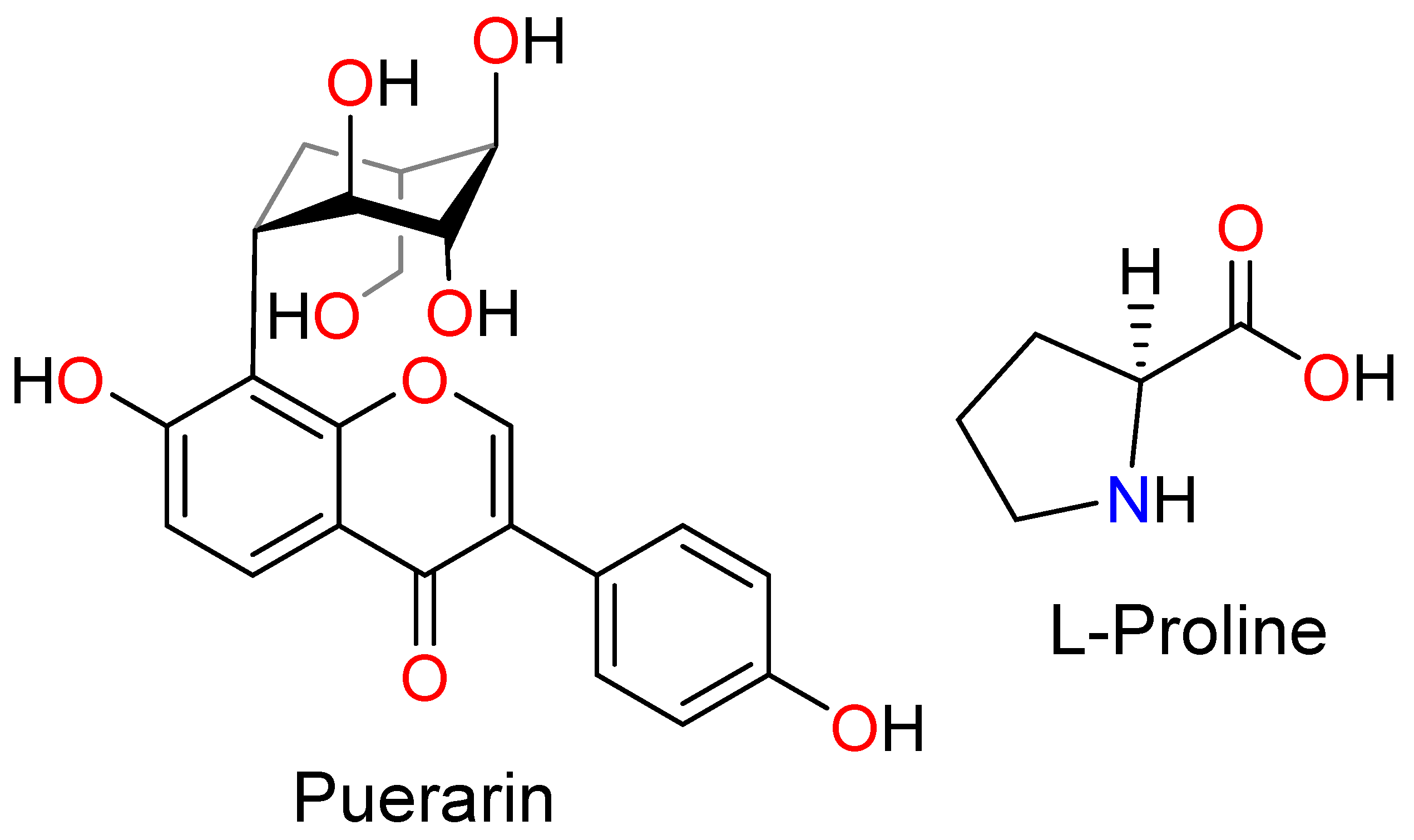
Enhancing the Physiochemical Properties of Puerarin via L-Proline Co-Crystallization: Synthesis, Characterization, and Dissolution Studies of Two Phases of Pharmaceutical Co-Crystals

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
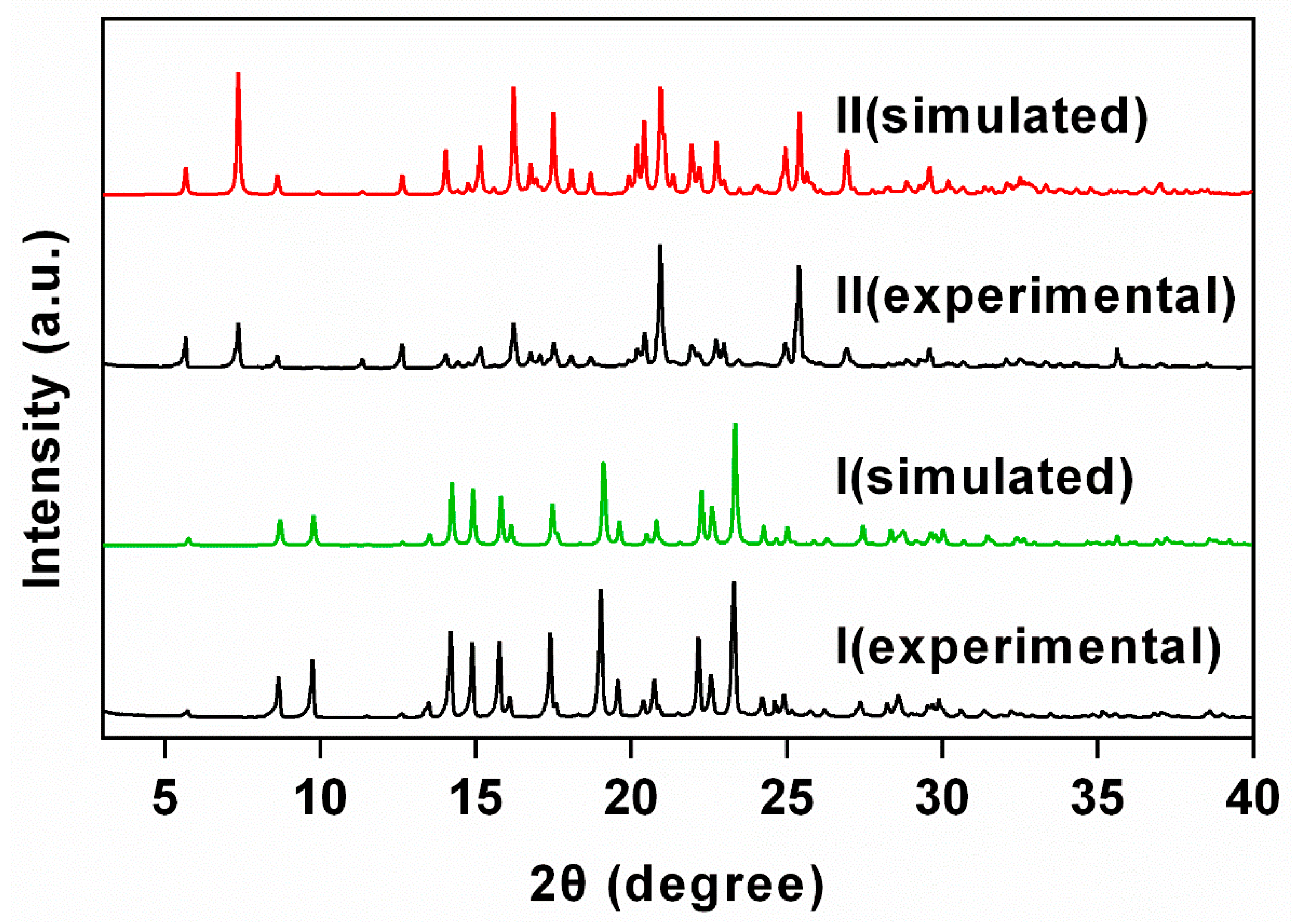
2.1. Synthesis and Co-Crystal Characterizations
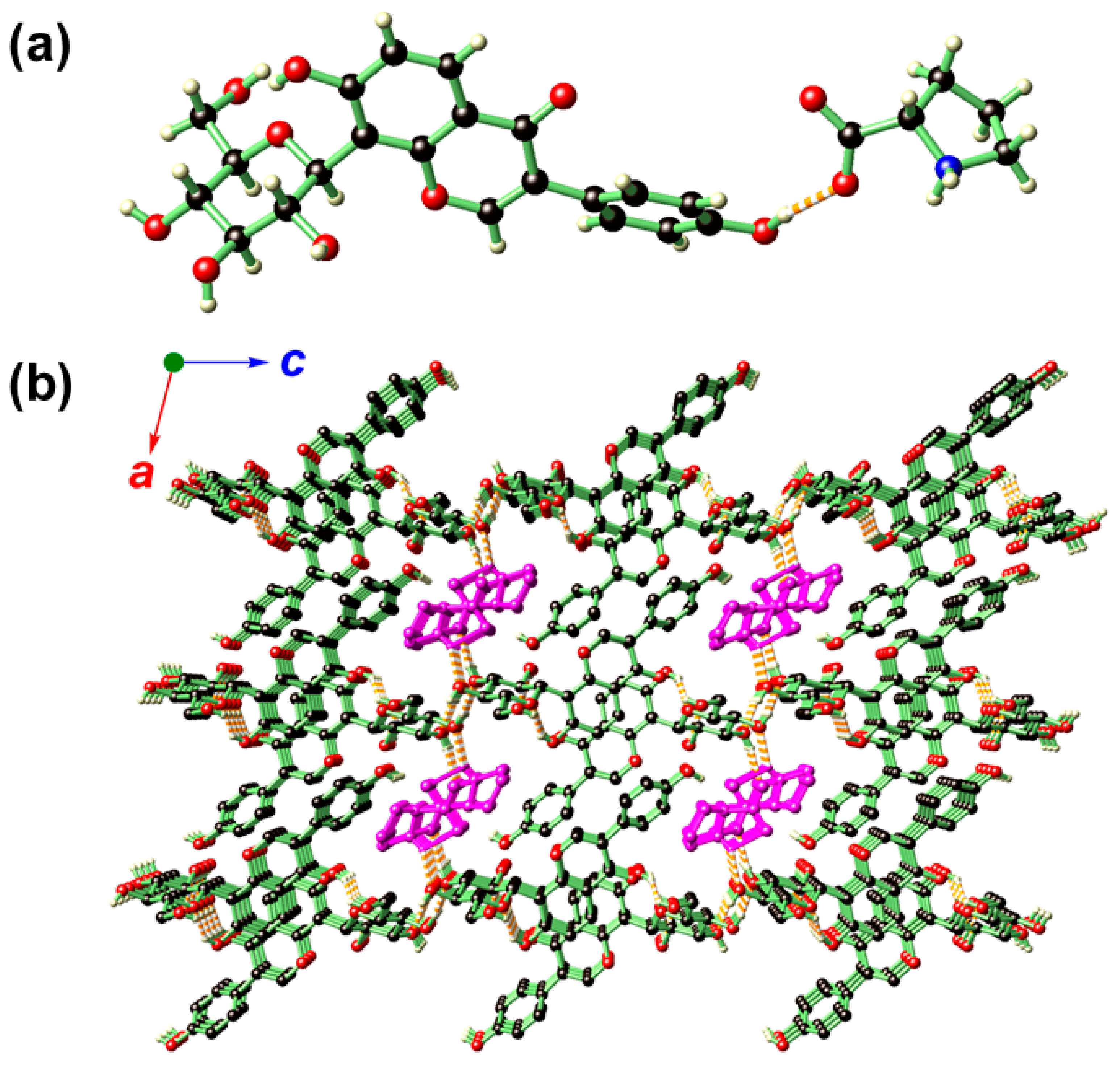
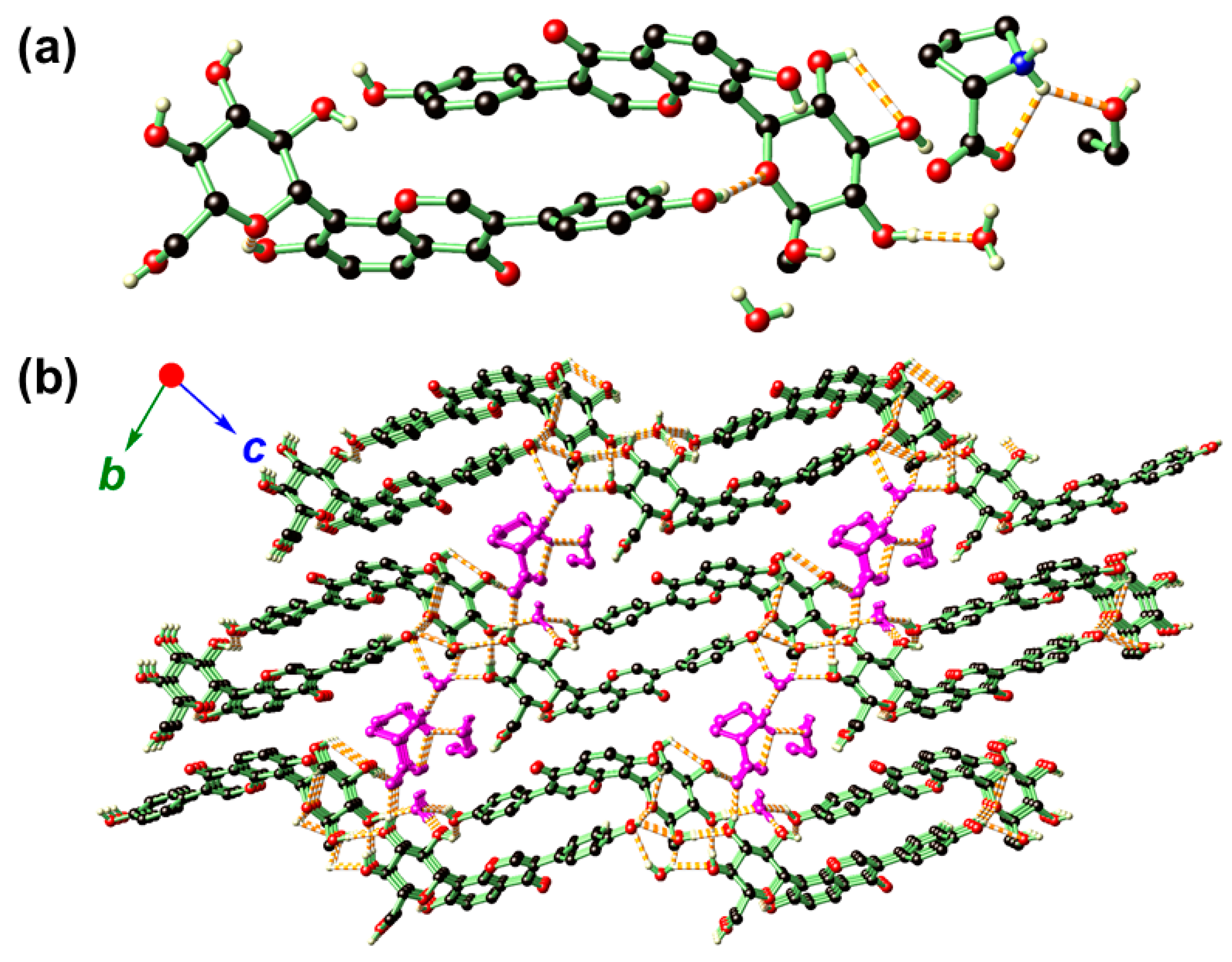
2.2. Crystal Structure Analysis
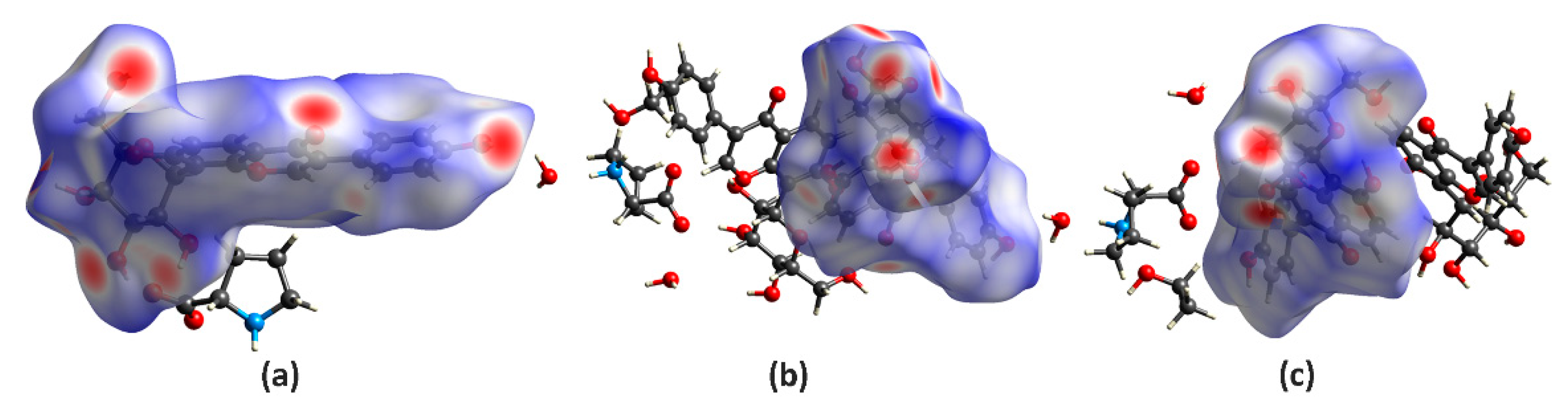
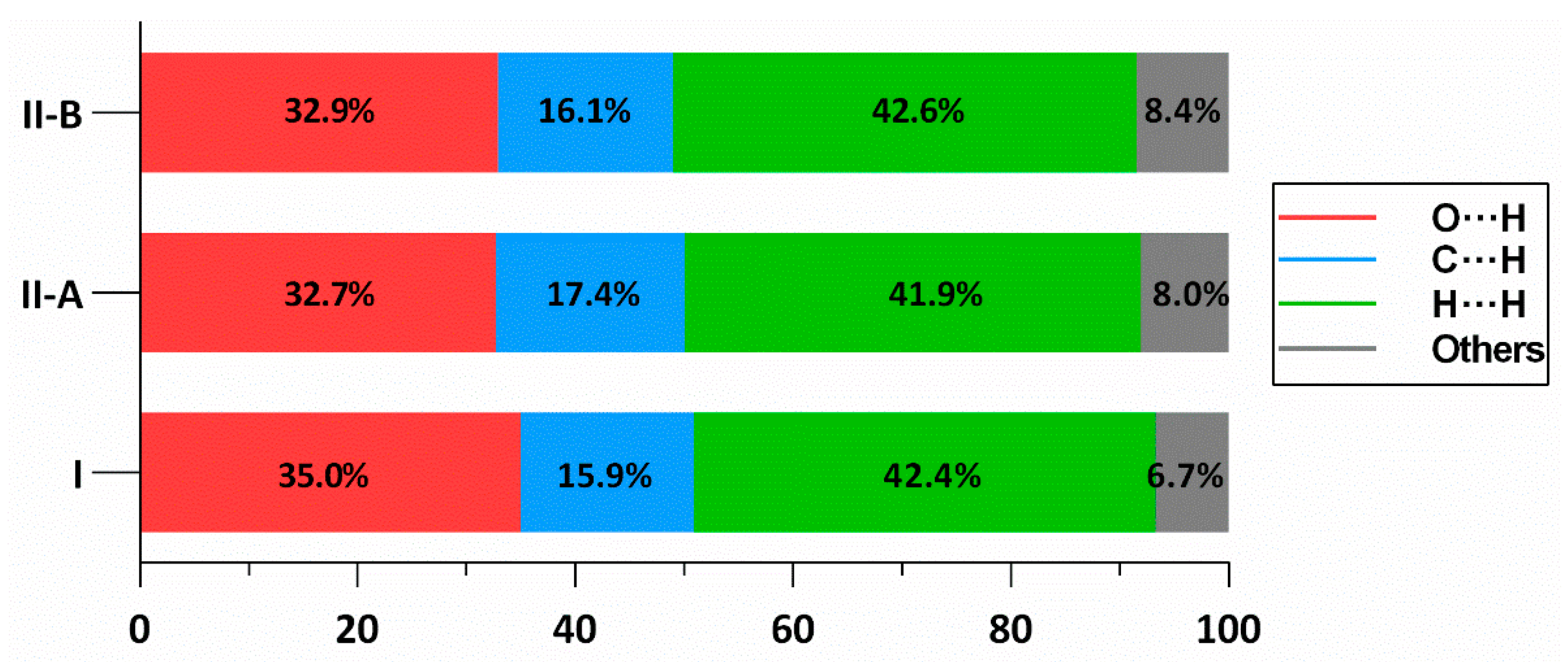
2.3. Hirshfeld Surface Analysis of TAF, I and II
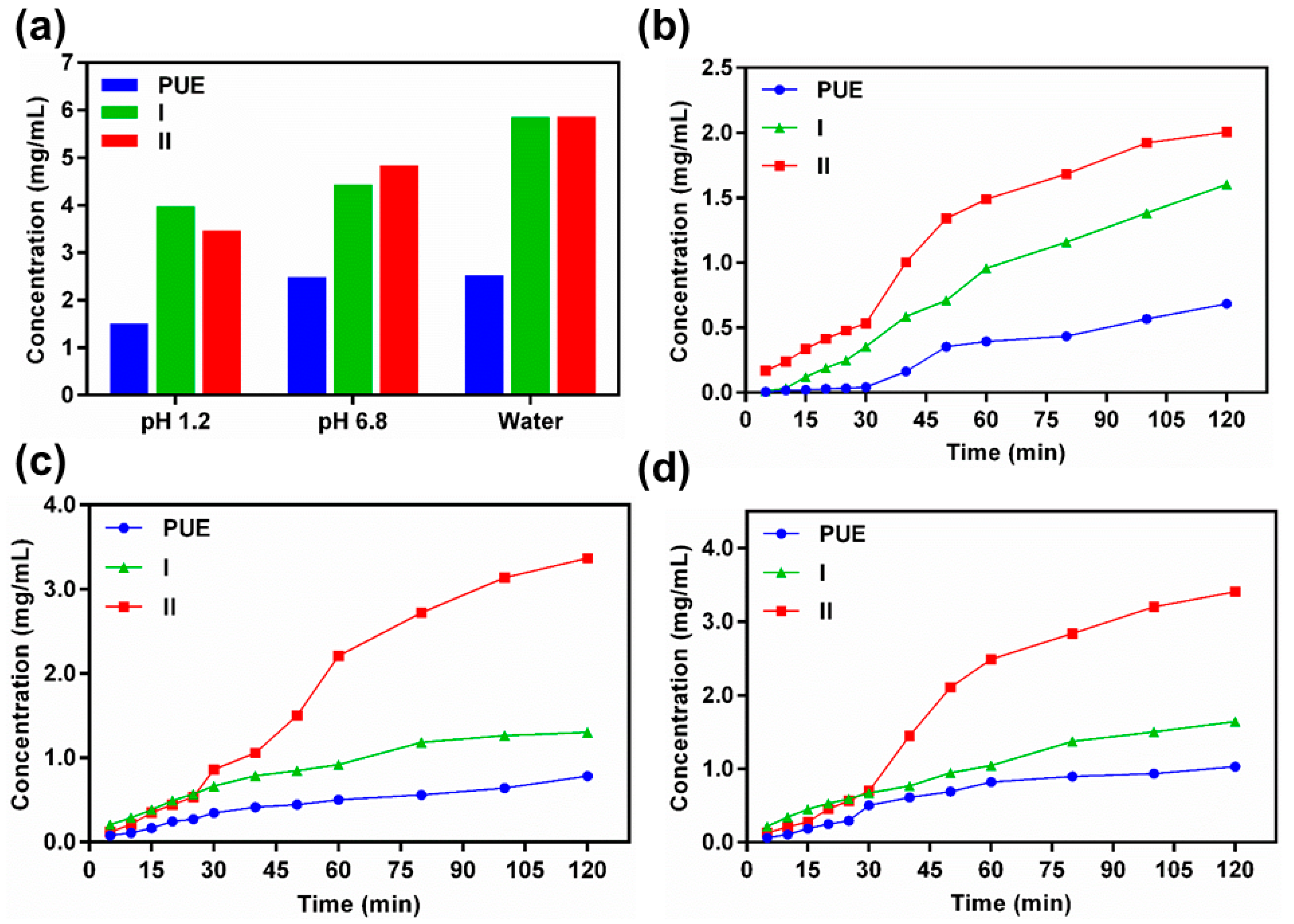
2.4. Solubility and Dissolution Rates of PUE, I and II
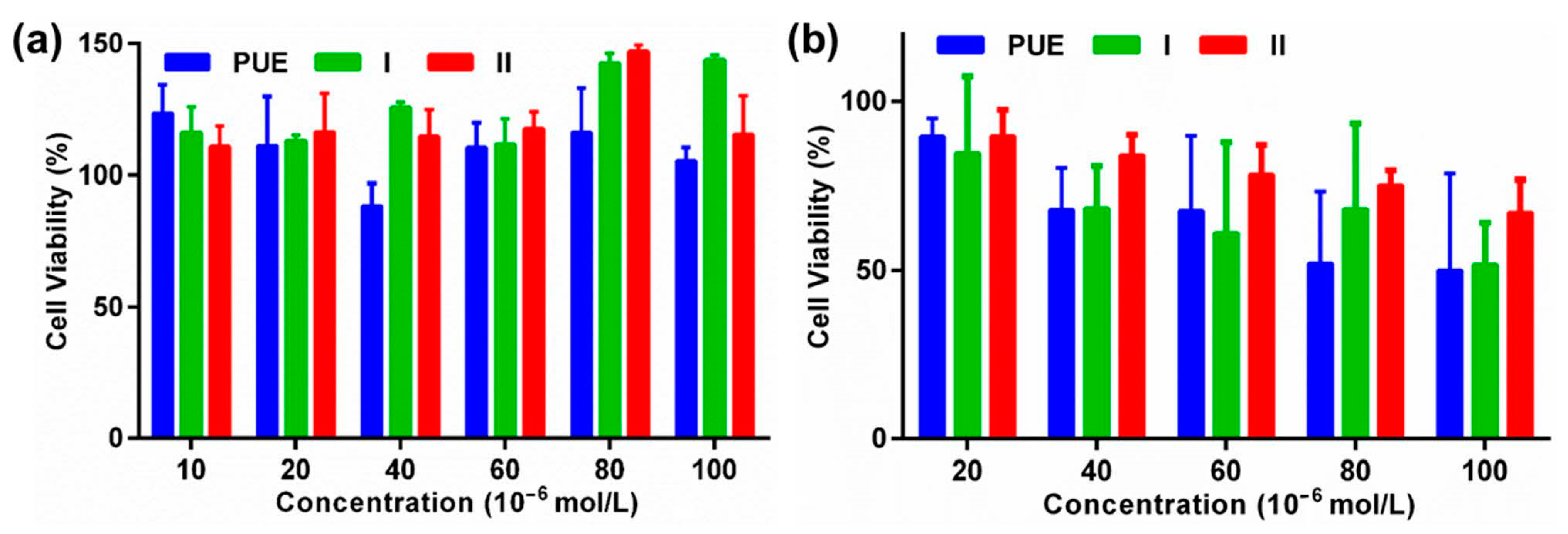
2.5. MTT Assays
3. Materials and Methods
3.1. General
3.2. Synthesis of Co-Crystals I and II
3.3. Single-Crystal X-ray Crystallography
3.4. Solubility and Dissolution Measurement
3.5. Cytotoxicity Evaluation by MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suzuki, H.; Yakushiji, K.; Matsunaga, S.; Yamauchi, Y.; Seto, Y.; Sato, H.; Onoue, S. Amorphous solid dispersion of meloxicam enhanced oral absorption in rats with impaired gastric motility. J. Pharm. Sci. 2018, 107, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, L.; Yao, J.; Ma, Y.-Y.; Chen, J.-M.; Lu, T.-B. Improving the solubility and bioavailability of apixaban via apixaban–oxalic acid cocrystal. Cryst. Growth Des. 2016, 16, 2923–2930. [Google Scholar] [CrossRef]
- Ulrich, J.; Frohberg, P. Problems, potentials and future of industrial crystallization. Front. Chem. Sci. Eng. 2013, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Manin, A.N.; Manin, N.G.; Kuzmina, L.G.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical cocrystals of diflunisal and diclofenac with theophylline. Mol. Pharm. 2014, 11, 3707–3715. [Google Scholar] [CrossRef] [PubMed]
- Steed, J.W. The role of co-crystals in pharmaceutical design. Trends Pharmacol. Sci. 2013, 34, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D. Creating cocrystals: A Review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Qiao, N.; Li, M.; Schlindwein, W.; Malek, N.; Davies, A.; Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 2011, 419, 1–11. [Google Scholar] [CrossRef]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. [Google Scholar] [CrossRef]
- Korotkova, E.I.; Kratochvil, B. Pharmaceutical cocrystals. Procedia Chem. 2014, 10, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C. Cocrystallization for successful drug delivery. Expert Opin. Drug Deliv. 2012, 10, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Perry, M.L.; Weyna, D.R.; Zaworotko, M.J. Impact of pharmaceutical cocrystals: The effects on drug pharma-cokinetics. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal engineering approach to forming cocrystals of amine hydrochlorides with organic acids. Molecular complexes of fluoxetine hydrochloride with benzoic, succinic, and fumaric acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagden, N.; De Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef]
- Elder, D.P.; Holm, R.; De Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.H.; Chow, P.S.; Tan, R.B. Novel solid forms of the anti-tuberculosis drug, isoniazid: Ternary and polymorphic cocrystals. CrystEngComm 2013, 15, 5877. [Google Scholar] [CrossRef]
- Tao, Q.; Chen, J.-M.; Ma, L.; Lu, T.-B. Phenazopyridine cocrystal and salts that exhibit enhanced solubility and stability. Cryst. Growth Des. 2012, 12, 3144–3152. [Google Scholar] [CrossRef]
- Zhou, Y.-X.; Zhang, H.; Peng, C. Puerarin: A review of pharmacological effects. Phytother. Res. 2014, 28, 961–975. [Google Scholar] [CrossRef]
- Kato, E.; Kawabata, J. Glucose uptake enhancing activity of puerarin and the role of C-glucoside suggested from activity of related compounds. Bioorg. Med. Chem. Lett. 2010, 20, 4333–4336. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Guo, C.; Lu, X.; Tan, W. Anti-colorectal cancer biotargets and biological mechanisms of puerarin: Study of molecular networks. Eur. J. Pharmacol. 2019, 858, 172483. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ma, X.; Song, Y.; Zhang, Y.; Xiong, W.; Li, L.; Zhou, L. Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. J. Cell. Biochem. 2019, 120, 11265–11273. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-Q.; Zhang, H.-B.; Wang, G.-F.; Xu, D.; Zhang, W.-Y.; Wang, Q.-S.; Cui, Y.-L. Colon-specific microspheres loaded with puerarin reduce tumorigenesis and metastasis in colitis-associated colorectal cancer. Int. J. Pharm. 2019, 570, 118644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, B.; Mo, J. Puerarin 6″-O-xyloside possesses significant antitumor activities on colon cancer through inducing apoptosis. Oncol. Lett. 2018, 16, 5557–5564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-G.; Yin, X.-C.; Liu, X.-F.; Meng, K.-W.; Tang, K.U.N.; Huang, F.-L.; Xu, G.; Gao, J.I.E. Puerarin induces hepa-tocellular carcinoma cell apoptosis modulated by mapk signaling pathways in a dose-dependent manner. Anticancer Res. 2017, 37, 4425–4431. [Google Scholar]
- Liu, X.; Zhao, W.; Wang, W.; Lin, S.; Yang, L. Puerarin suppresses LPS-induced breast cancer cell migration, invasion and adhesion by blockage NF-κB and Erk pathway. Biomed. Pharmacother. 2017, 92, 429–436. [Google Scholar] [CrossRef]
- Yan, J.; Guan, Z.; Zhu, W.; Zhong, L.; Qiu, Z.-Q.; Yue, P.; Wu, W.-T.; Liu, J.; Huang, X. Preparation of puerarin chitosan oral nanoparticles by ionic gelation method and its related kinetics. Pharmaceutics 2020, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Dong, L.; Liu, Y.; Wang, G.; Wang, G.; Qiao, Y. Biopharmaceutics classification of puerarin and comparison of perfusion approaches in rats. Int. J. Pharm. 2014, 466, 133–138. [Google Scholar] [CrossRef]
- Dong, Z.; Guo, J.; Xing, X.; Zhang, X.; Du, Y.; Lu, Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: Formulation, characterization and protective effects on acute myocardial ischemia model. Biomed. Pharmacother. 2017, 89, 297–304. [Google Scholar] [CrossRef]
- Tao, H.-Q.; Meng, Q.; Li, M.-H.; Yu, H.; Liu, M.-F.; Du, D.; Sun, S.-L.; Yang, H.-C.; Wang, Y.-M.; Ye, W.; et al. HP-β-CD-PLGA nanoparticles improve the penetration and bioavailability of puerarin and enhance the therapeutic effects on brain ischemia–reperfusion injury in rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 386, 61–70. [Google Scholar] [CrossRef]
- Luo, C.-F.; Yuan, M.; Chen, M.-S.; Liu, S.-M.; Zhu, L.; Huang, B.-Y.; Liu, X.-W.; Xiong, W. Pharmacokinetics, tissue dis-tribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int. J. Pharm. 2011, 410, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, L.-Y.; Liu, F.; Li, Y.-T.; Wu, Z.-Y.; Yan, C.-W. Simultaneously enhancing the In Vitro/In Vivo performances of acetazolamide using proline as a zwitterionic coformer for cocrystallization. CrystEngComm 2019, 21, 3064–3073. [Google Scholar] [CrossRef]
- Liu, M.; Hong, C.; Yao, Y.; Shen, H.; Ji, G.; Li, G.; Xie, Y. Development of a pharmaceutical cocrystal with solution crys-tallization technology: Preparation, characterization, and evaluation of myricetin-proline cocrystals. Eur. J. Pharm. Bio-pharm. 2016, 107, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Löbmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Tang, D.; Wang, F.; Zhang, J.; Zhang, J.; Wang, J.; Xu, X.; Zhang, J. Enhancing both oral bioavailability and brain penetration of puerarin using borneol in combination with preparation technologies. Drug Deliv. 2017, 24, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Dou, M.; Lu, C.; Sun, Z.; Rao, W. Natural cryoprotectants combinations of l-proline and trehalose for red blood cells cryopreservation. Cryobiology 2019, 91, 23–29. [Google Scholar] [CrossRef]
- Thangavel, P.; Ramachandran, B.; Kannan, R.; Muthuvijayan, V. Biomimetic hydrogel loaded with silk and l -proline for tissue engineering and wound healing applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 1401–1408. [Google Scholar] [CrossRef]
- Papageorgiou, S.K.; Kouvelos, E.P.; Favvas, E.P.; Sapalidis, A.A.; Romanos, G.E.; Katsaros, F.K. Metal–carboxylate inter-actions in metal–alginate complexes studied with FTIR spectroscopy. Carbohydr. Res. 2010, 345, 469–473. [Google Scholar] [CrossRef]
- Mary, Y.S.; Ushakumari, L.; Harikumar, B.; Varghese, H.T.; Panicker, C.Y. FT-IR, FT-raman and SERS spectra of L-proline. J. Iran. Chem. Soc. 2009, 6, 138–144. [Google Scholar] [CrossRef]
- Yuan, F.-L.; Yuan, Y.-Q.; Chao, M.-Y.; Young, D.J.; Zhang, W.-H.; Lang, J.-P. Deciphering the structural relationships of five cd-based metal–organic frameworks. Inorg. Chem. 2017, 56, 6522–6531. [Google Scholar] [CrossRef]
- Ratajczak, H.; Barycki, J.; Pietraszko, A.; Baran, J.; Debrus, S.; May, M.; Venturini, J. Preparation and structural study of a novel nonlinear molecular material: The l-histidinum dihydrogenarsenate orthoarsenic acid crystal. J. Mol. Struct. 2000, 526, 269–278. [Google Scholar] [CrossRef]
- Abu-Nawwas, A.-A.H.; Cano, J.; Christian, P.; Mallah, T.; Rajaraman, G.; Teat, S.J.; Winpenny, R.E.P.; Yukawa, Y. An Fe (III) wheel with a zwitterionic ligand: The structure and magnetic properties of [Fe(OMe)2(proline)]12[ClO4]12. Chem. Commun. 2004, 314–315. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Lan, Y.; Li, Y.; Wang, E.; Hao, N.; Xiao, D.; Duan, L.; Xu, L. A novel chain-like polymer constructed from heter-opolyanions covalently linked by lanthanide cations: (C5H9NO2)2[La(H2O)7CrMo6H6O24]·11H2O (Proline=C5H9NO2). Inorg. Chem. Commun. 2004, 7, 356–358. [Google Scholar] [CrossRef]
- Armaghan, M.; Shang, X.J.; Yuan, Y.Q.; Young, D.J.; Zhang, W.H.; Hor, T.S.A.; Lang, J.P. Metal−organic frameworks via emissive metal-carboxylate zwitterion intermediates. ChemPlusChem 2015, 80, 1231–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lin, S.-X.; Niu, R.-J.; Liu, Q.; Zhang, W.-H.; Young, D.J. Zinc and cadmium complexes of pyridinemethanol car-boxylates: Metal carboxylate zwitterions and metal–organic frameworks. ChemPlusChem 2020, 85, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Armaghan, M.; Young, D.J.; Wu, D.; Wei, Y.; Yuan, F.-L.; Ng, S.W.; Amini, M.M.; Zhang, W.-H.; Young, D.J.; Chen, H.W.; et al. Isolation of first row transition metal-carboxylate zwitterions. RSC Adv. 2015, 5, 42978–42989. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Wolff, S.; Grimwood, D.; McKinnon, J.; Turner, M.; Jayatilaka, D.; Spackman, M. Crystal Explorer; The University of Western Australia: Crawley, Australia, 2012. [Google Scholar]
- Yu, Z.; Li, W. Induction of apoptosis by puerarin in colon cancer HT-29 cells. Cancer Lett. 2006, 238, 53–60. [Google Scholar] [CrossRef]
- Sugandha, K.; Kaity, S.; Mukherjee, S.; Isaac, J.; Ghosh, A. Solubility enhancement of ezetimibe by a cocrystal engineering technique. Cryst. Growth Des. 2014, 14, 4475–4486. [Google Scholar] [CrossRef]
- Bruker. APEX2, SAINT and SADABS; Bruker ACS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for smallmolecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | I | II |
|---|---|---|
| Empirical formula | C21H20O9∙C5H9NO2 | 2(C21H20O9)·C5H9NO2·2H2O·C2H5OH |
| Formula Weight | 531.50 | 1029.97 |
| Crystal system | monoclinic | Triclinic |
| Space group | P21 | P1 |
| a/Å | 10.2463 (4) | 6.3123 (2) |
| b/Å | 7.8656 (3) | 12.1389 (4) |
| c/Å | 15.4520 (6) | 15.7274 (6) |
| α/° | 90 | 81.7330 (10) |
| β/° | 97.7830 (10) | 89.7330 (10) |
| γ/° | 90 | 87.4870 (10) |
| V/Å3 | 1233.86 (8) | 1191.43 (7) |
| Z | 2 | 1 |
| Dc/(g cm−3) | 1.431 | 1.436 |
| μ (Mo-Kα)/mm−1 | 0.952 | 0.974 |
| F(000) | 560 | 544 |
| Total reflections | 13,357 | 40,998 |
| Unique reflections | 4414 | 9385 |
| No observations | 4411 | 9322 |
| No parameters | 349 | 673 |
| Flack parameter | 0.03 (3) | −0.01 (3) |
| Rint | 0.0254 | 0.0293 |
| R a | 0.0293 | 0.0332 |
| wR b | 0.0774 | 0.0957 |
| GOF c | 1.057 | 1.080 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inam, M.; Liu, L.; Wang, J.-W.; Yu, K.-X.; Phan, C.-U.; Shen, J.; Zhang, W.-H.; Tang, G.; Hu, X. Enhancing the Physiochemical Properties of Puerarin via L-Proline Co-Crystallization: Synthesis, Characterization, and Dissolution Studies of Two Phases of Pharmaceutical Co-Crystals. Int. J. Mol. Sci. 2021, 22, 928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020928
Inam M, Liu L, Wang J-W, Yu K-X, Phan C-U, Shen J, Zhang W-H, Tang G, Hu X. Enhancing the Physiochemical Properties of Puerarin via L-Proline Co-Crystallization: Synthesis, Characterization, and Dissolution Studies of Two Phases of Pharmaceutical Co-Crystals. International Journal of Molecular Sciences. 2021; 22(2):928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020928
Chicago/Turabian StyleInam, Muhammad, Lu Liu, Jian-Wei Wang, Ka-Xi Yu, Chi-Uyen Phan, Jie Shen, Wen-Hua Zhang, Guping Tang, and Xiurong Hu. 2021. "Enhancing the Physiochemical Properties of Puerarin via L-Proline Co-Crystallization: Synthesis, Characterization, and Dissolution Studies of Two Phases of Pharmaceutical Co-Crystals" International Journal of Molecular Sciences 22, no. 2: 928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020928