TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism


1
Bernhard-Heine-Center for Locomotion Research, Department of Orthopedics, Julius-Maximilians-University Würzburg, 97076 Würzburg, Germany
2
Institute for Human Genetics, Biocenter, Julius-Maximilians-University Würzburg, 97074 Würzburg, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(2), 919; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020919
Submission received: 21 December 2020
/
Revised: 14 January 2021
/
Accepted: 14 January 2021
/
Published: 18 January 2021
(This article belongs to the Special Issue The Role of Tissue-Nonspecific Alkaline Phosphatase in Inflammation and Metabolic Syndrome)
{kind=link}
{kind=link}
Abstract
:This review summarizes important information on the ectoenzyme tissue-nonspecific alkaline phosphatase (TNAP) and gives a brief insight into the symptoms, diagnostics, and treatment of the rare disease Hypophosphatasia (HPP), which is resulting from mutations in the TNAP encoding ALPL gene. We emphasize the role of TNAP beyond its well-known contribution to mineralization processes. Therefore, above all, the impact of the enzyme on central molecular processes in the nervous system and on inflammation is presented here.
1. Structure, Function, and Substrates of TNAP
Tissue-nonspecific alkaline phosphatase (TNAP) or liver/bone/kidney alkaline phosphatase is an ectoenzyme that is anchored to the outer cell membrane (e.g. in osteoblasts) and to extracellular vesicles via its glycosyl-inositol-phosphate (GPI)-anchor [1,2]. TNAP belongs to the family of alkaline phosphatases (AP) that comprises in humans three additional tissue-specific isoforms: placental (PLAP, ALPP National Center for Biotechnology Information NCBI GeneID: 250), germ cell (GCAP, ALPG NCBI GeneID: 251), and intestinal alkaline phosphatase (IAP, ALPI NCBI GeneID: 248) [2,3].
As an ectophosphatase, TNAP catalyzes the dephosphorylation of its main substrates pyrophosphate (PPi) and pyridoxal-5′-phosphate (PLP) in the extracellular space [1,4,5]. In addition to the prominent mineralization inhibitor PPi, TNAP further dephosphorylates phosphorylated osteopontin (p-OPN, encoded by the SSP1 gene), which is preventing hydroxyapatite (HA) crystallization in its phosphorylated state [6]. Moreover, TNAP is also capable of dephosphorylating microbial products, such as lipopolysaccharides (LPS) as well as other phosphorylated small-molecule Toll-like-receptor (TLR) ligands [7]. Beyond that, TNAP is an ectonucleotidase that degrades adenosine triphosphate (ATP) in a stepwise manner to adenosine, with adenosine diphosphate (ADP) and adenosine monophosphate (AMP) as intermediates [1,8]. Consequently, TNAP’s enzymatic function is considerably influencing purinergic signaling cascades as it either provides or decreases the prevalence of substrates for several receptors (P1, P2X, P2Y).
Extrapolating from a structural analysis of the placental AP, a model for the protein structure of TNAP was developed, which implies that the enzyme contains two Zn2+ ions and one Mg2+ ion in its active center [9]. The function of an additionally identified Ca2+ site is still unclear [10]. The molecular weight of TNAP is approximately 80 kDa in its mature, post-translationally (above all, N-glycosylated) modified state [11,12]. Distinct protein domains have been characterized, namely the N-terminal α-helix, the crown domain, and the metal binding domain [9]. TNAP monomers form dimers via hydrophobic regions and the enzyme is mostly active as a GPI-anchored homodimer [13]. Altered dimerization can result in dominant negative effects of heterozygous mutations in the ALPL gene, which has already been described in the context of Hypophosphatasia (HPP) patients [14,15,16]. This implies that a deficient TNAP monomer is able to decrease the overall activity of its actually fully functional binding partner, either due to steric changes caused by dimerization or due to protein transport failure to the membrane [14,15,17].
The human ALPL gene (NCBI GeneID: 249) is located on chromosome 1 (1p36), contains 12 exons, including 11 coding exons as well as two alternative variants of exon 1 (1B and 1L, bone and liver variant), and has an overall gene size of approximately 50 kb [11,18]. Genetic variants resulting in disease-causing mutations have been described extensively [14,16,19,20,21,22] and to a certain extent, their molecular consequences can be predicted by bioinformatic tools [23]. As expected, mutations in essential functional regions that, for example, are coding for the active center or the dimerization domain, are predicted to result in more severe symptoms than mutations in sequences coding for less essential parts of the TNAP protein [23]. Nevertheless, it is important to mention that a definitive correlation between a certain genotype and a resulting phenotype (genotype–phenotype correlation) cannot be predicted and that additional modifying factors can strongly influence the disease manifestation [24]. One example was reported in a family with two affected daughters with the same ALPL mutation, where one girl developed a severe and the other a mild HPP onset [22]. These variable genotype–phenotype correlations pose difficulties for genetic counseling and an optimized disease management in HPP [25]. The majority of reported mutations in the human ALPL gene are missense mutations that are resulting in amino acid changes (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/?term=alpl%5Bgene%5D, accessed on 11 December 2020).
2. Heritable TNAP Dysfunction: The Rare Disease Hypophosphatasia
2.1. General Information on HPP
Genetically inheritable TNAP dysfunction, due to mutations in the ALPL gene, leads to the rare disease Hypophosphatasia (HPP) [1,26,27]. The prevalence of HPP is 1:300,000 for severe and approximately 1:6000 for mild courses of disease in the European population [28]. Rarely occurring severe manifestations of HPP are inherited in an autosomal recessive manner. Compound heterozygosity, implying that one patient carries two different mutations in the ALPL gene in a heterozygous manner, is very common in HPP patients and results in disease manifestations of very variable severity [14,16,29]. Mild to moderate forms follow an autosomal dominant or recessive trait [16]. Interestingly, all HPP patients displaying mild forms of HPP show a high variety of different genetic variants, including missense, splice site, frameshift, and nonsense mutations [16]. Mornet et al. only recently showed that dominant negative effects are resulting from missense mutations, which lead to moderate HPP forms, whereas haploinsufficiency is considered as the main cause for mild adult forms [16].
2.2. Clinical Subtypes
In general, HPP can lead to very heterogeneous outcomes and the patients can be categorized in six clinical subtypes, based on the age at diagnosis and/or of onset [1,30,31,32]. There are mild clinical subtypes, like Perinatal benign, Adult, or Odonto-HPP, with the latter only affecting the mineralization and the anchorage of teeth and leading to premature loss of deciduous teeth as the most prominent symptom [1,27,31,32,33,34,35]. Severe forms of HPP, like the Perinatal lethal or the Infantile subtypes, can both lead to a premature death [1,30,31,32]. Perinatal lethal forms often go along with epileptic seizures and respiratory failure, whereas Infantile HPP may go along with premature closure of the cranial sutures of the skull (craniosynostosis) [30,31,32]. The sixth described clinical subtype is Childhood HPP, which leads to symptoms like limb shortening, delayed walking, a characteristic waddling gait, and hypo-mineralization, and is not considered as life-threatening [27,30].
2.3. Diagnosis
Generally, the most prominent symptoms of HPP are hypo-mineralization of bones and teeth [1,26,31,36,37,38,39]. Nevertheless, lungs, kidneys, and the nervous system can also be affected, depending on the severity of the disease [21,27,31,36]. Additionally, chronic pain is an issue that goes along with HPP and that significantly reduces the patients’ quality of life [40]. Therefore, HPP is considered a multisystemic disease and should optimally be treated by an interdisciplinary team of physicians [32,40,41]. The diagnosis of HPP includes observance of characteristic symptoms, measurement of decreased serum AP activity (based on age- and sex-specific reference values), detection of increased PLP concentrations in the blood serum [42], as well as phosphoethanolamine (PEA) in the urine, and the detection of mutations in the ALPL gene sequence [43]. Radiologic imaging techniques may be used to further confirm the diagnosis [43,44]. Due to HPP being a rare disease with variable severity levels and symptoms, patients are often misdiagnosed or have to wait for years in order to get the correct diagnosis, which may lead to improper treatment in the meantime and can cause high psychological burden [45].
2.4. Treatment
Despite that functional and genetic understanding of HPP has greatly advanced in recent years, no curative treatment for HPP is available to date. Therefore, patients are still treated symptomatically, which includes, among others, treatment with non-steroidal anti-inflammatory drugs (NSAIDs) against inflammation [46,47], neurosurgery due to issues caused by craniosynostosis [48], treatment of dental problems by dentists [34,49], and eventually, orthopedic surgery after the occurrence of fractures [50,51,52]. An enzyme replacement therapy with a recombinant enzyme including a deca-aspartate anchor, called Asfotase alfa, has been approved for treatment of severely affected children since 2015 in Europe and the U.S. (https://www.ema.europa.eu/en/medicines/human/EPAR/strensiq; https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125513Orig1s000TOC.cfm, accessed on 14 December 2020) [53,54,55,56]. Several studies have analyzed the pharmacodynamics and safety of Asfotase alfa and additionally shown its positive effects on survival rates, dental health, skeletal development, motor functions, and respiratory functions in mouse models and HPP patients [53,54,55,56,57,58,59,60,61,62]. Furthermore, treatment of HPP patients with a sclerostin antibody and the osteoporosis medication teriparatide have already been described, but these approaches primarily target bone mass and fracture healing rather than substituting or supporting TNAP activity [63,64,65,66,67,68,69].
3. TNAP as an Ectophosphatase—The Molecular Role of TNAP in Mineralization Processes
The prominent skeletal and dental phenotype of HPP patients indicates a central role of TNAP within the mineralization process, including the crystallization of HA as a central step. Interestingly, TNAP is an important promoter of the mineralization process as it dephosphorylates central mineralization inhibitors, like PPi and p-OPN [6,70]. Generally, TNAP’s function is to propagate HA crystallization, rather than to initiate this process within the matrix vesicles [71]. In general, HA crystallization is controlled by coordinated activity of enzymes and channels, which regulate the availability of PPi and ATP in the extracellular microenvironment for the production of Pi as an essential partner for Ca2+ to form HA. Here, among others, ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase family member 1), ANKH (progressive ankylosis protein homolog), PHOSPHO1 (phosphatase orphan 1), and PANX1 (pannexin 1) are central molecular players [70]. An increase of PPi prevalence in the extracellular space is mediated by its transport from the inside of the cell to the extracellular matrix (ECM) through the channel ANKH and additionally, via the enzymatic activity of ENPP1 [70]. The transmembrane channel PANX1 promotes the transport of ATP to the extracellular microenvironment, where the enzymes ENPP1 and TNAP can subsequently degrade it [70,72]. PHOSPHO1 dephosphorylates PPi to Pi inside matrix vesicles in order to promote mineralization, however, in a non-redundant manner to TNAP that is membrane-bound and acting extracellularly [71,73].
4. The Molecular Role of TNAP in Neuronal Biology and Neurotransmitter Metabolism
Beyond their often more prominently manifesting mineralization phenotype, HPP patients show neurological symptoms, including epileptic seizures, depression, anxiety, and sleeping disorders (in an increased prevalence compared to the general U.S. population) [74]. Epileptic seizures occur in severely affected children but not in adult patients with milder onset [74,75,76]. Those seizures are often pyridoxine-dependent and have been linked to TNAP´s biochemical function to indirectly provide the brain with PLP, which is an important co-factor for central enzymes of the neurotransmitter metabolism in the central nervous system, e.g., glutamate decarboxylase (GAD) and aromatic L-amino acid decarboxylase (AADC) [75,77,78]. This seems paradoxical at first glance, but HPP patients, who display increased concentrations of PLP within their blood serum, exhibit a lack of the same molecule within the brain, as only the dephosphorylated form (PL), which is provided by TNAP, is able to cross the blood–brain barrier (BBB), and is subsequently re-phosphorylated [75]. Going along with this, TNAP-deficient mouse brains show aberrant concentrations of several neurotransmitters, e.g., gamma-aminobutyric acid (GABA) and adenosine, as well as of molecules that are involved in the myelin synthesis, e.g., N-acetyl-aspartate and N-acetyl-aspartyl-glutamate [79]. Further, evidence was provided for TNAP’s expression in neuronal cells, for its impact on the PLP-dependent synthesis of serotonin and catecholamines, and for its influence on the dephosphorylation of laminin [80].
Besides neurotransmission, TNAP has an influence on myelination and is exclusively expressed in myelin-free areas of the axon, which has already been described in mice and in primates [81,82]. Furthermore, the enzyme has an impact on the development and the functionality of synapses [81,82,83]. An additional observation is that the length of neurites is influenced by TNAP activity, as the purinergic receptor P2X7, which mediates an inhibitory effect on the axonal outgrowth and cannot be activated when TNAP degrades its ligand ATP to adenosine in a stepwise manner [84,85]. Furthermore, Akp2−/− mice (TNAP knockout mice) develop enhanced proliferation of neuronal precursor cells as well as changes concerning morphology and activity of neurons due to a downregulation of P2X7 receptor expression [86]. Interestingly, PLP is an antagonist of the P2X7 receptor, which provides a second option for TNAP to influence the activity of the receptor [87]. Taking a closer look at TNAP’s impact on purinergic signaling, effects on sleeping, epileptic seizures, and the perception of pain become apparent [8,86,88,89]. Both sleeping disorders and pain indeed occur in HPP patients [40,74].
TNAP’s molecular role in stem cells has been controversially discussed in the literature, which can eventually be reconciled by different roles on different levels of commitment. TNAP knockdown increases the proliferation of neuronal precursor cells in mice [86] and decreases the proliferation (and differentiation) of murine neural stem cells in vitro [90]. A case report describes that TNAP deficiency, due to two compound heterozygous null mutations in the ALPL gene, can result in severe and progressive encephalopathy with a fatal onset of the disease [21].
5. The Molecular Role of TNAP in Inflammation Processes
TNAP activity has recently been linked to new scientific fields, including inflammation and metabolism. In HPP, as a disease model for TNAP deficiency, inflammatory conditions, such as bone marrow edema/chronic non-bacterial osteomyelitis, myopathies, tendinitis, and increased predisposition for periodontitis, are very common in both children and adults [27,31,36,91]. Provided that TNAP deficiency contributes to inflammatory reactions, e.g., in bone and muscle in HPP patients, research groups from different fields teamed up to unravel the molecular mechanisms of TNAP activity that modulate inflammatory processes. Both the ectophosphatase- and the ectonucleotidase-related functions contribute to the initiation and modulation of inflammation in variable tissues and conditions. Moreover, either accumulation of substrates as well as delivery of products can be involved in pro- and anti-inflammatory scenarios. Especially, TNAP’s role concerning the balance between pro-inflammatory ATP effects and anti-inflammatory effects of its breakdown product adenosine has recently received attention (see Figure 1 and Figure 2); hence, chemical agonists and antagonists have been developed and are currently being studied in clinical trials [92,93,94,95,96,97].
Impaired TNAP ectophosphatase activity causes PPi accumulation, which can initiate the generation of calcium pyrophosphate dehydrate (CPPD) crystals. Crystals accumulate in the joints and tissues of HPP patients and consequently initiate inflammation processes [98]. Those crystals stimulate transmembrane scavenger receptors and/or are phagocytosed, which triggers both the NLRP3 inflammasome and a process called necroinflammation [99]. The latter is a process of inflammation-related regulated necrosis, which can either occur in the phagocytosing cell (e.g., primarily macrophages) or in any neighboring cells via danger-associated molecular pattern (DAMP) signaling that activates the innate immune reaction [99]. Osteopontin, a protein substrate of TNAP, is not only involved in mineralization but also in a broad variety of processes, above all in inflammation, cytokine secretion and consecutive insulin resistance, and even inflammation-related tumorigenesis [100,101,102]. It is generally assigned as a putatively “proinflammatory protein”, however, both pro- and anti-inflammatory attitudes have been described and the role of phosphorylation and dephosphorylation has rarely been addressed in this context (see Figure 2). Recent work describes that recombinant osteopontin mediates anti-inflammatory effects in brain microglia [103]. Other reports render it a candidate orchestrator of chronic inflammatory processes in almost any tissue and organ, among others, in muscle, joints, kidney, and liver [104,105]. More research is definitely needed to dissect whether TNAP-related dephosphorylation balances anti-inflammatory vs. pro-inflammatory effects of osteopontin and to clarify its impact on inflammation.
TNAP’s ectophosphatase activity is also involved in the modulation of TLR ligands, such as the double-stranded RNA mimic poly-inosine:cytosine (pI:C, TLR3 agonist) and microbial LPS (ligand of TLR4) [7]. Ligand dephosphorylation mitigates TLR-related stimulation of the inflammasome and cytokine secretion. Such effects appear to be clinically meaningful, e.g., in preterm neonates with late-onset sepsis [7,106]. The impact of TLR ligand processing on chronic inflammatory processes that trigger aging and related degenerative diseases, including Alzheimer’s disease, remains to be unraveled in more detail [107,108]. The role of TNAP in inflammatory processes in the intestine, especially in chronic inflammatory intestinal conditions like colitis, is of putative relevance, but the molecular mechanisms involved are largely unknown. In preclinical models of colitis, TNAP is characterized as a modulator of T-cell function [109]. However, often, the overlapping roles of TNAP and its intestinal isoenzyme IAP cannot clearly be separated in the intestine, although experiments in artificial systems document an unequivocal effect of TNAP on T-cell biology. In this context, another function of TNAP and its intestinal counterpart IAP has to be taken into account and this is dephosphorylation of microbial LPS, as already mentioned above (see Figure 2) [7]. This function is an ancient one that appears to be evolutionarily conserved in the phosphatase family and is important for the interface with the intestinal microbiome. As such, APs may have an anti-inflammatory effect by dephosphorylating the lipid A moiety of LPS, that is important for binding to the TLR receptor 4/MD-2 innate immune receptor complex and downstream pro-inflammatory signaling. Both IAP and TNAP are capable of catalyzing this reaction but their respective allotment in this performance in health and disease is hard to dissect in humans and mice [7,110].
TNAP’s ectonucleotidase activity controls the level of inflammation by converting the pro-inflammatory ATP to the anti-inflammatory nucleotide adenosine (see Figure 1 and Figure 2). High expression or overexpression of TNAP have documented anti-inflammatory effects via ATP degradation and production of adenosine, however, as an unwanted side effect, dystopic calcification can be stimulated, which can again trigger and support inflammatory calcifying conditions, such as calcifying tendinitis or inflammatory atherosclerotic plaques [111]. Generally, the balance between P1 and P2 receptor signaling is key for the net outcome of the level of inflammatory activity (see Figure 1).
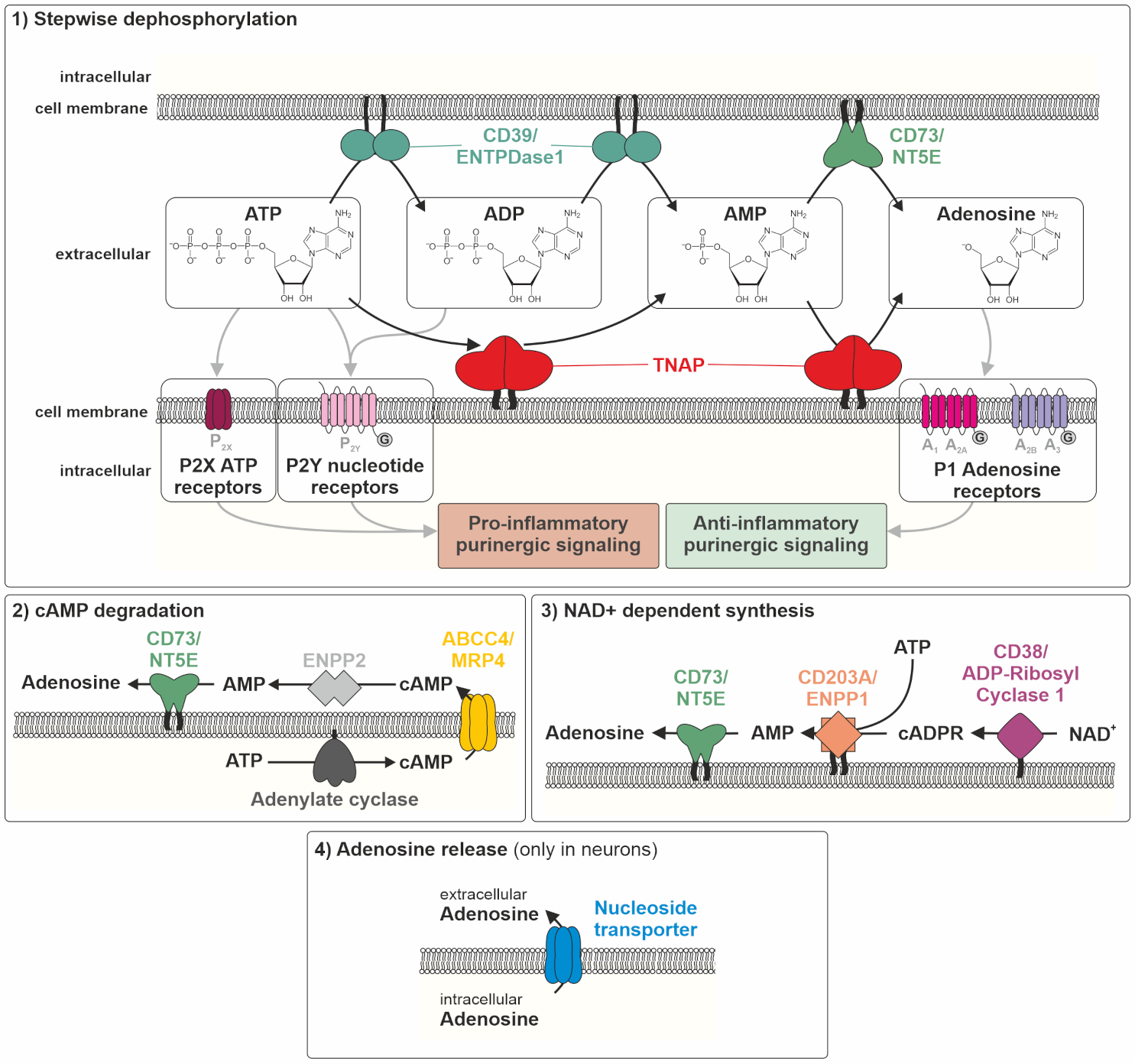
Thus, talking about adenosine delivery by nucleotidases, one has to take into account that besides TNAP, there are several efficient membrane-associated nucleotidases contributing to the adenosine pool. At least four different sources contribute to the adenosine pool, (1) ATP dephosphorylation, (2) cAMP degradation, (3) nicotinamide adenine dinucleotide (NAD+)-dependent synthesis of cADPR (cyclic adenosine diphosphate ribose) and its degradation, and (4) adenosine release from the intracellular pool to the extracellular space via nucleoside transporters (only in neurons, see Figure 1).
Apart from TNAP, the enzymes ectonucleotide pyrophosphatases/phosphodiesterases 1–3 (CD203A–C, ENPP1–3), ectonucleoside triphosphate diphosphohydrolase 1 (CD39, ENTPDase1), and ecto-5′-nucleotidase (CD73, NT5E) are involved in the pre-receptor metabolism of nucleotides [112,113]. The stepwise dephosphorylation of ATP towards adenosine is mediated by TNAP, ENPP1, CD39, and CD73 (Figure 1) [112,113]. The second contribution to the adenosine pool comes from adenylate cyclase (AC) activation and the generation of intracellular cyclic AMP (cAMP), cAMP efflux through multidrug resistance protein 4 (ABCC4, MRP4), and the hydrolysis of released cAMP to AMP by ENPP2, followed by dephosphorylation to adenosine again by CD73 and putatively, TNAP [114]. The third contribution is based on a synthesis pathway, where cADPR, generated by the enzyme ecto-NAD-glycohydrolase (CD38, ADP-ribosyl cyclase 1) via NAD+, can be converted to AMP (by ENPP1) to then be dephosphorylated to adenosine by CD73 and again, putatively, TNAP [112,113]. Exclusively in the nervous system, adenosine can be released via nucleoside transporters into the extracellular space [115]. This, again with the exception of the neuronal system, renders CD73 a gatekeeper of adenosine generation. The question is, how much and under which conditions or tissue context does TNAP substantially contribute to this gatekeeper function (see Figure 1). In vitro enzymatic assays provide unequivocal evidence for efficient TNAP activity in the purinergic metabolic cascade from ATP to adenosine at any step [112,113]. However, its impact on the cellular microenvironment and finally on the in vivo situation in tissues may still be different and may vary between homeostatic and challenge situations [112,113].
Given its role as a dominant gatekeeper of the anti-inflammatory adenosine signaling system, CD73 is anti-nociceptive and protects against inflammatory damage, while also contributing to age-dependent decline in cortical plasticity [116]. CD73 preserves barrier function in multiple tissues, a role that is most evident in the respiratory system [116]. While the expression of CD73 on endothelia is documented across species, the expression of CD73 on immune cells is species-specific [117].
Adenosine downstream signaling is mediated via a set of P1 adenosine receptors (A1R, A2AR, A2BR, and A3R), which initiate G protein-coupled signaling and modulation of AC activity [97,118]. Adenosine receptors modulate cAMP production with differential effects, as A1R and A3R inhibit, whereas A2AR and A2BR enhance, cAMP production [95,119]. However, phosphorylation cascades downstream cAMP production may again converge into ERK1/2 activation and this may be stimulatory with all four receptors [95]. The biological effects of all receptors, with respect to cytokine release and inflammation, are uniformly anti-inflammatory, as it has been reported for the intestinal effects of adenosine signaling. The dark side of these effects, if chronically sustained, may be the support of immunosuppression and eventually the susceptibility to develop cancer [95]. Adenosine receptors are expressed in cells of the bone marrow and show effects on osteo- and adipogenesis [119]. Similar effects have been reported in other organs, such as liver and kidney [120,121]. Additionally, adenosine directly regulates cells of the immune system, such as myeloid cells [122].
Addressing the question whether TNAP contributes to the modulation of pro- vs. anti-inflammatory signaling via pre-receptor metabolism of ligands for P1 or P2 receptors under physiological or challenge conditions is a matter of partially controversial discussions. Some evidence that CD73 activity is sufficient to provide adequate adenosine levels comes from research addressing the ACDC syndrome (arterial calcification due to deficiency of CD73) [123]. This autosomal recessive disease is caused by loss-of-function mutations in the CD73 gene and manifests as severe early atherosclerosis and calcifications in affected patients [123]. As a compensation for the prevalent CD73 deficit, TNAP was upregulated and produced adenosine simultaneously, supporting the calcification effects in mesenchymal stem cells from patients, but not from healthy controls [123]. In CD73 knockout mice, TNAP is upregulated and can compensate for the deficiency in adenosine production, although with an obvious difference between male vs. female animals [124].
In summary, TNAP can exert influence on inflammatory processes in variable ways, which are schematically depicted in Figure 2.
6. Conclusions
TNAP is an ancient, multi-systemically active enzyme with a broad number of substrates. Its activity is mainly composed of general ectophosphatase and specific ectonucleotidase components, where the latter, of course, also comprise dephosphorylation steps. It is involved in networks of enzymatic activities in physiology and pathology. Under certain conditions, TNAP function seems to be more or less redundantly backed up by enzymes with similar functions, but in case of bone mineralization, it appears to be one of two indispensable enzymes (TNAP and PHOSPHO1). Hence, the model disease Hypophosphatasia shows primarily a skeletal phenotype. This may have led to an unbalanced view on this enzyme in the past. More recently, the impact of TNAP on inflammatory activity is more and more apparent on multiple levels. TNAP and its substrates modulate a series of chronic inflammatory conditions, including aging-related degenerative diseases, such as atherosclerosis and Alzheimer’s disease, but also metabolic conditions, like insulin resistance and type 2 diabetes, occurring in the wake of chronic inflammation. Thus, based on the knowledge that has been accumulated about the role of TNAP in bone, it appears timely to widen the focus towards inflammation and metabolism in order to finally evaluate the therapeutic potential of agonists and antagonists in these complex systems.
Author Contributions
Conceptualization, methodology, validation, formal analysis, investigation, writing: original draft preparation, and writing: review and editing: S.G., D.L., and F.J.; visualization: D.L.; supervision: S.G. and F.J. All authors have read and agreed to the published version of the manuscript.
Funding
S.G. and D.L. are funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project Number: 397519724, and financially supported by the German Hypophosphatasia patients’ organization “Hypophosphatasie Deutschland e.V.”. This publication was supported by the Open Access Publication Fund of the University of Wuerzburg.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We further thank Christine Hofmann for scientific discussions and Eva Klopocki for scientific discussions and proofreading of the manuscript.
Conflicts of Interest
F.J. received honoraria for lectures and advice from Alexion Pharma. Apart from this, no competing interests need to be declared.
References
- Millan, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millan, J.L. Alkaline Phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H. The human alkaline phosphatases: What we know and what we don’t know. Clin. Chim. Acta 1990, 186, 133–150. [Google Scholar] [CrossRef]
- Simao, A.M.; Bolean, M.; Hoylaerts, M.F.; Millan, J.L.; Ciancaglini, P. Effects of pH on the production of phosphate and pyrophosphate by matrix vesicles’ biomimetics. Calcif. Tissue Int. 2013, 93, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedde, K.N.; Whyte, M.P. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5′-phosphate ectophosphatase: Normal and hypophosphatasia fibroblast study. Am. J. Hum. Genet. 1990, 47, 767–775. [Google Scholar] [PubMed]
- Narisawa, S.; Yadav, M.C.; Millan, J.L. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Pettengill, M.; Matute, J.D.; Tresenriter, M.; Hibbert, J.; Burgner, D.; Richmond, P.; Millan, J.L.; Ozonoff, A.; Strunk, T.; Currie, A.; et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS ONE 2017, 12, e0175936. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- Le Du, M.H.; Stigbrand, T.; Taussig, M.J.; Menez, A.; Stura, E.A. Crystal structure of alkaline phosphatase from human placenta at 1.8 A resolution. Implication for a substrate specificity. J. Biol. Chem. 2001, 276, 9158–9165. [Google Scholar] [CrossRef] [Green Version]
- Hoylaerts, M.F.; Van Kerckhoven, S.; Kiffer-Moreira, T.; Sheen, C.; Narisawa, S.; Millan, J.L. Functional significance of calcium binding to tissue-nonspecific alkaline phosphatase. PLoS ONE 2015, 10, e0119874. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Martins, L.; Rodrigues, T.L.; Ribeiro, M.M.; Saito, M.T.; Giorgetti, A.P.; Casati, M.Z.; Sallum, E.A.; Foster, B.L.; Somerman, M.J.; Nociti, F.H., Jr. Novel ALPL genetic alteration associated with an odontohypophosphatasia phenotype. Bone 2013, 56, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Du, M.H.; Millan, J.L. Structural evidence of functional divergence in human alkaline phosphatases. J. Biol. Chem. 2002, 277, 49808–49814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauvert, D.; Brun-Heath, I.; Lia-Baldini, A.S.; Bellazi, L.; Taillandier, A.; Serre, J.L.; de Mazancourt, P.; Mornet, E. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med. Genet. 2009, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Muller, H.L.; Yamazaki, M.; Michigami, T.; Kageyama, T.; Schonau, E.; Schneider, P.; Ozono, K. Asp361Val Mutant of alkaline phosphatase found in patients with dominantly inherited hypophosphatasia inhibits the activity of the wild-type enzyme. J. Clin. Endocrinol. Metab. 2000, 85, 743–747. [Google Scholar] [CrossRef]
- Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun, E.; Lavaud, N.; Wallon, F.; Rousseau, N.; Charle, C.; Guberto, M.; et al. Hypophosphatasia: A genetic-based nosology and new insights in genotype-phenotype correlation. Eur. J. Hum. Genet. 2020, 1–11. [Google Scholar] [CrossRef]
- Lia-Baldini, A.S.; Brun-Heath, I.; Carrion, C.; Simon-Bouy, B.; Serre, J.L.; Nunes, M.E.; Mornet, E. A new mechanism of dominance in hypophosphatasia: The mutated protein can disturb the cell localization of the wild-type protein. Hum. Genet. 2008, 123, 429–432. [Google Scholar] [CrossRef]
- Weiss, M.J.; Henthorn, P.S.; Lafferty, M.A.; Slaughter, C.; Raducha, M.; Harris, H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 7182–7186. [Google Scholar] [CrossRef] [Green Version]
- Bhadada, S.K.; Pal, R.; Dhiman, V.; Alonso, N.; Ralston, S.H.; Kaur, S.; Gupta, R. Adult hypophosphatasia with a novel ALPL mutation: Report of an Indian kindred. Bone Rep. 2020, 12, 100247. [Google Scholar] [CrossRef]
- Michigami, T.; Tachikawa, K.; Yamazaki, M.; Kawai, M.; Kubota, T.; Ozono, K. Hypophosphatasia in Japan: ALPL Mutation Analysis in 98 Unrelated Patients. Calcif. Tissue Int. 2020, 106, 221–231. [Google Scholar] [CrossRef]
- Hofmann, C.; Liese, J.; Schwarz, T.; Kunzmann, S.; Wirbelauer, J.; Nowak, J.; Hamann, J.; Girschick, H.; Graser, S.; Dietz, K.; et al. Compound heterozygosity of two functional null mutations in the ALPL gene associated with deleterious neurological outcome in an infant with hypophosphatasia. Bone 2013, 55, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Girschick, H.; Mornet, E.; Schneider, D.; Jakob, F.; Mentrup, B. Unexpected high intrafamilial phenotypic variability observed in hypophosphatasia. Eur. J. Hum. Genet. 2014, 22, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Silvent, J.; Gasse, B.; Mornet, E.; Sire, J.Y. Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J. Biol. Chem. 2014, 289, 24168–24179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mornet, E. Molecular Genetics of Hypophosphatasia and Phenotype-Genotype Correlations. Subcell Biochem. 2015, 76, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Huggins, E.; Ong, R.; Rockman-Greenberg, C.; Flueckinger, L.B.; Dahir, K.M.; Kishnani, P.S. Multigenerational case examples of hypophosphatasia: Challenges in genetic counseling and disease management. Mol. Genet. Metab. Rep. 2020, 25, 100661. [Google Scholar] [CrossRef]
- Rathbun, J.C. Hypophosphatasia; A new developmental anomaly. Am. J. Dis. Child. 1948, 75, 822–831. [Google Scholar] [CrossRef]
- Hofmann, C.; Girschick, H.J.; Mentrup, B.; Graser, S.; Seefried, L.; Liese, J.; Jakob, F. Clinical Aspects of Hypophosphatasia: An Update. Clinic Rev. Bone Miner. Metab. 2013, 11, 60–70. [Google Scholar] [CrossRef]
- Mornet, E.; Yvard, A.; Taillandier, A.; Fauvert, D.; Simon-Bouy, B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann. Hum. Genet. 2011, 75, 439–445. [Google Scholar] [CrossRef]
- Fukushi-Irie, M.; Ito, M.; Amaya, Y.; Amizuka, N.; Ozawa, H.; Omura, S.; Ikehara, Y.; Oda, K. Possible interference between tissue-non-specific alkaline phosphatase with an Arg54-->Cys substitution and acounterpart with an Asp277-->Ala substitution found in a compound heterozygote associated with severe hypophosphatasia. Biochem. J. 2000, 348 Pt 3, 633–642. [Google Scholar] [CrossRef]
- Vogt, M.; Girschick, H.; Schweitzer, T.; Benoit, C.; Holl-Wieden, A.; Seefried, L.; Jakob, F.; Hofmann, C. Pediatric hypophosphatasia: Lessons learned from a retrospective single-center chart review of 50 children. Orphanet J. Rare Dis. 2020, 15, 212. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E.; Hofmann, C.; Bloch-Zupan, A.; Girschick, H.; Le Merrer, M. Clinical utility gene card for: Hypophosphatasia—Update 2013. Eur. J. Hum. Genet. 2014, 22. [Google Scholar] [CrossRef] [PubMed]
- Eberle, F.; Hartenfels, S.; Pralle, H.; Kabisch, A. Adult hypophosphatasia without apparent skeletal disease: “odontohypophosphatasia” in four heterozygote members of a family. Klin. Wochenschr. 1984, 62, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Vaysse, F. Hypophosphatasia: Oral cavity and dental disorders. Arch. Pediatr. 2017, 24, 5S80–5S84. [Google Scholar] [CrossRef]
- Reibel, A.; Maniere, M.C.; Clauss, F.; Droz, D.; Alembik, Y.; Mornet, E.; Bloch-Zupan, A. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J. Rare Dis. 2009, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.L.; Bishop, N.J.; Guanabens, N.; Hofmann, C.; Jakob, F.; Roux, C.; Zillikens, M.C.; Rare Bone Disease Action Group of the European Calcified Tissue Society. Hypophosphatasia in adolescents and adults: Overview of diagnosis and treatment. Osteoporos. Int. 2020, 31, 1445–1460. [Google Scholar] [CrossRef]
- Lopez-Delgado, L.; Riancho-Zarrabeitia, L.; Garcia-Unzueta, M.T.; Tenorio, J.A.; Garcia-Hoyos, M.; Lapunzina, P.; Valero, C.; Riancho, J.A. Abnormal bone turnover in individuals with low serum alkaline phosphatase. Osteoporos. Int. 2018, 29, 2147–2150. [Google Scholar] [CrossRef] [Green Version]
- Berkseth, K.E.; Tebben, P.J.; Drake, M.T.; Hefferan, T.E.; Jewison, D.E.; Wermers, R.A. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone 2013, 54, 21–27. [Google Scholar] [CrossRef]
- Kramer, K.; Chavez, M.B.; Tran, A.T.; Farah, F.; Tan, M.H.; Kolli, T.N.; Dos Santos, E.J.L.; Wimer, H.F.; Millan, J.L.; Suva, L.J.; et al. Dental defects in the primary dentition associated with hypophosphatasia from biallelic ALPL mutations. Bone 2020, 143, 115732. [Google Scholar] [CrossRef]
- Seefried, L.; Dahir, K.; Petryk, A.; Hogler, W.; Linglart, A.; Martos-Moreno, G.A.; Ozono, K.; Fang, S.; Rockman-Greenberg, C.; Kishnani, P.S. Burden of Illness in Adults with Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J. Bone Miner. Res. 2020, 35, 2171–2178. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Rush, E.T.; Arundel, P.; Bishop, N.; Dahir, K.; Fraser, W.; Harmatz, P.; Linglart, A.; Munns, C.F.; Nunes, M.E.; et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol. Genet. Metab. 2017, 122, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; Mahuren, J.D.; Vrabel, L.A.; Coburn, S.P. Markedly increased circulating pyridoxal-5’-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J. Clin. Investig. 1985, 76, 752–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michigami, T.; Ohata, Y.; Fujiwara, M.; Mochizuki, H.; Adachi, M.; Kitaoka, T.; Kubota, T.; Sawai, H.; Namba, N.; Hasegawa, K.; et al. Clinical Practice Guidelines for Hypophosphatasia. Clin. Pediatr. Endocrinol. 2020, 29, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Offiah, A.C.; Vockley, J.; Munns, C.F.; Murotsuki, J. Differential diagnosis of perinatal hypophosphatasia: Radiologic perspectives. Pediatr. Radiol. 2019, 49, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Hogler, W.; Langman, C.; da Silva, H.G.; Fang, S.; Linglart, A.; Ozono, K.; Petryk, A.; Rockman-Greenberg, C.; Seefried, L.; Kishnani, P.S. Diagnostic delay is common among patients with hypophosphatasia: Initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord. 2019, 20, 80. [Google Scholar] [CrossRef] [Green Version]
- Girschick, H.J.; Seyberth, H.W.; Huppertz, H.I. Treatment of childhood hypophosphatasia with nonsteroidal antiinflammatory drugs. Bone 1999, 25, 603–607. [Google Scholar] [CrossRef]
- Girschick, H.J.; Schneider, P.; Haubitz, I.; Hiort, O.; Collmann, H.; Beer, M.; Shin, Y.S.; Seyberth, H.W. Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia. Orphanet J. Rare Dis. 2006, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Collmann, H.; Mornet, E.; Gattenlohner, S.; Beck, C.; Girschick, H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv. Syst. 2009, 25, 217–223. [Google Scholar] [CrossRef]
- Feeney, C.; Stanford, N.; Lee, S.; Barry, S. Hypophosphatasia and the importance of the general dental practitioner—A case series and discussion of upcoming treatments. Br. Dent. J. 2018, 224, 937–943. [Google Scholar] [CrossRef]
- Weber, T.J.; Sawyer, E.K.; Moseley, S.; Odrljin, T.; Kishnani, P.S. Burden of disease in adult patients with hypophosphatasia: Results from two patient-reported surveys. Metabolism 2016, 65, 1522–1530. [Google Scholar] [CrossRef]
- Genest, F.; Seefried, L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia—Not atypical at all. Osteoporos. Int. 2018, 29, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Klidaras, P.; Severt, J.; Aggers, D.; Payne, J.; Miller, P.D.; Ing, S.W. Fracture Healing in Two Adult Patients with Hypophosphatasia After Asfotase Alfa Therapy. JBMR Plus 2018, 2, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, C.; Seefried, L.; Jakob, F. Asfotase alfa: Enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today 2016, 52, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.E.; Harmatz, P.; Vockley, J.; Hogler, W.; Nakayama, H.; Bishop, N.; Martos-Moreno, G.A.; Moseley, S.; Fujita, K.P.; Liese, J.; et al. Efficacy and Safety of Asfotase Alfa in Infants and Young Children With Hypophosphatasia: A Phase 2 Open-Label Study. J. Clin. Endocrinol. Metab. 2019, 104, 2735–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; Rockman-Greenberg, C.; Ozono, K.; Riese, R.; Moseley, S.; Melian, A.; Thompson, D.D.; Bishop, N.; Hofmann, C. Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J. Clin. Endocrinol. Metab. 2016, 101, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.C.; de Oliveira, R.C.; Foster, B.L.; Fong, H.; Cory, E.; Narisawa, S.; Sah, R.L.; Somerman, M.; Whyte, M.P.; Millan, J.L. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J. Bone Miner. Res. 2012, 27, 1722–1734. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P.; Simmons, J.H.; Moseley, S.; Fujita, K.P.; Bishop, N.; Salman, N.J.; Taylor, J.; Phillips, D.; McGinn, M.; McAlister, W.H. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes Endocrinol. 2019, 7, 93–105. [Google Scholar] [CrossRef]
- Seefried, L.; Kishnani, P.S.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P.; Dahir, K.M. Pharmacodynamics of asfotase alfa in adults with pediatric-onset hypophosphatasia. Bone 2020, 142, 115664. [Google Scholar] [CrossRef]
- Magdaleno, A.L.; Singh, S.; Venkataraman, S.; Perilli, G.A.; Lee, Y.Y. Adult-Onset Hypophosphatasia: Before and after Treatment with Asfotase Alfa. AACE Clin. Case Rep. 2019, 5, e344–e348. [Google Scholar] [CrossRef]
- Remde, H.; Cooper, M.S.; Quinkler, M. Successful Asfotase Alfa Treatment in an Adult Dialysis Patient with Childhood-Onset Hypophosphatasia. J. Endocr. Soc. 2017, 1, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- Bowden, S.A.; Foster, B.L. Profile of asfotase alfa in the treatment of hypophosphatasia: Design, development, and place in therapy. Drug Des. Devel. Ther. 2018, 12, 3147–3161. [Google Scholar] [CrossRef] [Green Version]
- Seefried, L.; Baumann, J.; Hemsley, S.; Hofmann, C.; Kunstmann, E.; Kiese, B.; Huang, Y.; Chivers, S.; Valentin, M.A.; Borah, B.; et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J. Clin. Investig. 2017, 127, 2148–2158. [Google Scholar] [CrossRef] [Green Version]
- Camacho, P.M.; Painter, S.; Kadanoff, R. Treatment of adult hypophosphatasia with teriparatide. Endocr. Pract. 2008, 14, 204–208. [Google Scholar] [CrossRef]
- Doshi, K.B.; Hamrahian, A.H.; Licata, A.A. Teriparatide treatment in adult hypophosphatasia in a patient exposed to bisphosphonate: A case report. Clin. Cases Miner. Bone Metab. 2009, 6, 266–269. [Google Scholar]
- Laroche, M. Failure of teriparatide in treatment of bone complications of adult hypophosphatasia. Calcif. Tissue Int. 2012, 90, 250. [Google Scholar] [CrossRef] [Green Version]
- Righetti, M.; Wach, J.; Desmarchelier, R.; Coury, F. Teriparatide treatment in an adult patient with hypophosphatasia exposed to bisphosphonate and revealed by bilateral atypical fractures. Jt. Bone Spine 2018, 85, 365–367. [Google Scholar] [CrossRef]
- Schmidt, T.; Rolvien, T.; Linke, C.; Jandl, N.M.; Oheim, R.; Amling, M.; Barvencik, F. Outcome of Teriparatide Treatment on Fracture Healing Complications and Symptomatic Bone Marrow Edema in Four Adult Patients with Hypophosphatasia. JBMR Plus 2019, 3, e10215. [Google Scholar] [CrossRef] [Green Version]
- Schalin-Jantti, C.; Mornet, E.; Lamminen, A.; Valimaki, M.J. Parathyroid hormone treatment improves pain and fracture healing in adult hypophosphatasia. J. Clin. Endocrinol. Metab. 2010, 95, 5174–5179. [Google Scholar] [CrossRef]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millan, J.L. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: An integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Millan, J.L. The role of phosphatases in the initiation of skeletal mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Sebastian-Serrano, A.; de Diego-Garcia, L.; Henshall, D.C.; Engel, T.; Diaz-Hernandez, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Huesa, C.; Houston, D.; Kiffer-Moreira, T.; Yadav, M.M.; Millan, J.L.; Farquharson, C. The Functional co-operativity of Tissue-Nonspecific Alkaline Phosphatase (TNAP) and PHOSPHO1 during initiation of Skeletal Mineralization. Biochem. Biophys. Rep. 2015, 4, 196–201. [Google Scholar] [CrossRef] [Green Version]
- Colazo, J.M.; Hu, J.R.; Dahir, K.M.; Simmons, J.H. Neurological symptoms in Hypophosphatasia. Osteoporos. Int. 2019, 30, 469–480. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Bowling, F.; Carpenter, K.; Earl, J.; Chaitow, J.; Pitt, J.; Mornet, E.; Sillence, D.; Ellaway, C. Perinatal hypophosphatasia presenting as neonatal epileptic encephalopathy with abnormal neurotransmitter metabolism secondary to reduced co-factor pyridoxal-5′-phosphate availability. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S25–S33. [Google Scholar] [CrossRef]
- Nunes, M.L.; Mugnol, F.; Bica, I.; Fiori, R.M. Pyridoxine-dependent seizures associated with hypophosphatasia in a newborn. J. Child. Neurol. 2002, 17, 222–224. [Google Scholar] [CrossRef]
- Guzel Nur, B.; Celmeli, G.; Manguoglu, E.; Soyucen, E.; Bircan, I.; Mihci, E. Pyridoxine-Responsive Seizures in Infantile Hypophosphatasia and a Novel Homozygous Mutation in ALPL Gene. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 360–364. [Google Scholar] [CrossRef]
- Demirbilek, H.; Alanay, Y.; Alikasifoglu, A.; Topcu, M.; Mornet, E.; Gonc, N.; Ozon, A.; Kandemir, N. Hypophosphatasia presenting with pyridoxine-responsive seizures, hypercalcemia, and pseudotumor cerebri: Case report. J. Clin. Res. Pediatr. Endocrinol. 2012, 4, 34–38. [Google Scholar] [CrossRef]
- Cruz, T.; Gleizes, M.; Balayssac, S.; Mornet, E.; Marsal, G.; Millan, J.L.; Malet-Martino, M.; Nowak, L.G.; Gilard, V.; Fonta, C. Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis. J. Neurochem. 2017, 140, 919–940. [Google Scholar] [CrossRef] [Green Version]
- Ermonval, M.; Baudry, A.; Baychelier, F.; Pradines, E.; Pietri, M.; Oda, K.; Schneider, B.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O. The cellular prion protein interacts with the tissue non-specific alkaline phosphatase in membrane microdomains of bioaminergic neuronal cells. PLoS ONE 2009, 4, e6497. [Google Scholar] [CrossRef] [Green Version]
- Fonta, C.; Negyessy, L.; Renaud, L.; Barone, P. Postnatal development of alkaline phosphatase activity correlates with the maturation of neurotransmission in the cerebral cortex. J. Comp. Neurol. 2005, 486, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Hanics, J.; Barna, J.; Xiao, J.; Millan, J.L.; Fonta, C.; Negyessy, L. Ablation of TNAP function compromises myelination and synaptogenesis in the mouse brain. Cell Tissue Res. 2012, 349, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonta, C.; Negyessy, L.; Renaud, L.; Barone, P. Areal and subcellular localization of the ubiquitous alkaline phosphatase in the primate cerebral cortex: Evidence for a role in neurotransmission. Cereb. Cortex 2004, 14, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Zaera, M.; Diaz-Hernandez, J.I.; Hernandez-Alvarez, E.; Zimmermann, H.; Diaz-Hernandez, M.; Miras-Portugal, M.T. Tissue-nonspecific alkaline phosphatase promotes axonal growth of hippocampal neurons. Mol. Biol. Cell 2011, 22, 1014–1024. [Google Scholar] [CrossRef]
- Graser, S.; Mentrup, B.; Schneider, D.; Klein-Hitpass, L.; Jakob, F.; Hofmann, C. Overexpression of tissue-nonspecific alkaline phosphatase increases the expression of neurogenic differentiation markers in the human SH-SY5Y neuroblastoma cell line. Bone 2015, 79, 150–161. [Google Scholar] [CrossRef]
- Sebastian-Serrano, A.; Engel, T.; de Diego-Garcia, L.; Olivos-Ore, L.A.; Arribas-Blazquez, M.; Martinez-Frailes, C.; Perez-Diaz, C.; Millan, J.L.; Artalejo, A.R.; Miras-Portugal, M.T.; et al. Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum. Mol. Genet. 2016, 25, 4143–4156. [Google Scholar] [CrossRef] [Green Version]
- Theriault, O.; Poulin, H.; Thomas, G.R.; Friesen, A.D.; Al-Shaqha, W.A.; Chahine, M. Pyridoxal-5′-phosphate (MC-1), a vitamin B6 derivative, inhibits expressed P2X receptors. Can. J. Physiol. Pharmacol. 2014, 92, 189–196. [Google Scholar] [CrossRef]
- Street, S.E.; Sowa, N.A. TNAP and Pain Control. Subcell Biochem. 2015, 76, 283–305. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic receptors and pain. Curr. Pharm. Des. 2009, 15, 1717–1735. [Google Scholar] [CrossRef] [Green Version]
- Kermer, V.; Ritter, M.; Albuquerque, B.; Leib, C.; Stanke, M.; Zimmermann, H. Knockdown of tissue nonspecific alkaline phosphatase impairs neural stem cell proliferation and differentiation. Neurosci. Lett. 2010, 485, 208–211. [Google Scholar] [CrossRef]
- Hedrich, C.M.; Morbach, H.; Reiser, C.; Girschick, H.J. New Insights into Adult and Paediatric Chronic Non-bacterial Osteomyelitis CNO. Curr. Rheumatol. Rep. 2020, 22, 52. [Google Scholar] [CrossRef]
- Bessueille, L.; Briolay, A.; Como, J.; Mebarek, S.; Mansouri, C.; Gleizes, M.; El Jamal, A.; Buchet, R.; Dumontet, C.; Matera, E.L.; et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 2020, 133, 115262. [Google Scholar] [CrossRef]
- Jacobson, K.A.; AP, I.J.; Muller, C.E. Medicinal chemistry of P2 and adenosine receptors: Common scaffolds adapted for multiple targets. Biochem. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Muller, C.E.; Baqi, Y.; Namasivayam, V. Agonists and Antagonists for Purinergic Receptors. Methods Mol. Biol. 2020, 2041, 45–64. [Google Scholar] [CrossRef]
- D’Antongiovanni, V.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Blandizzi, C.; Antonioli, L. The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia. Int. J. Mol. Sci. 2020, 21, 5089. [Google Scholar] [CrossRef]
- Gratal, P.; Lamuedra, A.; Medina, J.P.; Bermejo-Alvarez, I.; Largo, R.; Herrero-Beaumont, G.; Mediero, A. Purinergic System Signaling in Metainflammation-Associated Osteoarthritis. Front. Med. 2020, 7, 506. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Beck, C.; Morbach, H.; Richl, P.; Stenzel, M.; Girschick, H.J. How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol. Int. 2009, 29, 229–238. [Google Scholar] [CrossRef]
- Mulay, S.R.; Steiger, S.; Shi, C.; Anders, H.J. A guide to crystal-related and nano- or microparticle-related tissue responses. FEBS J. 2020, 287, 818–832. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef] [PubMed]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Zhang, X.; Shu, Q.; Liu, Z.; Gao, C.; Wang, Z.; Xing, Z.; Song, J. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. 2020, 1751, 147170. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Jeong, S.; Xia, Q.; Kong, X. Role of Osteopontin in Liver Diseases. Int. J. Biol. Sci. 2016, 12, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Iida, T.; Wagatsuma, K.; Hirayama, D.; Nakase, H. Is Osteopontin a Friend or Foe of Cell Apoptosis in Inflammatory Gastrointestinal and Liver Diseases? Int. J. Mol. Sci. 2017, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Ni, H.; Herington, J.; Reese, J.; Paria, B.C. Alkaline phosphatase protects lipopolysaccharide-induced early pregnancy defects in mice. PLoS ONE 2015, 10, e0123243. [Google Scholar] [CrossRef] [Green Version]
- Kellett, K.A.; Hooper, N.M. The Role of Tissue Non-specific Alkaline Phosphatase (TNAP) in Neurodegenerative Diseases: Alzheimer’s Disease in the Focus. Subcell Biochem. 2015, 76, 363–374. [Google Scholar] [CrossRef]
- Moskalev, A.; Stambler, I.; Caruso, C. Innate and Adaptive Immunity in Aging and Longevity: The Foundation of Resilience. Aging Dis. 2020, 11, 1363–1373. [Google Scholar] [CrossRef]
- Hernandez-Chirlaque, C.; Gamez-Belmonte, R.; Ocon, B.; Martinez-Moya, P.; Wirtz, S.; Sanchez de Medina, F.; Martinez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohns Colitis 2017, 11, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Rader, B.A. Alkaline Phosphatase, an Unconventional Immune Protein. Front. Immunol. 2017, 8, 897. [Google Scholar] [CrossRef] [Green Version]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification—A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, H. History of ectonucleotidases and their role in purinergic signaling. Biochem. Pharmacol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Pleli, T.; Mondorf, A.; Ferreiros, N.; Thomas, D.; Dvorak, K.; Biondi, R.M.; Heringdorf, D.M.Z.; Zeuzem, S.; Geisslinger, G.; Zimmermann, H.; et al. Activation of Adenylyl Cyclase Causes Stimulation of Adenosine Receptors. Cell Physiol. Biochem. 2018, 45, 2516–2528. [Google Scholar] [CrossRef]
- Yang, D.; Ding, C.; Qi, G.; Feldmeyer, D. Cholinergic and Adenosinergic Modulation of Synaptic Release. Neuroscience 2020, in press. [Google Scholar] [CrossRef]
- Minor, M.; Alcedo, K.P.; Battaglia, R.A.; Snider, N.T. Cell type- and tissue-specific functions of ecto-5′-nucleotidase (CD73). Am. J. Physiol. Cell Physiol. 2019, 317, C1079–C1092. [Google Scholar] [CrossRef]
- Schneider, E.; Rissiek, A.; Winzer, R.; Puig, B.; Rissiek, B.; Haag, F.; Mittrucker, H.W.; Magnus, T.; Tolosa, E. Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 2019, 10, 1729. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Eisenstein, A.; Chitalia, S.V.; Ravid, K. Bone Marrow and Adipose Tissue Adenosine Receptors Effect on Osteogenesis and Adipogenesis. Int. J. Mol. Sci. 2020, 21, 7470. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Kishore, B.K.; Robson, S.C. Conversion of extracellular ATP into adenosine: A master switch in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 509–524. [Google Scholar] [CrossRef]
- Wang, S.; Gao, S.; Zhou, D.; Qian, X.; Luan, J.; Lv, X. The role of the CD39-CD73-adenosine pathway in liver disease. J. Cell Physiol. 2021, 236, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C. Modulation of myeloid cells by adenosine signaling. Curr. Opin. Pharmacol. 2020, 53, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; St Hilaire, C.; Huang, Y.; Yang, D.; Dmitrieva, N.I.; Negro, A.; Schwartzbeck, R.; Liu, Y.; Yu, Z.; Walts, A.; et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal. 2016, 9, ra121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Copeland, J.; Shin, M.; Chang, Y.; Venton, B.J. CD73 or CD39 Deletion Reveals Different Mechanisms of Formation for Spontaneous and Mechanically Stimulated Adenosine and Sex Specific Compensations in ATP Degradation. ACS Chem. Neurosci. 2020, 11, 919–928. [Google Scholar] [CrossRef]
Figure 1.
Balance between pro- and anti-inflammatory signaling depending on purine metabolizing enzymes. ABCC4/MRP4: ATP binding cassette subfamily C member 4/multidrug resistance protein 4, NAD+: nicotinamide adenine dinucleotide, ATP/ADP/AMP: adenosine tri-/di-/monophosphate, cADPR: cyclic adenosine 5′-diphosphate ribose, cAMP: cyclic adenosine monophosphate, CD39/ENTPDase 1: ectonucleoside triphosphate diphosphohydrolase 1, CD73/NT5E: ecto-5’-nucleotidase, ENPP1/2: ectonucleotide pyrophosphatase/phosphodiesterase 1/2, TNAP: tissue-nonspecific alkaline phosphatase. Note: Adenosine release via nucleoside transporters, depicted in (4), is only prevalent in neurons.
Figure 1.
Balance between pro- and anti-inflammatory signaling depending on purine metabolizing enzymes. ABCC4/MRP4: ATP binding cassette subfamily C member 4/multidrug resistance protein 4, NAD+: nicotinamide adenine dinucleotide, ATP/ADP/AMP: adenosine tri-/di-/monophosphate, cADPR: cyclic adenosine 5′-diphosphate ribose, cAMP: cyclic adenosine monophosphate, CD39/ENTPDase 1: ectonucleoside triphosphate diphosphohydrolase 1, CD73/NT5E: ecto-5’-nucleotidase, ENPP1/2: ectonucleotide pyrophosphatase/phosphodiesterase 1/2, TNAP: tissue-nonspecific alkaline phosphatase. Note: Adenosine release via nucleoside transporters, depicted in (4), is only prevalent in neurons.

Figure 2.
TNAP’s influence on the regulation of inflammation processes. ATP: adenosine triphosphate, CPPD: calcium pyrophosphate dehydrate, dsRNA: double-stranded ribonucleic acid, LPS: lipopolysaccharide, OPN: osteopontin, poly(I:C): poly-inosine:cytosine, p-OPN: phosphorylated osteopontin, PPi: inorganic pyrophosphate, TLR3: toll-like receptor 3, TLR4: toll-like receptor 4, TNAP: tissue-nonspecific alkaline phosphatase. Note: All mechanisms depicted are solid in terms of data, except the role of p-OPN vs. OPN on anti-/pro-inflammatory-effects. Green: anti-inflammatory effects, red: pro-inflammatory effects.
Figure 2.
TNAP’s influence on the regulation of inflammation processes. ATP: adenosine triphosphate, CPPD: calcium pyrophosphate dehydrate, dsRNA: double-stranded ribonucleic acid, LPS: lipopolysaccharide, OPN: osteopontin, poly(I:C): poly-inosine:cytosine, p-OPN: phosphorylated osteopontin, PPi: inorganic pyrophosphate, TLR3: toll-like receptor 3, TLR4: toll-like receptor 4, TNAP: tissue-nonspecific alkaline phosphatase. Note: All mechanisms depicted are solid in terms of data, except the role of p-OPN vs. OPN on anti-/pro-inflammatory-effects. Green: anti-inflammatory effects, red: pro-inflammatory effects.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020919
AMA Style
Graser S, Liedtke D, Jakob F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. International Journal of Molecular Sciences. 2021; 22(2):919. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020919
Chicago/Turabian StyleGraser, Stephanie, Daniel Liedtke, and Franz Jakob. 2021. "TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism" International Journal of Molecular Sciences 22, no. 2: 919. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020919
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.