Repetitive Traumatic Brain Injury Causes Neuroinflammation before Tau Pathology in Adolescent P301S Mice


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
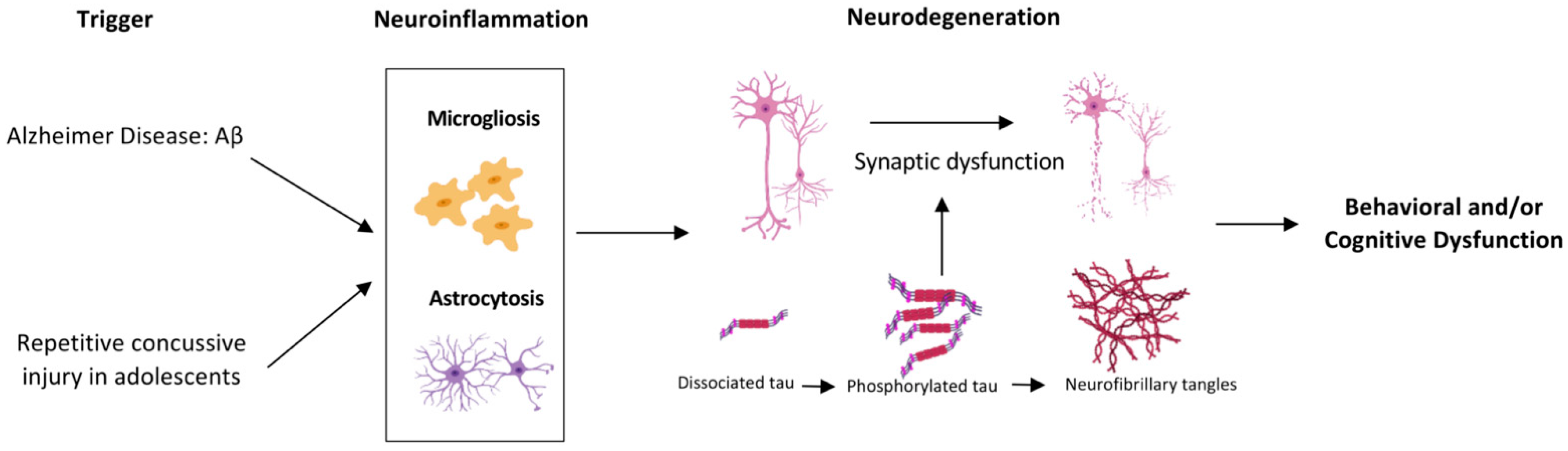
:1. Introduction
2. Results
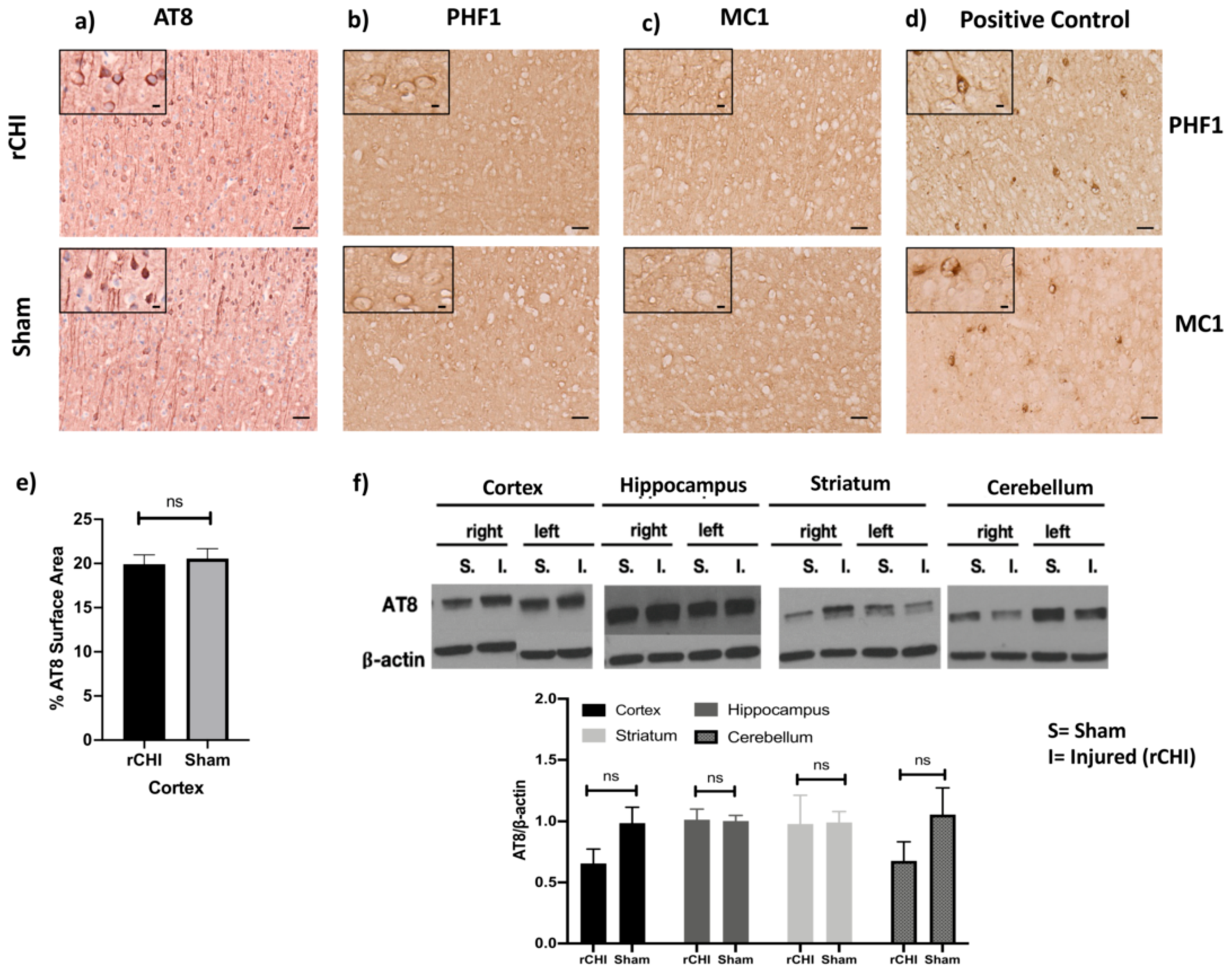
2.1. Repetitive Closed-Head Injury (rCHI) Did Not Alter Tau Pathology in Adolescent P301S Mice 40 Days Post Injury
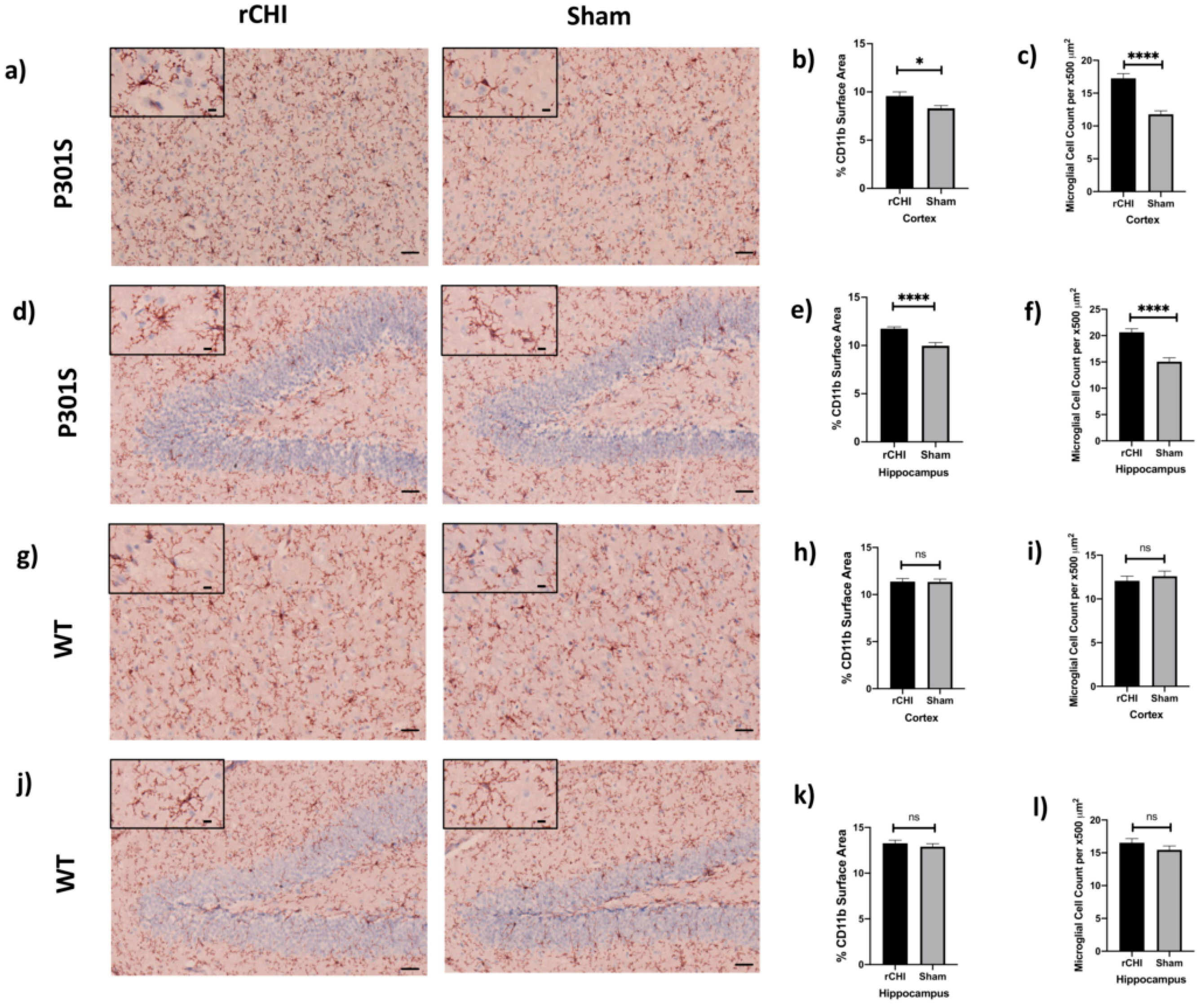
2.2. Repetitive Closed-Head Injury (rCHI) Induced Microgliosis in Adolescent P301S but Not WT Mice at 40 Days Post Injury
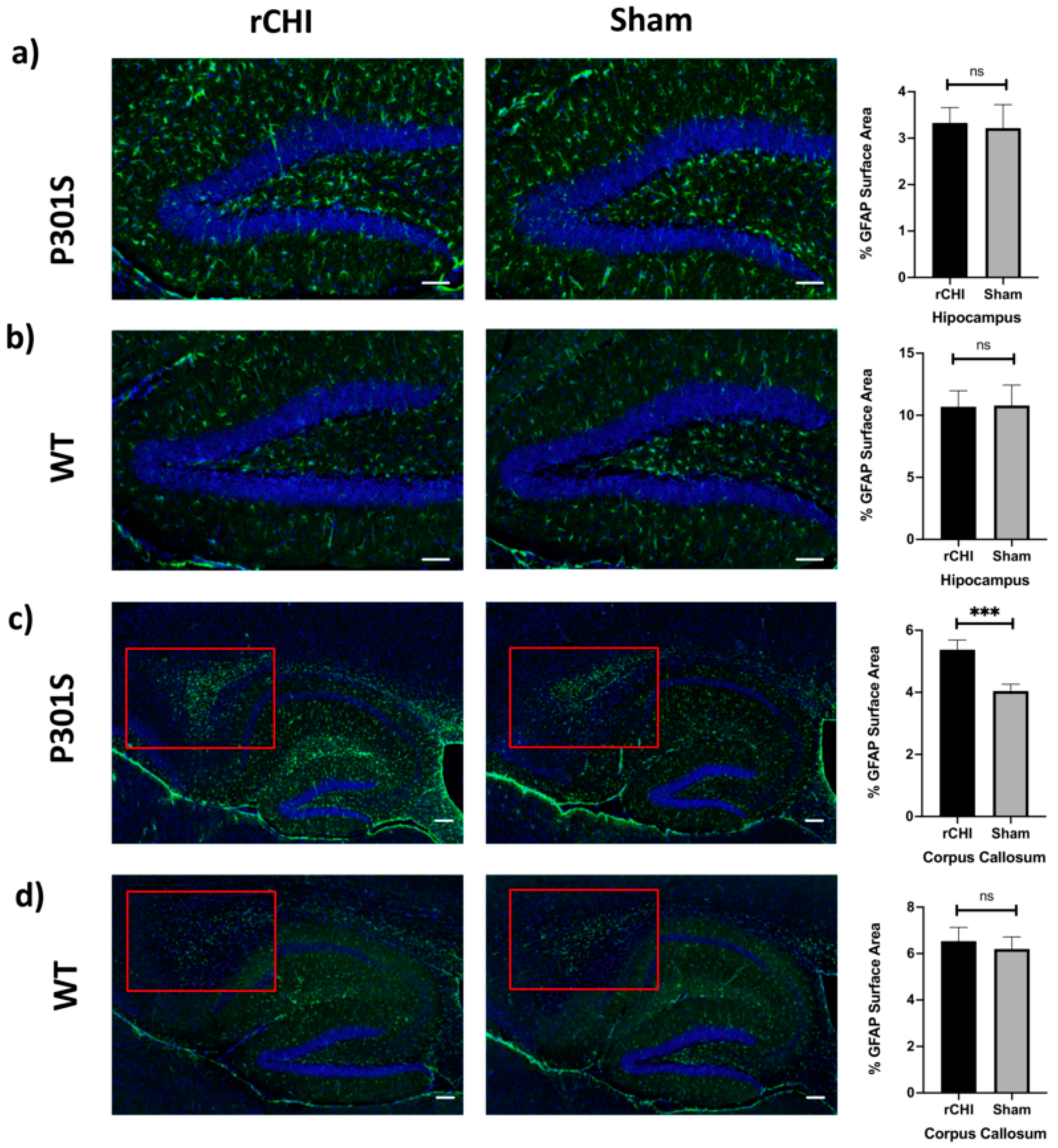
2.3. Repetitive Closed-Head Injury (rCHI) Increased Astrocytosis in Adolescent P301S but Not in WT Mice at 40 Days Post Injury
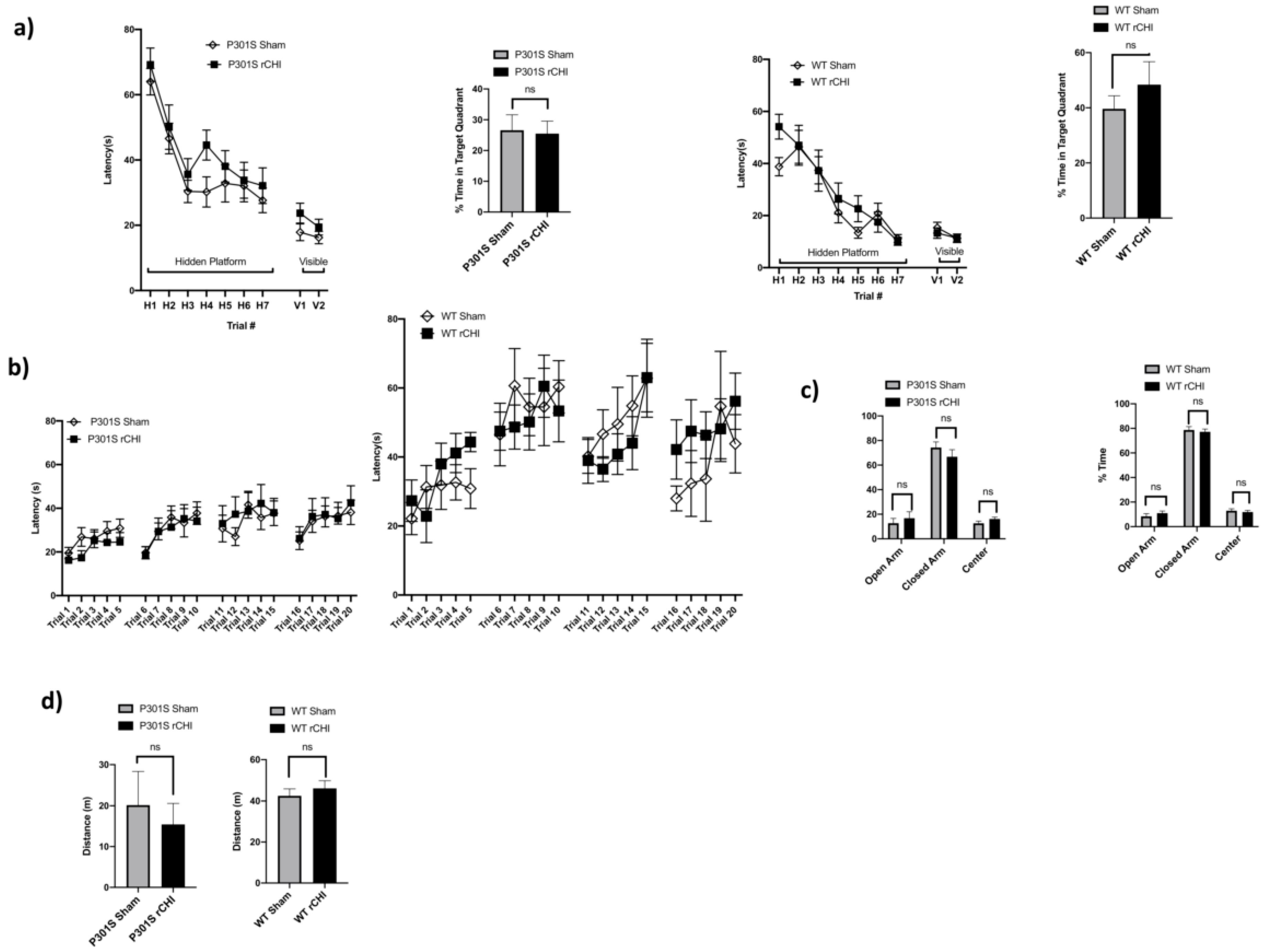
2.4. No Behavioral Deficits Were Detected 40 Days after Repetitive Closed-Head Injury (rCHI) in Adolescent P301S and WT Mice
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Repetitive Closed Head Injury Model (rCHI)
4.3. Preparation of Brain Tissue for Immunohistochemistry
4.4. CD11b Immunohistochemistry
4.5. AT-8 Immunohistochemistry
4.6. MC1 and PHF1 Immunohistochemistry
4.7. GFAP Immunofluorescence Staining
4.8. Image Analysis
4.9. Western Blot
4.10. Behavioral Studies
4.10.1. Open Field Testing
4.10.2. Plus Maze
4.10.3. Rotarod
4.10.4. Morris Water Maze
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bakhos, L.L.; Lockhart, G.R.; Myers, R.; Linakis, J.G. Emergency department visits for concussion in young child athletes. Pediatrics 2010, 126, 550–556. [Google Scholar] [CrossRef] [Green Version]
- Mitka, M. Reports of concussions from youth sports rise along with awareness of the problem. JAMA 2010, 304, 1775–1776. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.J.; MacDougall, R.; Quatman-Yates, C.C.; Myer, G.D.; Sugimoto, D.; Dennison, R.J.; Meehan, W.P., 3rd. Young athletes’ concerns about sport-related concussion: The patient’s perspective. Clin. J. Sport Med. 2016, 26, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Fishman, M.; Taranto, E.; Perlman, M.; Quinlan, K.; Benjamin, H.J.; Ross, L.F. Attitudes and counseling practices of pediatricians regarding youth sports participation and concussion risks. J. Pediatr. 2017, 184, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ropper, A.H.; Gorson, K.C. Clinical practice. Concussion. N. Engl. J. Med. 2007, 356, 166–172. [Google Scholar] [CrossRef]
- Smith, D.H.; Johnson, V.E.; Stewart, W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 2013, 9, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, M.W.; Yeates, K.O.; Wilson, P.E. Pediatric sport-related concussion: A review of the clinical management of an oft-neglected population. Pediatrics 2006, 117, 1359–1371. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Baugh, C.M.; Stamm, J.M.; Riley, D.O.; Gavett, B.E.; Shenton, M.E.; Lin, A.; Nowinski, C.J.; Cantu, R.C.; McKee, A.C.; Stern, R.A. Chronic traumatic encephalopathy: Neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav. 2012, 6, 244–254. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Johnson, V.E.; Stewart, W.; Trojanowski, J.Q.; Smith, D.H. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 2011, 122, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.A.; Zhang, X.L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 2018, 141, 422–458. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.F.; Vowles, G.H.; Robinson, S.F.; Sutcliffe, J.C. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol. Appl. Neurobiol. 1996, 22, 12–16. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.F.; Vowles, G.H.; Nicoll, J.A.; Revesz, T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999, 98, 171–178. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G. Tau biology, tauopathy, traumatic brain injury, and diagnostic challenges. J. Alzheimers Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef] [Green Version]
- Bieniek, K.F.; Ross, O.A.; Cormier, K.A.; Walton, R.L.; Soto-Ortolaza, A.; Johnston, A.E.; DeSaro, P.; Boylan, K.B.; Graff-Radford, N.R.; Wszolek, Z.K.; et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015, 130, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.T.; LaFerla, F.M.; Holtzman, D.M.; Brody, D.L. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 2011, 31, 9513–9525. [Google Scholar] [CrossRef] [Green Version]
- Mouzon, B.; Bachmeier, C.; Ojo, J.; Acker, C.; Ferguson, S.; Crynen, G.; Davies, P.; Mullan, M.; Stewart, W.; Crawford, F. Chronic white matter degeneration, but no tau pathology at one-year post-repetitive mild traumatic brain injury in a tau transgenic model. J. Neurotrauma 2019, 36, 576–588. [Google Scholar] [CrossRef]
- Petraglia, A.L.; Plog, B.A.; Dayawansa, S.; Dashnaw, M.L.; Czerniecka, K.; Walker, C.T.; Chen, M.; Hyrien, O.; Iliff, J.J.; Deane, R.; et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg. Neurol. Int. 2014, 5, 184. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.C.; Crawford, F. Repetitive head trauma, chronic traumatic encephalopathy and tau: Challenges in translating from mice to men. Exp. Neurol. 2016, 275, 389–404. [Google Scholar] [CrossRef]
- Winston, C.N.; Noel, A.; Neustadtl, A.; Parsadanian, M.; Barton, D.J.; Chellappa, D.; Wilkins, T.E.; Alikhani, A.D.; Zapple, D.N.; Villapol, S.; et al. Dendritic spine loss and chronic white matter inflammation in a mouse model of highly repetitive head trauma. Am. J. Pathol. 2016, 186, 552–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, J.O.; Mouzon, B.; Greenberg, M.B.; Bachmeier, C.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J. Neuropathol. Exp. Neurol. 2013, 72, 137–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, Y.; Uryu, K.; Higuchi, M.; Longhi, L.; Hoover, R.; Fujimoto, S.; McIntosh, T.; Lee, V.M.; Trojanowski, J.Q. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J. Neurotrauma 2005, 22, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Gangolli, M.; Benetatos, J.; Esparza, T.J.; Fountain, E.M.; Seneviratne, S.; Brody, D.L. Repetitive concussive and subconcussive injury in a human tau mouse model results in chronic cognitive dysfunction and disruption of white matter tracts, but not tau pathology. J. Neurotrauma 2019, 36, 735–755. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ryu, J.; Nguyen, J.V.; Arena, J.; Rha, E.; Vranis, P.; Hitt, D.; Marsh-Armstrong, N.; Koliatsos, V.E. Evidence for accelerated tauopathy in the retina of transgenic P301S tau mice exposed to repetitive mild traumatic brain injury. Exp. Neurol. 2015, 273, 168–176. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C.; Abdolmohammadi, B.; Stein, T.D. The neuropathology of chronic traumatic encephalopathy. Handb. Clin. Neurol. 2018, 158, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, W.L.; MacKinnon, M.A.; Stewart, J.E.; Graham, D.I. Stereology of cerebral cortex after traumatic brain injury matched to the Glasgow outcome score. Brain 2010, 133, 139–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giunta, B.; Obregon, D.; Velisetty, R.; Sanberg, P.R.; Borlongan, C.V.; Tan, J. The immunology of traumatic brain injury: A prime target for Alzheimer’s disease prevention. J. Neuroinflamm. 2012, 9, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erturk, A.; Mentz, S.; Stout, E.E.; Hedehus, M.; Dominguez, S.L.; Neumaier, L.; Krammer, F.; Llovera, G.; Srinivasan, K.; Hansen, D.V.; et al. Interfering with the chronic immune response rescues chronic degeneration after traumatic brain injury. J. Neurosci. 2016, 36, 9962–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Alquraini, A.; El Khoury, J. Scavenger receptors. Curr. Biol. 2020, 30, R790–R795. [Google Scholar] [CrossRef]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Leung, E.; Guo, L.; Bu, J.; Maloof, M.; El Khoury, J.; Geula, C. Microglia activation mediates fibrillar amyloid-beta toxicity in the aged primate cortex. Neurobiol. Aging 2011, 32, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Sardari, M.; Skuljec, J.; Yin, D.; Zec, K.; de Carvalho, T.S.; Albers, D.; Wang, C.; Pul, R.; Popa-Wagner, A.; Doeppner, T.R.; et al. Lipopolysaccharide-induced sepsis-like state compromises post-ischemic neurological recovery, brain tissue survival and remodeling via mechanisms involving microvascular thrombosis and brain T cell infiltration. Brain Behav. Immun. 2021, 91, 627–638. [Google Scholar] [CrossRef]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [Green Version]
- Scattoni, M.L.; Gasparini, L.; Alleva, E.; Goedert, M.; Calamandrei, G.; Spillantini, M.G. Early behavioural markers of disease in P301S tau transgenic mice. Behav. Brain Res. 2010, 208, 250–257. [Google Scholar] [CrossRef]
- Takeuchi, H.; Iba, M.; Inoue, H.; Higuchi, M.; Takao, K.; Tsukita, K.; Karatsu, Y.; Iwamoto, Y.; Miyakawa, T.; Suhara, T.; et al. P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS ONE 2011, 6, e21050. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Currer, M.J.; Harrison, D.E. The Mouse in Aging Research, 2nd ed.; American College Laboratory Animal Medicine (Elsevier): Burlington, MA, USA, 2007; pp. 637–672. [Google Scholar]
- Zhang, Z.; Ma, Z.; Zou, W.; Guo, H.; Liu, M.; Ma, Y.; Zhang, L. The appropriate marker for astrocytes: Comparing the distribution and expression of three astrocytic markers in different mouse cerebral regions. Biomed. Res. Int. 2019, 2019, 9605265. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.D.; Alvarez, V.E.; McKee, A.C. Concussion in chronic traumatic encephalopathy. Curr. Pain Headache Rep. 2015, 19, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G., 3rd; Moreno-Gonzalez, I.; Soto, C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 2017, 483, 1137–1142. [Google Scholar] [CrossRef]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Edwards, G., 3rd; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Deaton, L.M.; Qiu, M.; Ha, S.; Pacoma, R.; Lao, J.; Tolley, V.; Moran, R.; Keeton, A.; Lamb, J.R.; et al. Tau overexpression exacerbates neuropathology after repeated mild head impacts in male mice. Neurobiol. Dis. 2020, 134, 104683. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Morganti, J.M.; Bodnar, C.N.; Webster, S.J.; Higgins, E.K.; Roberts, K.N.; Snider, H.; Meier, S.E.; Nation, G.K.; Goulding, D.S.; et al. The effects of mild closed head injuries on tauopathy and cognitive deficits in rodents: Primary results in wild type and rTg4510 mice, and a systematic review. Exp. Neurol. 2020, 326, 113180. [Google Scholar] [CrossRef]
- Namjoshi, D.R.; Cheng, W.H.; McInnes, K.A.; Martens, K.M.; Carr, M.; Wilkinson, A.; Fan, J.; Robert, J.; Hayat, A.; Cripton, P.A.; et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Mol. Neurodegener 2014, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Qiu, J.; Alcon, S.; Hashim, J.; Rotenberg, A.; Sun, Y.; Meehan, W.P., 3rd; Mannix, R. Memantine improves outcomes after repetitive traumatic brain injury. Behav. Brain Res. 2018, 340, 195–204. [Google Scholar] [CrossRef]
- Wu, L.; Chung, J.Y.; Saith, S.; Tozzi, L.; Buckley, E.M.; Sanders, B.; Franceschini, M.A.; Lule, S.; Izzy, S.; Lok, J.; et al. Repetitive head injury in adolescent mice: A role for vascular inflammation. J. Cereb. Blood Flow. Metab. 2019, 39, 2196–2209. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzy, S.; Liu, Q.; Fang, Z.; Lule, S.; Wu, L.; Chung, J.Y.; Sarro-Schwartz, A.; Brown-Whalen, A.; Perner, C.; Hickman, S.E.; et al. Time-dependent changes in microglia transcriptional networks following traumatic brain injury. Front. Cell Neurosci. 2019, 13, 307. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.J.; et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 2018, 141, 2740–2754. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Clayton, E.L.; Mancuso, R.; Nielsen, T.T.; Mizielinska, S.; Holmes, H.; Powell, N.; Norona, F.; Larsen, J.O.; Milioto, C.; Wilson, K.M.; et al. Early microgliosis precedes neuronal loss and behavioural impairment in mice with a frontotemporal dementia-causing CHMP2B mutation. Hum. Mol. Genet. 2017, 26, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Khuman, J.; Meehan, W.P., 3rd; Zhu, X.; Qiu, J.; Hoffmann, U.; Zhang, J.; Giovannone, E.; Lo, E.H.; Whalen, M.J. Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J. Cereb. Blood Flow. Metab. 2011, 31, 778–789. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izzy, S.; Brown-Whalen, A.; Yahya, T.; Sarro-Schwartz, A.; Jin, G.; Chung, J.Y.; Lule, S.; Morsett, L.M.; Alquraini, A.; Wu, L.; et al. Repetitive Traumatic Brain Injury Causes Neuroinflammation before Tau Pathology in Adolescent P301S Mice. Int. J. Mol. Sci. 2021, 22, 907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020907
Izzy S, Brown-Whalen A, Yahya T, Sarro-Schwartz A, Jin G, Chung JY, Lule S, Morsett LM, Alquraini A, Wu L, et al. Repetitive Traumatic Brain Injury Causes Neuroinflammation before Tau Pathology in Adolescent P301S Mice. International Journal of Molecular Sciences. 2021; 22(2):907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020907
Chicago/Turabian StyleIzzy, Saef, Alexander Brown-Whalen, Taha Yahya, Aliyah Sarro-Schwartz, Gina Jin, Joon Yong Chung, Sevda Lule, Liza M. Morsett, Ali Alquraini, Limin Wu, and et al. 2021. "Repetitive Traumatic Brain Injury Causes Neuroinflammation before Tau Pathology in Adolescent P301S Mice" International Journal of Molecular Sciences 22, no. 2: 907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020907