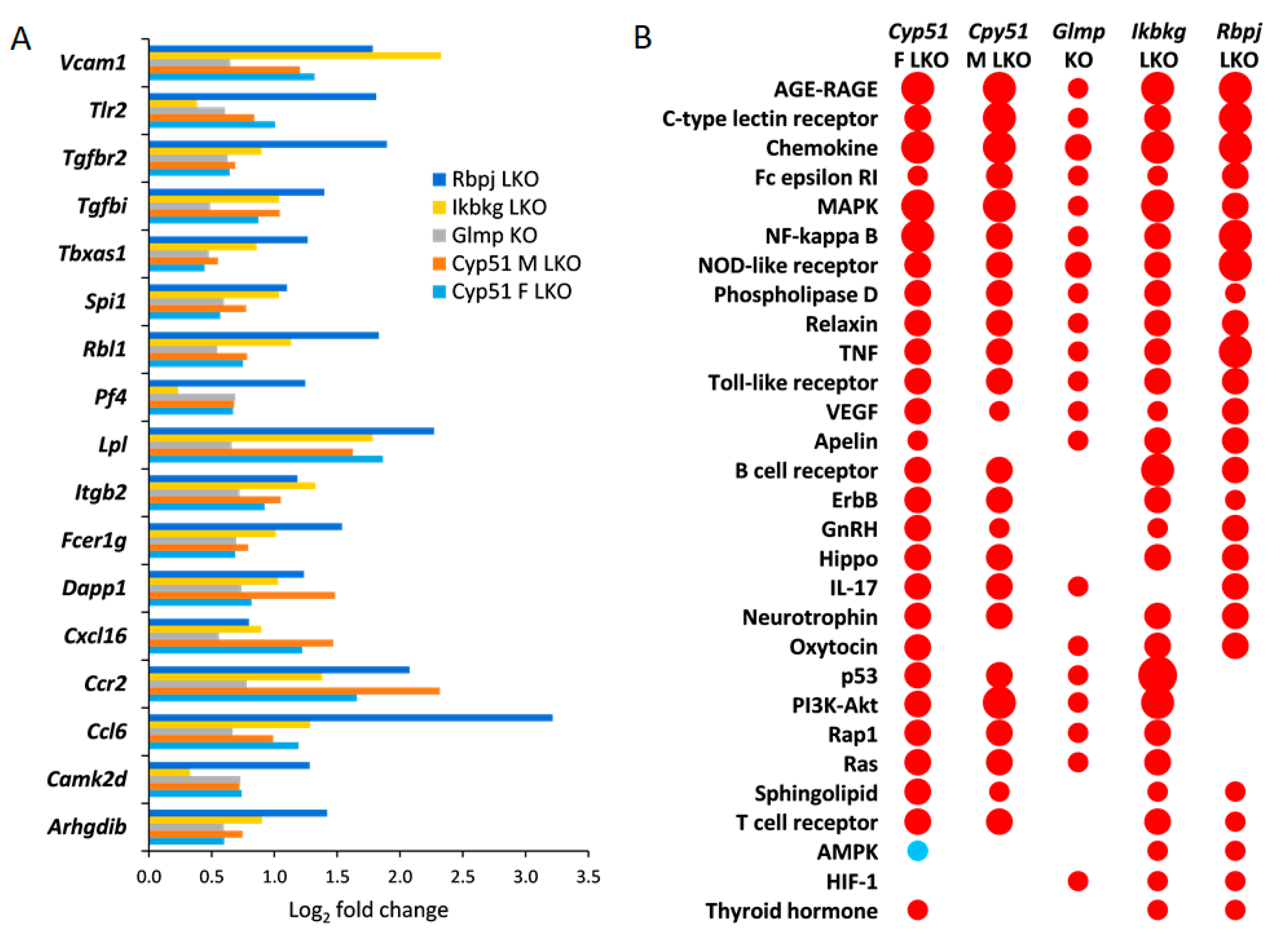
2.2. Common KEGG and Reactome Pathways in Different Fibrotic Models
2.3. Mouse Genetic Models Show Overlapping Transcriptome Signatures with Human NAFLD and NASH
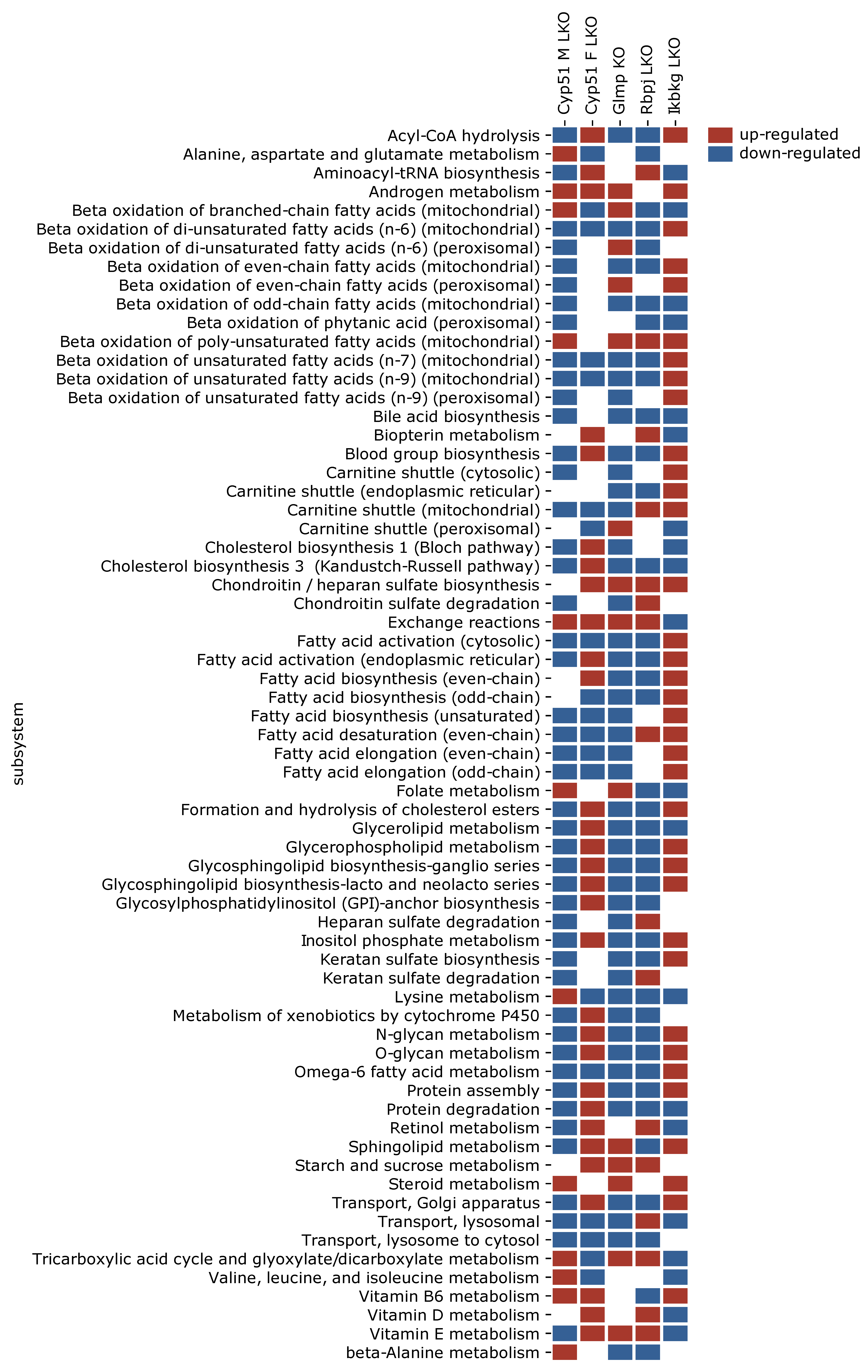
2.4. Negative Enrichment of Metabolic Pathways in Fibrosis
2.5. Genome-Scale Metabolic Models Confirmed Rearrangements in Lipid Metabolism Pathways
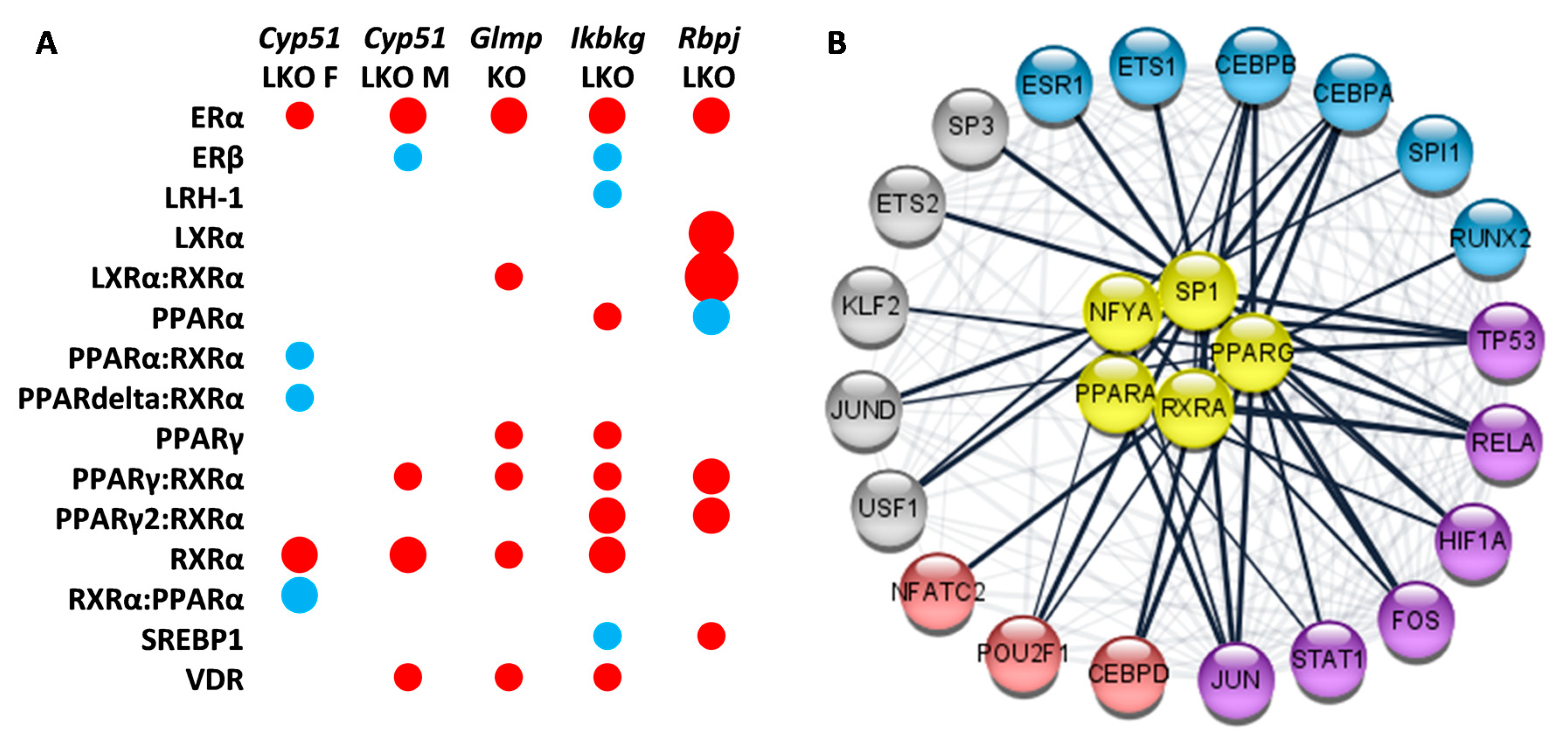
2.6. Enrichment of Transcription Factors Exposed Variability in Metabolic Regulators
3. Discussion
4. Materials and Methods
4.1. Microarray-Based Gene Expression Analysis
4.2. Genome–Scale Metabolic Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AASLD | American Association for the Study of Liver Diseases |
| AMPK | AMP-activated protein kinase |
| CYP51 | Cytochrome P450, family 51 |
| DEGs | Differentially expressed genes |
| E2F1 | E2F transcription factor 1 |
| EASL | The European Association for the Study of the Liver |
| ECM | Extracellular matrix |
| ERα | Estrogen receptor alpha |
| ErbB | Erb-B2 receptor tyrosine kinase |
| F | Female |
| FXR | Farnesoid X receptor |
| GEM | Genome-scale metabolic model |
| GLMP | Glycosylated lysosomal membrane protein |
| HCC | Hepatocellular carcinoma |
| HDL | High-density lipoprotein |
| IGF1R | Insulin like growth factor 1 receptor |
| IKBKG | Inhibitor of nuclear factor kappa B kinase regulatory subunit gamma |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | Knock-out |
| LKO | Liver knock-out |
| LXR | Liver X receptor |
| M | Male |
| MAFLD | Metabolic Associated Fatty Liver Disease |
| MAPK | Mitogen-activated protein kinase |
| MFSD1 | Major facilitator superfamily domain containing 1 |
| mTOR | The mammalian target of rapamycin |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| pGSEA | Parametric gene set enrichment analysis |
| PPAR | Peroxisome proliferator activated receptor |
| RBPJ | Recombination signal binding protein for immunoglobulin kappa J region |
| RXR | Retinoid X receptor |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| STRING | Search tool for recurring instances of neighboring genes |
| TAG | Triglycerides |
| TCA | Tricarboxylic acid cycle |
| TF | Transcription factor |
| TGF-β | Transforming growth factor beta |
| TGFBI | Transforming growth factor beta induced |
| TRANSFAC | TRANScription FACtor |
| VDR | Vitamin D receptor |
| VEGF | Vascular endothelial growth factor |
| WT | Wild type |
References
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Luedde, T.; Sauerbruch, T.; Scholten, D.; Streetz, K.; Tacke, F.; Tolba, R.; Trautwein, C.; Trebicka, J.; Weiskirchen, R. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenes. Tissue Repair 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Sircana, A.; Paschetta, E.; Saba, F.; Molinaro, F.; Musso, G. Recent insight into the role of fibrosis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Barb, D.; Lomonaco, R.; Lai, J.; Cusi, K. Change in hepatic fat content measured by MRI does not predict treatment-induced histological improvement of steatohepatitis. J. Hepatol. 2020, 72, 401–410. [Google Scholar] [CrossRef]
- Rinella, M.E.; Tacke, F.; Sanyal, A.J.; Anstee, Q.M. Report on the AASLD/EASL joint workshop on clinical trial endpoints in NAFLD. J. Hepatol. 2019, 71, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e7. [Google Scholar] [CrossRef] [Green Version]
- Lorbek, G.; Perše, M.; Jeruc, J.; Juvan, P.; Gutierrez-mariscal, F.M.; Lewinska, M.; Gebhardt, R.; Horvat, S.; Bjorkhem, I.; Rozman, D.; et al. Lessons from Hepatocyte-Specific Cyp51 Knockout Mice: Impaired Cholesterol Synthesis Leads to Oval Cell-Driven Liver Injury. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.Y.; Nesset, C.K.; Damme, M.; Lbøerg, E.M.; Lubke, T.; Mhælen, J.; Andersson, K.B.; Lorenzo, P.I.; Roos, N.; Thoresen, G.H.; et al. Loss of lysosomal membrane protein NCU-G1 in mice results in spontaneous liver fibrosis with accumulation of lipofuscin and iron. DMM Dis. Model. Mech. 2014, 7, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Nesset, C.K.; Kong, X.Y.; Damme, M.; Schjalm, C.; Roos, N.; Løberg, E.M.; Eskild, W. Age-dependent development of liver fibrosis in Glmpgt/gt mice. Fibrogenes. Tissue Repair 2016, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.Y.; Kase, E.T.; Herskedal, A.; Schjalm, C.; Damme, M.; Nesset, C.K.; Thoresen, G.H.; Rustan, A.C.; Eskild, W. Lack of the lysosomal membrane protein, GLMP, in mice results in metabolic dysregulation in liver. PLoS ONE 2015, 10, e0129402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharehalli, U.; Svinarenko, M.; Kraus, J.M.; Kühlwein, S.D.; Szekely, R.; Kiesle, U.; Scheffold, A.; Barth, T.F.E.; Kleger, A.; Schirmbeck, R.; et al. YAP activation drives liver regeneration after cholestatic damage induced by Rbpj deletion. Int. J. Mol. Sci. 2018, 19, 3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beraza, N.; Malato, Y.; Sander, L.E.; Al-Masaoudi, M.; Freimuth, J.; Riethmacher, D.; Gores, G.J.; Roskams, T.; Liedtke, C.; Trautwein, C. Hepatocyte-specific NEMO deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. J. Exp. Med. 2009, 206, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKγ in Liver Parenchymal Cells Causes Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, 1–16. [Google Scholar] [CrossRef]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.L.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.H.; Mcgilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, P.; Klag, T.; Malek, N.P.; Heikenwalder, M. Platelets in chronic liver disease, from bench to bedside. JHEP Reports 2019, 1, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naguib, G.; Morris, N.; Yang, S.; Fryzek, N.; Haynes-Williams, V.; Huang, W.C.A.; Norman-Wheeler, J.; Rotman, Y. Dietary fatty acid oxidation is decreased in non-alcoholic fatty liver disease: A palmitate breath test study. Liver Int. 2020, 40, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Satriano, L.; Lewinska, M.; Rodrigues, P.M.; Banales, J.M.; Andersen, J.B. Metabolic rearrangements in primary liver cancers: Cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 2019, 16. [Google Scholar] [CrossRef]
- Yoo, H.J.; Jung, K.J.; Kim, M.M.; Kim, M.M.; Kang, M.; Jee, S.H.; Choi, Y.; Lee, J.H. Liver Cirrhosis Patients Who Had Normal Liver Function Before Liver Cirrhosis Development Have the Altered Metabolic Profiles Before the Disease Occurrence Compared to Healthy Controls. Front. Physiol. 2019, 10, 1421. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1–11. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Cubero, F.J.; Singh, A.; Borkham-Kamphorst, E.; Nevzorova, Y.A.; Al Masaoudi, M.; Haas, U.; Boekschoten, M.V.; Gassler, N.; Weiskirchen, R.; Muller, M.; et al. TNFR1 determines progression of chronic liver injury in the IKKγ/Nemo genetic model. Cell Death Differ. 2013, 20, 1580–1592. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.R.; Sidney Barritt, A. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Kahlan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma Metabolomic Profile in Non-Alcoholic Fatty Liver Disease. Metabolism 2011, 60, 404–413. [Google Scholar]
- Chen, J.; Zheng, M.; Liu, J.; Luo, Y.; Yang, W.; Yang, J.; Liu, J.; Zhou, J.; Xu, C.; Zhao, F.; et al. Ratio of Conjugated Chenodeoxycholic to Muricholic Acids is Associated with Severity of Nonalcoholic Steatohepatitis. Obesity 2019, 27, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Jahnel, J.; Zöhrer, E.; Alisi, A.; Ferrari, F.; Ceccarelli, S.; De Vito, R.; Scharnagl, H.; Stojakovic, T.; Fauler, G.; Trauner, M.; et al. Serum Bile Acid Levels in Children with Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Bullò, M.; Tinahones, F.J.; Martínez-González, M.Á.; Corella, D.; Fragkiadakis, G.A.; López-Miranda, J.; Estruch, R.; Fitó, M.; Salas-Salvadó, J. Serum metabolites in non-alcoholic fatty-liver disease development or reversion; A targeted metabolomic approach within the PREDIMED trial. Nutr. Metab. 2017, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Caussy, C.; Hsu, C.; Singh, S.; Bassirian, S.; Kolar, J.; Faulkner, C.; Sinha, N.; Bettencourt, R.; Gara, N.; Valasek, M.A.; et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment. Pharmacol. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef]
- Spahis, S.; Alvarez, F.; Ahmed, N.; Dubois, J.; Jalbout, R.; Paganelli, M.; Grzywacz, K.; Delvin, E.; Peretti, N.; Levy, E. Non-alcoholic fatty liver disease severity and metabolic complications in obese children: Impact of omega-3 fatty acids. J. Nutr. Biochem. 2018, 58, 28–36. [Google Scholar] [CrossRef]
- Lou, D.J.; Zhu, Q.Q.; Si, X.W.; Guan, L.L.; You, Q.Y.; Yu, Z.M.; Zhang, A.Z.; Li, D. Serum phospholipid omega-3 polyunsaturated fatty acids and insulin resistance in type 2 diabetes mellitus and non-alcoholic fatty liver disease. J. Diabetes Complicat. 2014, 28, 711–714. [Google Scholar] [CrossRef]
- Elizondo, A.; Araya, J.; Rodrigo, R.; Sgherri, C.; Comporti, M.; Poniachik, J.; Videla, L.A. Effects of weight loss on liver and erythrocyte polyunsaturated fatty acid pattern and oxidative stress status in obese patients with non-alcoholic fatty liver disease. Biol. Res. 2008, 41, 59–68. [Google Scholar] [CrossRef]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.F.; Yang, B.; Tang, J.; Li, D. Fatty acid and non-alcoholic fatty liver disease: Meta-analyses of case-control and randomized controlled trials. Clin. Nutr. 2018, 37, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Molina, B.; Castellano-Castillo, D.; Pastor, O.; Ocaña-Wilhelmi, L.; Fernández-García, D.; Romero-Gómez, M.; Cardona, F.; Tinahones, F.J. A Pilot Study of Serum Sphingomyelin Dynamics in Subjects with Severe Obesity and Non-alcoholic Steatohepatitis after Sleeve Gastrectomy. Obes. Surg. 2019, 29, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schönauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects with Non-alcoholic Fatty Liver. Am. J. Gastroenterol. 2017, 112, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Männistö, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Käkelä, P.; Kärjä, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 2015, 35, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Yang, Z.X.; Shen, W.; Sun, H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol. Int. 2010, 4, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sire, J.; Hamsten, A.; Fisher, R.M.; Yki-ja, H. Genes involved in fatty acid partitioning and binding, inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [Green Version]
- Bozic, M.; Guzmán, C.; Benet, M.; Sánchez-Campos, S.; García-Monzón, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016, 65, 748–757. [Google Scholar] [CrossRef]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: Relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef]
- Dubois, V.; Gheeraert, C.; Vankrunkelsven, W.; Dubois-Chevalier, J.; Dehondt, H.; Bobowski-Gerard, M.; Vinod, M.; Zummo, F.P.; Güiza, F.; Ploton, M.; et al. Endoplasmic reticulum stress actively suppresses hepatic molecular identity in damaged liver. Mol. Syst. Biol. 2020, 16, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, J.; Jung, Y. Potential Therapeutic Application of Estrogen in Gender Disparity of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Cells 2019, 8, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggieri, A.; Gagliardi, M.C.; Anticoli, S. Sex-Dependent Outcome of Hepatitis B and C Viruses Infections: Synergy of Sex Hormones and Immune Responses? Front. Immunol. 2018, 9, 2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.; Kim, W.; Joo, S.K.; Park, S.; Park, J.H.; Kang, Y.K.; Jin, S.-Y.; Chang, M.S. Expression patterns of STAT3, ERK and estrogen-receptor α are associated with development and histologic severity of hepatic steatosis: A retrospective study. Diagn. Pathol. 2018, 13, 23. [Google Scholar] [CrossRef] [Green Version]
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone signaling and fatty liver in females: Analysis of estrogen receptor α mutant mice. Int. J. Obes. 2017, 41, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Vazquez, J.T.; Boulger, E.; Liu, H.; Xue, P.; Hussain, M.A.; Wolfe, A. Hepatic estrogen receptor α is critical for regulation of gluconeogenesis and lipid metabolism in males. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Hevener, A.L.; Clegg, D.J.; Mauvais-Jarvis, F. Impaired estrogen receptor action in the pathogenesis of the metabolic syndrome. Mol. Cell. Endocrinol. 2015, 418, 306–321. [Google Scholar] [CrossRef] [Green Version]
- Urlep, Ž.; Lorbek, G.; Perše, M.; Jeruc, J.; Juvan, P.; Matz-Soja, M.; Gebhardt, R.; Björkhem, I.; Hall, J.A.; Bonneau, R.; et al. Disrupting hepatocyte Cyp51 from cholesterol synthesis leads to progressive liver injury in the developing mouse and decreases RORC signalling. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Ke, P.Y. Diverse functions of autophagy in liver physiology and liver diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef] [Green Version]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar]
- López, D.M.; Thelen, M.; Stahl, F.; Thiel, C.; Linhorst, A.; Sylvester, M.; Hermanns-Borgmeyer, I.; Lüllmann-Rauch, R.; Eskild, W.; Saftig, P.; et al. The lysosomal transporter mfsd1 is essential for liver homeostasis and critically depends on its accessory subunit glmp. eLife 2019, 8. [Google Scholar] [CrossRef]
- Perland, E.; Bagchi, S.; Klaesson, A.; Fredriksson, R. Characteristics of 29 novel atypical solute carriers of major facilitator superfamily type: Evolutionary conservation, predicted structure and neuronal co-expression. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, W.W.Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavallard, V.J.; Gual, P. Autophagy and Non-Alcoholic Fatty Liver Disease. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics—A bioconductor package for quality assessment of microarray data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Matys, V. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [Green Version]
- Furge, K.K.D. PGSEA: Parametric Gene Set Enrichment Analysis. R Package Version 2006, 1, 44. [Google Scholar]
- Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R Package Version 2018, 1, 1. [Google Scholar]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst. Biol. 2011, 5, 147. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
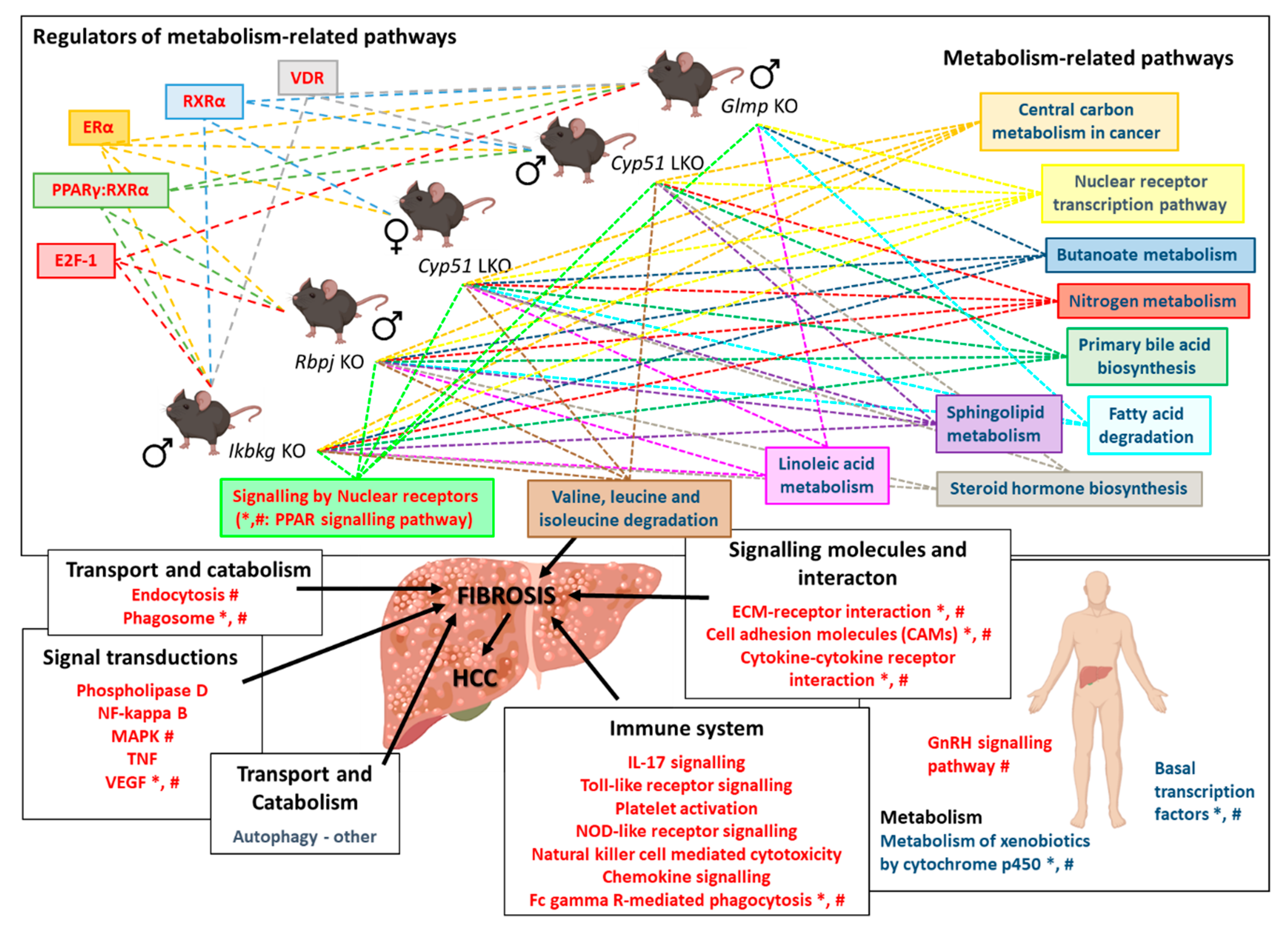{kind=link}
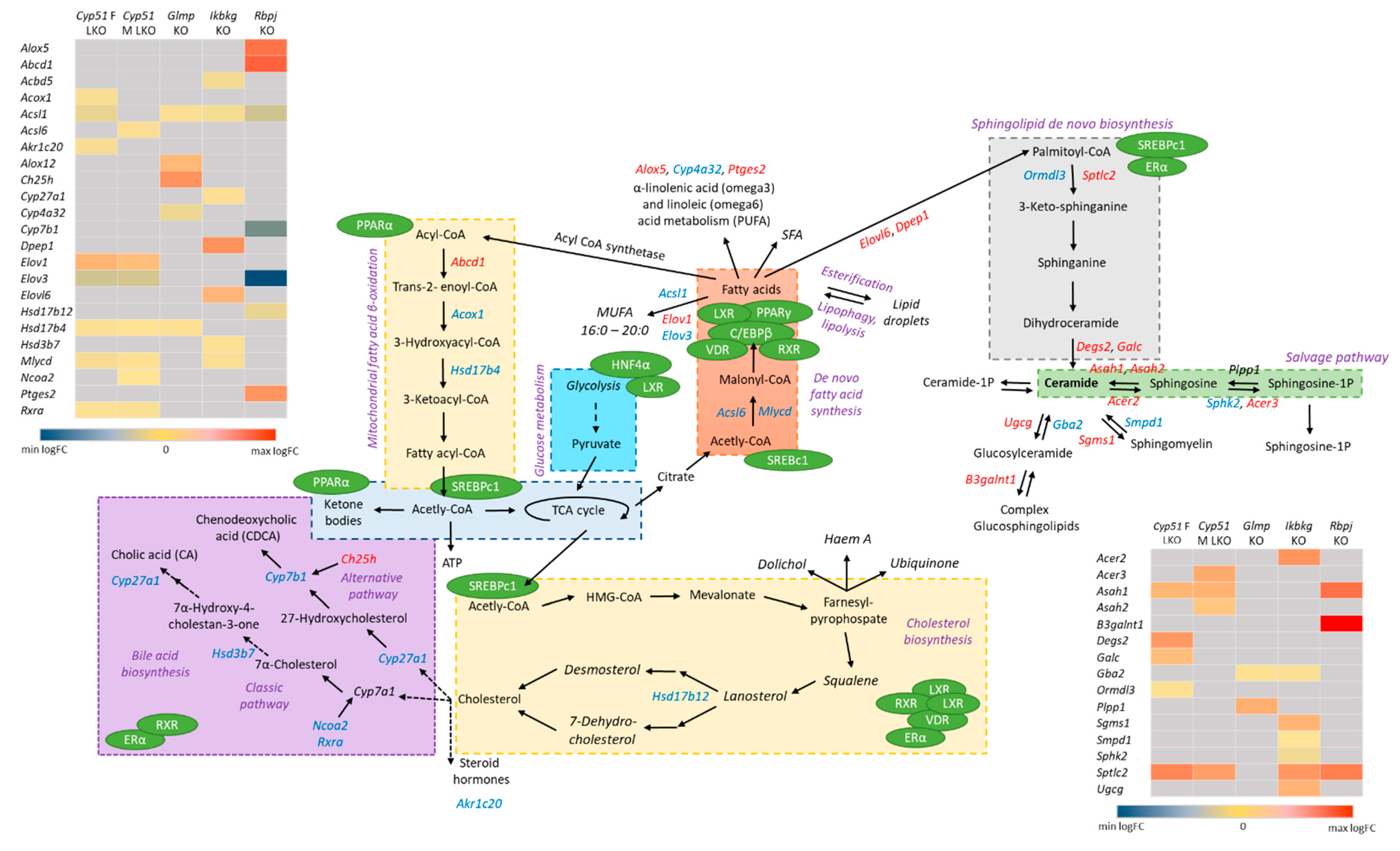{kind=link}
| KEGG Pathway | NAFLD | NASH | Cyp51 F LKO | Cyp51 M LKO | Glmp KO | Ikbkg LKO | Rbpj LKO |
|---|---|---|---|---|---|---|---|
| Antigen processing and presentation | + | + | 1.00 | ns | 0.84 | 1.05 | ns |
| B cell receptor signaling pathway | + | + | 0.89 | 0.77 | ns | 1.06 | 0.91 |
| Cell adhesion molecules (CAMs) | + | + | 1.43 | 1.12 | 1.02 | 1.34 | 0.60 |
| Cell cycle | ns | + | 1.25 | ns | 0.50 | 2.40 | 1.40 |
| Chemokine signaling pathway | ns | + | 1.06 | 0.78 | 0.89 | 1.17 | 1.39 |
| Colorectal cancer | ns | + | 0.87 | 0.73 | ns | 0.84 | 0.47 |
| Cytokine–cytokine receptor interaction | ns | + | 0.76 | 0.77 | 0.71 | 0.93 | 1.21 |
| DNA replication | + | + | 0.99 | ns | ns | 1.01 | 1.23 |
| ECM-receptor interaction | + | + | 1.67 | 1.87 | 0.54 | 1.50 | 0.50 |
| Endocytosis | ns | + | 1.20 | 0.72 | ns | 0.93 | 0.65 |
| ErbB signaling pathway | ns | + | 0.80 | 0.56 | ns | 0.78 | 0.49 |
| Fc epsilon RI signaling pathway | + | + | 0.67 | 0.40 | ns | 0.50 | 0.75 |
| Fc gamma R-mediated phagocytosis | + | + | 1.08 | 0.93 | ns | 1.06 | 0.97 |
| Focal adhesion | + | + | 1.96 | 1.85 | ns | 1.65 | 0.67 |
| Hematopoietic cell lineage | + | + | 1.20 | 1.59 | 1.23 | 1.05 | 0.95 |
| Leukocyte transendothelial migration | + | + | 1.52 | 1.11 | 0.70 | 1.08 | 0.90 |
| MAPK signaling pathway | ns | + | 1.09 | 1.08 | ns | 1.26 | 0.70 |
| Natural killer cell mediated cytotoxicity | ns | + | 0.95 | ns | 0.54 | 0.87 | 0.80 |
| Neurotrophin signaling pathway | ns | + | 0.98 | 0.53 | ns | 0.63 | 0.60 |
| Pancreatic cancer | + | + | 1.01 | 0.76 | ns | 0.95 | 0.42 |
| Pathways in cancer | ns | + | 1.42 | 1.18 | ns | 1.31 | 1.21 |
| Phagosome | + | + | 1.71 | 1.48 | 1.10 | 1.70 | 1.53 |
| Regulation of actin cytoskeleton | ns | + | 1.77 | 1.39 | ns | 1.27 | 0.63 |
| Small cell lung cancer | + | + | 1.21 | 1.18 | 0.44 | 1.02 | 0.64 |
| T cell receptor signaling pathway | ns | + | 0.73 | 0.52 | ns | 0.63 | 0.40 |
| Toll-like receptor signaling pathway | ns | + | 0.85 | 0.70 | ns | 0.69 | 0.65 |
| VEGF signaling pathway | + | + | 0.48 | 0.28 | ns | 0.49 | 0.54 |
| Reactome Pathway | Cyp51 F LKO | Cyp51 M LKO | Glmp KO | Ikbkg LKO | Rbpj LKO |
|---|---|---|---|---|---|
| Metabolism | −1.79 | ns | ns | −0.84 | ns |
| Metabolism of carbohydrates | ns | ns | ns | 0.45 | 0.49 |
| Glycosaminoglycan metabolism | ns | ns | ns | 0.55 | ns |
| Hyaluronan metabolism | ns | ns | 0.29 | 0.48 | 0.50 |
| Chondroitin sulfate biosynthesis | ns | ns | 0.21 | ns | 0.36 |
| Chondroitin sulfate/dermatan sulfate metabolism | ns | ns | ns | 0.29 | ns |
| Formation of xylulose-5-phosphate | ns | −0.38 | −0.16 | ns | ns |
| Fructose biosynthesis | 0.33 | 0.51 | ns | 0.75 | ns |
| Fructose catabolism | −0.30 | ns | ns | ns | ns |
| Fructose metabolism | ns | ns | ns | 0.50 | ns |
| Gluconeogenesis | ns | ns | ns | ns | 0.45 |
| Glucose metabolism | ns | ns | ns | ns | 0.55 |
| Glycolysis | ns | ns | ns | ns | 0.45 |
| Glycogen breakdown (glycogenolysis) | 0.17 | 0.22 | ns | ns | ns |
| Glycogen synthesis | ns | ns | ns | 0.18 | ns |
| Pentose phosphate pathway | ns | 0.39 | ns | ns | ns |
| Metabolism of steroids | ns | ns | ns | −0.72 | ns |
| Metabolism of steroid hormones | ns | ns | ns | −0.24 | ns |
| Androgen biosynthesis | ns | ns | ns | −0.77 | −0.90 |
| Glucocorticoid biosynthesis | ns | −0.99 | −0.50 | −0.84 | ns |
| Mineralocorticoid biosynthesis | ns | −0.99 | −0.72 | −0.88 | ns |
| Pregnenolone biosynthesis | 0.34 | 0.49 | ns | 0.41 | ns |
| Estrogen biosynthesis | ns | ns | ns | ns | 0.48 |
| Cholesterol biosynthesis | ns | 1.73 | 0.70 | ns | ns |
| Bile acid and bile salt metabolism | ns | ns | −0.20 | −0.54 | −0.75 |
| Recycling of bile acids and salts | ns | −0.36 | −0.29 | −0.36 | −0.32 |
| Synthesis of bile acids and bile salts | ns | ns | ns | −0.53 | −0.69 |
| Synthesis of bile acids and bile salts via 24-hydroxycholesterol | −0.80 | ns | −0.22 | −0.45 | −0.77 |
| Synthesis of bile acids and bile salts via 27-hydroxycholesterol | −0.79 | ns | ns | −0.59 | −0.96 |
| Synthesis of bile acids and bile salts via 7alpha-hydroxycholesterol | −0.82 | ns | ns | −0.72 | −1.00 |
| Fatty acid metabolism | −1.31 | ns | ns | ns | ns |
| Fatty acyl-CoA biosynthesis | ns | 0.81 | ns | ns | ns |
| alpha-linoleic (omega3) and linoleic (omega6) acid metabolism | −0.70 | ns | ns | −0.36 | −1.49 |
| Mitochondrial Fatty Acid Beta-Oxidation | −1.18 | −0.69 | −0.46 | ns | ns |
| Mitochondrial fatty acid beta-oxidation of saturated fatty acids | −0.68 | −0.41 | −0.33 | ns | ns |
| Propionyl-CoA catabolism | −0.37 | ns | ns | −0.33 | ns |
| Beta oxidation of decanoyl-CoA to octanoyl-CoA-CoA | −0.56 | −0.30 | −0.29 | ns | ns |
| Beta oxidation of hexanoyl-CoA to butanoyl-CoA | −0.58 | −0.33 | −0.27 | ns | ns |
| Beta oxidation of lauroyl-CoA to decanoyl-CoA-CoA | −0.59 | −0.37 | −0.26 | ns | ns |
| Beta oxidation of octanoyl-CoA to hexanoyl-CoA | −0.57 | −0.34 | −0.29 | ns | ns |
| Peroxisomal lipid metabolism | −1.12 | ns | −0.36 | ns | −0.82 |
| Alpha-oxidation of phytanate | −0.61 | ns | −0.21 | −0.33 | ns |
| Beta-oxidation of pristanoyl-CoA | ns | ns | ns | ns | −0.62 |
| Beta-oxidation of very long chain fatty acids | −0.87 | −0.46 | −0.38 | ns | ns |
| Sphingolipid metabolism | 0.49 | 0.34 | ns | 0.38 | 0.77 |
| Glycosphingolipid metabolism | 0.56 | 0.46 | ns | 0.45 | 0.76 |
| Triglyceride metabolism | ns | ns | 0.62 | ns | ns |
| Triglyceride biosynthesis | −0.39 | ns | ns | ns | 0.54 |
| Triglyceride catabolism | ns | ns | 0.92 | ns | ns |
| Wax and plasmalogen biosynthesis | ns | 0.31 | ns | ns | ns |
| Ketone body metabolism | −0.40 | ns | ns | ns | −0.43 |
| Synthesis of Ketone Bodies | −0.59 | ns | −0.28 | −0.22 | −0.44 |
| The citric acid (TCA) cycle and respiratory electron transport | −1.31 | ns | ns | −0.84 | ns |
| Pyruvate metabolism and Citric Acid (TCA) cycle | −0.75 | ns | ns | −0.43 | ns |
| Metabolism of vitamins and cofactors | −1.35 | ns | ns | ns | ns |
| Metabolism of water-soluble vitamins and cofactors | −1.40 | ns | ns | −0.76 | ns |
| Metabolism of fat-soluble vitamins | −0.52 | ns | ns | ns | 0.43 |
| Metabolism of amino acids and derivatives | −1.38 | ns | ns | −1.14 | ns |
| Metabolism of amine-derived hormones | −0.43 | ns | ns | −0.37 | ns |
| Aspartate and asparagine metabolism | −0.51 | ns | ns | −0.22 | ns |
| Branched-chain amino acid catabolism | −0.98 | ns | −0.42 | −0.73 | −0.59 |
| Choline catabolism | ns | ns | ns | −0.32 | ns |
| Degradation of cysteine and homocysteine | −0.59 | ns | ns | −0.41 | ns |
| Glutamate and glutamine metabolism | −0.24 | ns | ns | ns | ns |
| Glyoxylate metabolism and glycine degradation | −0.92 | ns | −0.43 | −0.63 | ns |
| Histidine catabolism | −0.24 | −0.27 | ns | −0.24 | ns |
| Lysine catabolism | −0.65 | ns | −0.25 | −0.37 | ns |
| Phenylalanine and tyrosine metabolism | −0.68 | ns | ns | −0.25 | ns |
| Phenylalanine metabolism | −0.51 | ns | ns | ns | ns |
| Tyrosine catabolism | −0.44 | ns | ns | −0.21 | ns |
| Serine biosynthesis | 0.32 | ns | ns | ns | ns |
| Sulfur amino acid metabolism | −0.63 | ns | ns | −0.49 | ns |
| Threonine catabolism | −0.43 | −0.33 | ns | −0.36 | ns |
| Tryptophan catabolism | −0.62 | ns | ns | −0.44 | ns |
| Urea cycle | −0.37 | ns | ns | −0.29 | ns |
| Factor | Stage of Disease | Change | Reference |
|---|---|---|---|
| Metabolites | |||
| Bile acids | NASH | Primary and secondary bile acids are increased | [26] |
| NASH | C4 increased | [29] | |
| NASH | Primary and secondary bile acids increased | [30] | |
| NASH | Bile acids increase with NASH progression | [31] | |
| NASH | Bile acids increased | [32] | |
| NASH | Primary bile acids increased, secondary decreased | [33] | |
| NAFLD | Total bile acid are decreased but major difference is in composition, bile acid level increases with fibrosis progression | [34] | |
| NAFLD | Bile acids change with disease progression, direction depended on the type of bile acid | [35] | |
| NASH | Primary conjugated bile acid increase with fibrosis, unconjugated bile acids decrease | [36] | |
| NAFLD | Primary and secondary bile acids are increased in higher fibrotic stages, but no change in C4 | [27] | |
| Polyunsaturated fatty acids (PUFA) | NAFLD | Decreased | [35] |
| Severe NAFLD | Total PUFA decreased in red blood cell membrane, n-3 all decreased, n-6 majority increased, except linoleic acid decreased | [37] | |
| NAFLD | Total PUFA n-3 decreased in serum | [38] | |
| NAFLD | Total PUFA decreased in erythrocytes | [39] | |
| NASH | PUFA (18:3n-3) decreased | [9] | |
| NASH | Eicosapentaenoate (20:5n-3), docosahexaenoate (22:6n-3), arachidonate (20:4n-6) are decreased | [32] | |
| NASH | PUFA are altered | [40] | |
| Monounsaturated fatty acids (MUFA) | NASH | Total MUFA increased | [40] |
| Severe NAFLD | Total MUFA increased in red blood cell membrane | [37] | |
| NAFLD | Total MUFA increased, docosahexaenoic acid (C22:6) and arachidonic acid (C20:4) decreased in blood | [41] | |
| Sphingolipids | NASH | Sphingomyelin (36:0) increased | [42] |
| NAFLD | Sphingomyelins decreased | [43] | |
| NASH | Sphingomyelin increased | [9] | |
| Ketones | NASH | Decreased | [44] |
| Branched amino acids | NAFLD | All three increased | [43] |
| NASH | All three increased | [32] | |
| Transcription factors | |||
| PPARα | NASH | Decreased mRNA expression in liver, negative correlation with NASH progression | [45,46] |
| LXR, SREBPC1 | NAFLD | Increased mRNA and protein expression in liver | [47] |
| PPARγ | NAFLD | PPARγ2 mRNA is increased in liver | [48] |
| VDR | steatosis | mRNA is increased in liver | [49] |
| NASH | Protein is decreased in liver | [50] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagotinšek Cokan, K.; Urlep, Ž.; Moškon, M.; Mraz, M.; Kong, X.Y.; Eskild, W.; Rozman, D.; Juvan, P.; Režen, T. Common Transcriptional Program of Liver Fibrosis in Mouse Genetic Models and Humans. Int. J. Mol. Sci. 2021, 22, 832. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020832
Blagotinšek Cokan K, Urlep Ž, Moškon M, Mraz M, Kong XY, Eskild W, Rozman D, Juvan P, Režen T. Common Transcriptional Program of Liver Fibrosis in Mouse Genetic Models and Humans. International Journal of Molecular Sciences. 2021; 22(2):832. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020832
Chicago/Turabian StyleBlagotinšek Cokan, Kaja, Žiga Urlep, Miha Moškon, Miha Mraz, Xiang Yi Kong, Winnie Eskild, Damjana Rozman, Peter Juvan, and Tadeja Režen. 2021. "Common Transcriptional Program of Liver Fibrosis in Mouse Genetic Models and Humans" International Journal of Molecular Sciences 22, no. 2: 832. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020832