Steroidal Regulation of Oviductal microRNAs Is Associated with microRNA-Processing in Beef Cows

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Animal Model
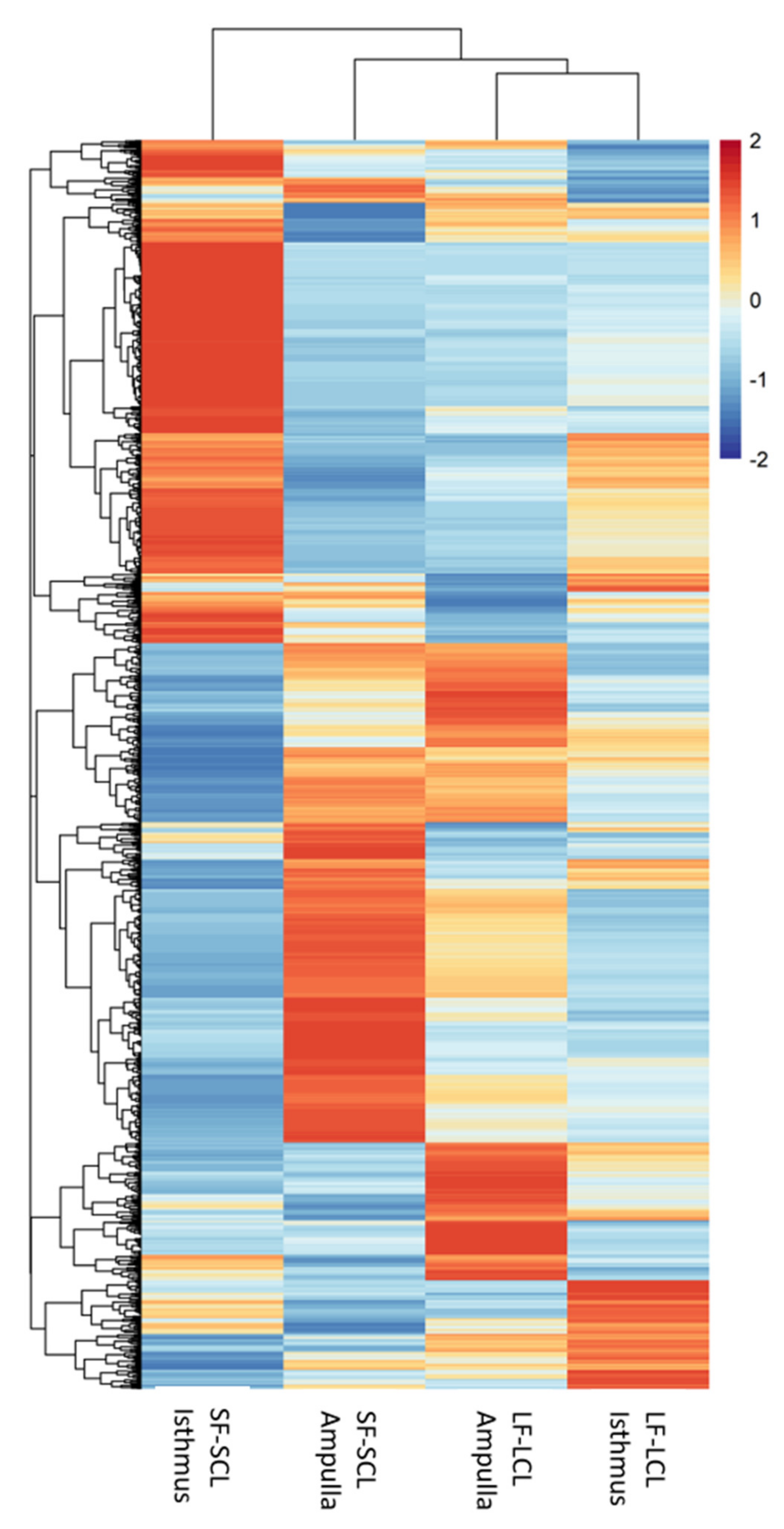
2.2. RNA-Seq Analyses
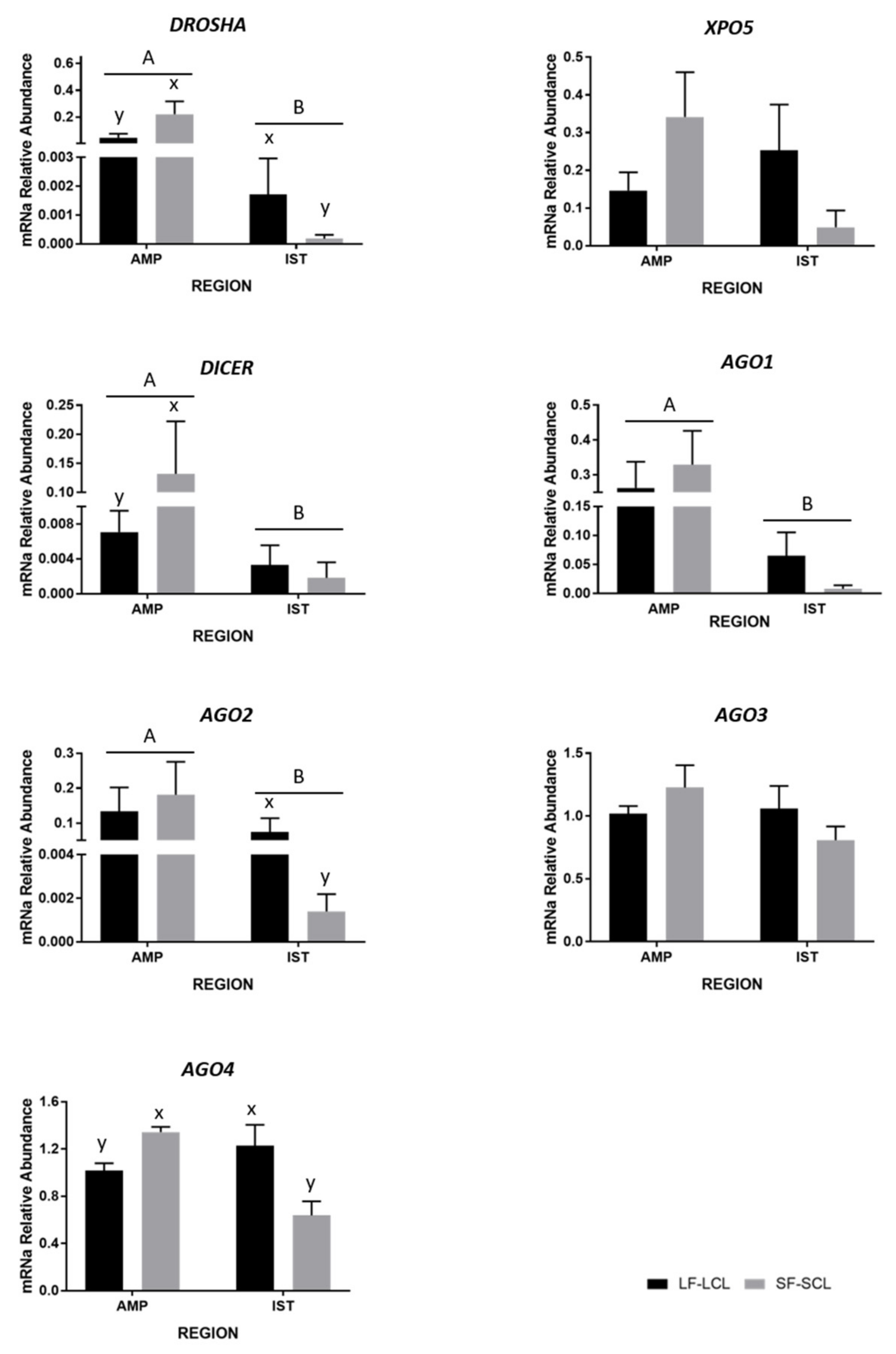
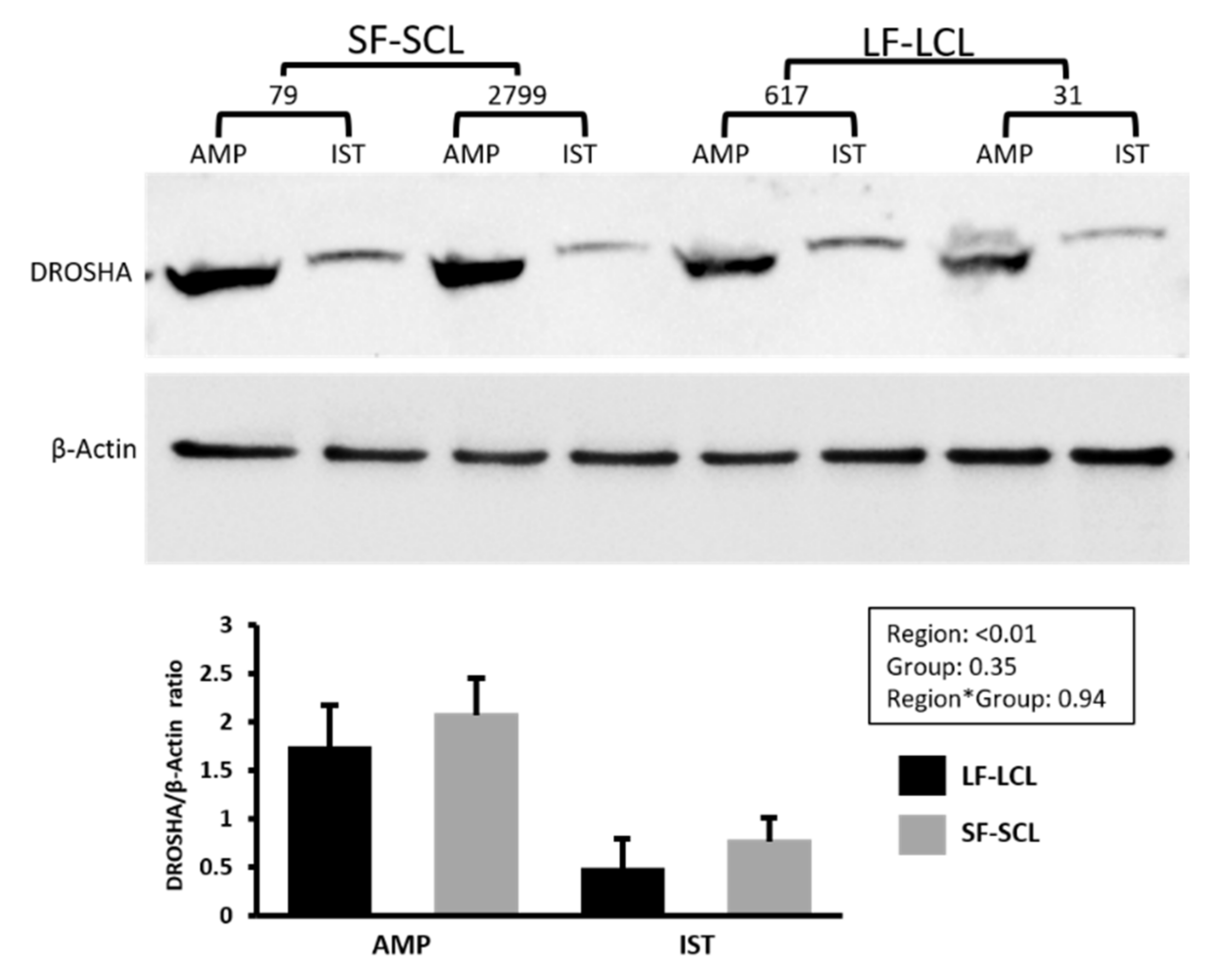
2.3. Abundance of miRNA Processing Pathway-Components
2.4. miRNA Abundance Profiles in Ampulla and Isthmus
2.5. Selected miRNAs Abundance in LF-LCL and SF-SCL Groups
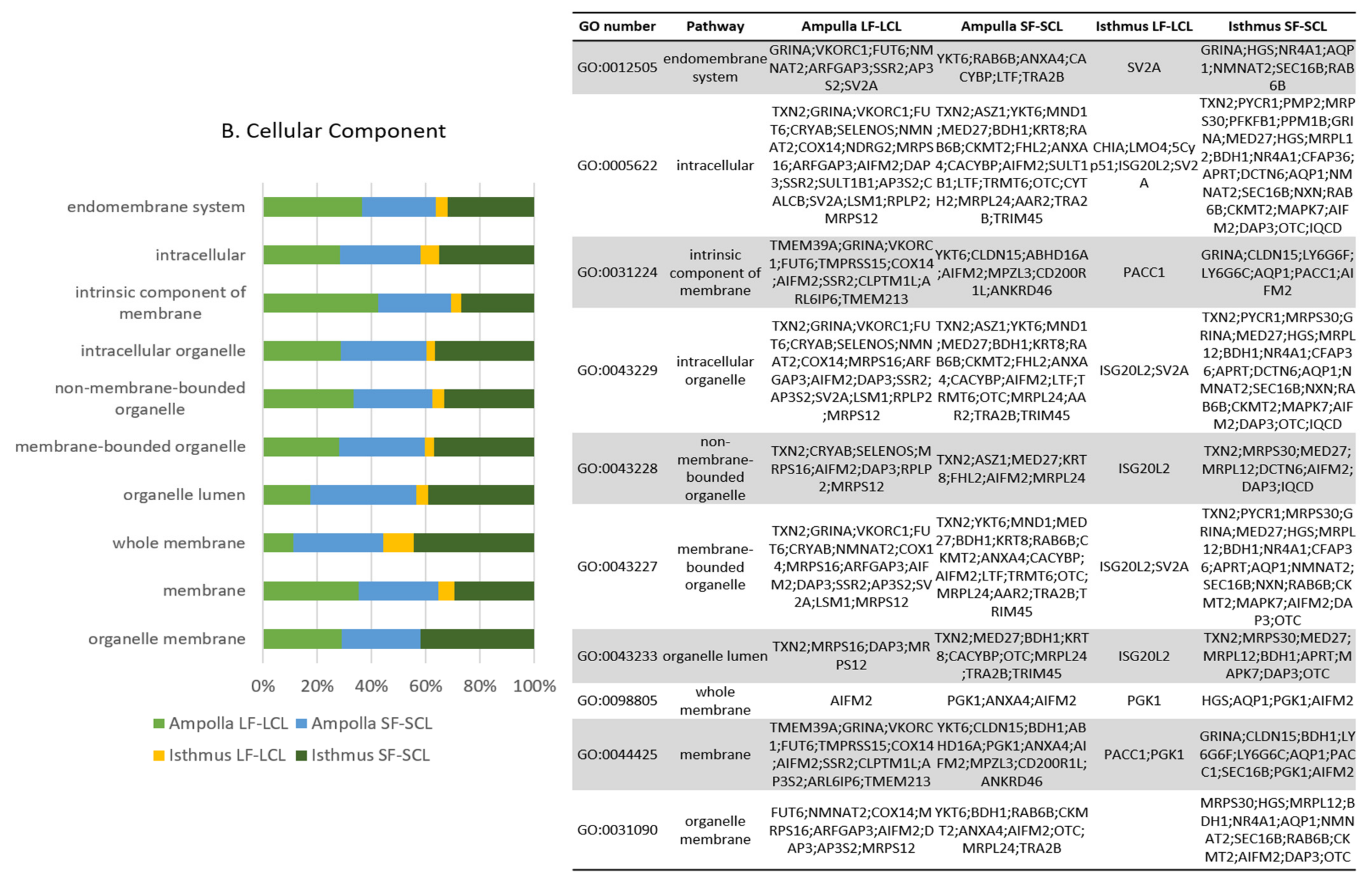
2.6. Integration of mRNAs and miRNAs Data
3. Discussion
4. Materials and Methods
4.1. Animals, Reproductive Management, and Tissue Processing
4.2. RNA Extraction
4.3. mRNA Libraries, Sequencing, and Bioinformatics
4.4. Reverse Transcription and qPCR of mRNA Molecules
4.5. Protein Quantification by Western Blotting
4.6. qPCR Analysis of miRNAs
4.7. microRNA-Targeted Transcript Modulation
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spencer, T.E.; Bazer, F.W. Temporal and Spatial Alterations in Uterine Estrogen-Receptor and Progesterone-Receptor Gene-Expression during the Estrous-Cycle and Early-Pregnancy in the Ewe. Biol. Reprod. 1995, 53, 1527–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Krebs, S.; Bauersachs, S.; Blum, H.; Wolf, E.; Miyamoto, A. Actions and interactions of progesterone and estrogen on transcriptome profiles of the bovine endometrium. Physiol. Genomics 2010, 42A, 290–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbalik, M.E.; Sagsoz, H.; Saruhan, B.G. Localization of Estrogen Receptor Alpha and Progesterone Receptor B in the Bovine Ovary During the Follicular and Luteal Phase of the Sexual Cycle. Kafkas. Univ. Vet. Fak. 2011, 17, 795–802. [Google Scholar]
- Araujo, E.R.; Sponchiado, M.; Pugliesi, G.; Van Hoeck, V.; Mesquita, F.S.; Membrive, C.M.B.; Binelli, M. Spatio-specific regulation of endocrine-responsive gene transcription by periovulatory endocrine profiles in the bovine reproductive tract. Reprod. Fertil. Dev. 2014, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerny, K.L.; Garrett, E.; Walton, A.J.; Anderson, L.H.; Bridges, P.J. A transcriptomal analysis of bovine oviductal epithelial cells collected during the follicular phase versus the luteal phase of the estrous cycle. Reprod. Biol. Endocrinol. 2015, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonella-Diaza, A.M.; Mesquita, F.S.; Ribeiro da Silva, K.; Balieiro, J.C.C.; Dos Santos, N.P.; Pugliesi, G.; Strefezzi, R.F.; Binelli, M. Sex steroids modulate morphological and functional features of the bovine oviduct. Cell. Tissue Res. 2017, 370, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Pugliesi, G.; Scolari, S.C.; França, M.R.; Ramos, R.S.; Oliveira, M.; Papa, P.C.; Bressan, F.F.; Meirelles, F.V.; Silva, L.A.; et al. Manipulation of the periovulatory sex steroidal milieu affects endometrial but not luteal gene expression in early diestrus Nelore cows. Theriogenology 2014, 81, 861–869. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Ramos, R.S.; Pugliesi, G.; Andrade, S.C.S.; Van Hoeck, V.; Langbeen, A.; Oliveira, M.L.; Gonella-Diaza, A.M.; Gasparin, G.; Fukumasu, H.; et al. The Receptive Endometrial Transcriptomic Signature Indicates an Earlier Shift from Proliferation to Metabolism at Early Diestrus in the Cow. Biol. Reprod. 2015, 93, 52. [Google Scholar] [CrossRef]
- Gonella-Diaza, A.M.; da Silva Andrade, S.C.; Sponchiado, M.; Pugliesi, G.; Mesquita, F.S.; Van Hoeck, V.; de Francisco Strefezzi, R.; Gasparin, G.R.; Coutinho, L.L.; Binelli, M. Size of the Ovulatory Follicle Dictates Spatial Differences in the Oviductal Transcriptome in Cattle. PLoS ONE 2015, 10, e0145321. [Google Scholar] [CrossRef] [Green Version]
- Gonella-Diaza, A.M.; Silveira Mesquita, F.; Lopes, E.; Ribeiro da Silva, K.; Cogliati, B.; De Francisco Strefezzi, R.; Binelli, M. Sex Steroids Drive the Remodeling of Oviductal Extracellular Matrix in Cattle. Biol. Reprod. 2018, 99, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Davis-Dusenbery, B.N.; Hata, A. Mechanisms of control of microRNA biogenesis. J. Biochem. 2010, 148, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveto, S.; Mancino, M.; Manfrini, N.; Biffo, S. Role of microRNAs in translation regulation and cancer. World J. Biol. Chem. 2017, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; de Castro, I.P.; Malumbres, M. Control of cell proliferation pathways by microRNAs. Cell Cycle 2008, 7, 3143–3148. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan micrornas. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, A.K.; Andreu-Vieyra, C.; Franco, H.L.; Ma, L.; Chen, R.; Han, D.Y.; Zhu, H.; Agno, J.E.; Gunaratne, P.H.; DeMayo, F.J.; et al. Deletion of Dicer in somatic cells of the female reproductive tract causes sterility. Mol. Endocrinol. 2008, 22, 2336–2352. [Google Scholar] [CrossRef] [Green Version]
- Nothnick, W.B.; Healy, C.; Hong, X. Steroidal regulation of uterine miRNAs is associated with modulation of the miRNA biogenesis components Exportin-5 and Dicer1. Endocrine 2010, 37, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, D.R.; Spoelstra, N.S.; Richer, J.K. The role of miRNAs in progesterone action. Mol. Cell. Endocrinol. 2012, 357, 50–59. [Google Scholar] [CrossRef]
- Klinge, C.M. miRNAs and estrogen action. Trends Endocrinol. Metab. 2012, 23, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, D.R.; Cittelly, D.M.; Howe, E.N.; Spoelstra, N.S.; McKinsey, E.L.; LaPara, K.; Elias, A.; Yee, D.; Richer, J.K. MicroRNAs link estrogen receptor alpha status and Dicer levels in breast cancer. Horm. Cancer 2010, 1, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Gonella-Diaza, A.M.; da Silva Andrade, S.C.; Sponchiado, M.; Pugliesi, G.; Mesquita, F.S.; Van Hoeck, V.; de Francisco Strefezzi, R.; Gasparin, G.R.; Coutinho, L.L.; Binelli, M. Oviductal transcriptional profiling of a bovine fertility model by next-generation sequencing. Genomics Data 2017, 13, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, J.; Meister, G. Argonaute regulation: Two roads to the same destination. Dev. Cell 2013, 25, 553–554. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Chandradoss, S.D.; Depken, M.; Joo, C. Why Argonaute is needed to make microRNA target search fast and reliable. Semin. Cell. Dev. Biol. 2017, 65, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Pugliesi, G.; Santos, F.B.; Lopes, E.; Nogueira, É.; Maio, J.R.G.; Binelli, M. Improved fertility in suckled beef cows ovulating large follicles or supplemented with long-acting progesterone after timed-AI. Theriogenology 2016, 85, 1239–1248. [Google Scholar] [CrossRef]
- Sperber, H.; Beem, A.; Shannon, S.; Jones, R.; Banik, P.; Chen, Y.; Ku, S.; Varani, G.; Yao, S.; Ruohola-Baker, H. miRNA sensitivity to Drosha levels correlates with pre-miRNA secondary structure. RNA 2014, 20, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhang, X.; Song, Q.; Li, T.; Zeng, Y. Drosha processing controls the specificity and efficiency of global microRNA expression. Biochim. Biophys. Acta 2011, 1809, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Bhat-Nakshatri, P.; Wang, G.; Collins, N.R.; Thomson, M.J.; Geistlinger, T.R.; Carroll, J.S.; Brown, M.; Hammond, S.; Srour, E.F.; Liu, Y.; et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 2009, 37, 4850–4861. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Fu, X.; Alves, P.; Gerstein, M. mRNA expression profiles show differential regulatory effects of microRNAs between estrogen receptor-positive and estrogen receptor-negative breast cancer. Genome Biol. 2009, 10, R90. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.; Bae, H.; Lim, W.; Song, G. Dicer1, AGO3, and AGO4 microRNA machinery genes are differentially expressed in developing female reproductive organs and overexpressed in cancerous ovaries of chickens. J. Anim. Sci. 2017, 95, 4857–4868. [Google Scholar] [CrossRef] [PubMed]
- Almiñana, C.; Tsikis, G.; Labas, V.; Uzbekov, R.; da Silveira, J.C.; Bauersachs, S.; Mermillod, P. Deciphering the oviductal extracellular vesicles content across the estrous cycle: Implications for the gametes-oviduct interactions and the environment of the potential embryo. BMC Genomics 2018, 19, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fereshteh, Z.; Schmidt, S.A.; Al-Dossary, A.A.; Accerbi, M.; Arighi, C.; Cowart, J.; Song, J.L.; Green, P.J.; Choi, K.; Yoo, S.; et al. Murine Oviductosomes (OVS) microRNA profiling during the estrous cycle: Delivery of OVS-borne microRNAs to sperm where miR-34c-5p localizes at the centrosome. Sci. Rep. 2018, 8, 16094. [Google Scholar] [CrossRef]
- Johnson, M.H.; Nasresfahani, M.H. Radical solutions and cultural problems: Could free oxygen radicals be responsible for the impaired development of preimplantation mammalian embryos in vitro? Bioessays 1994, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Marei, W.F.; Van den Bosch, L.; Pintelon, I.; Mohey-Elsaeed, O.; Bols, P.E.; Leroy, J.L. Mitochondria-targeted therapy rescues development and quality of embryos derived from oocytes matured under oxidative stress conditions: A bovine in vitro model. Human Reprod. 2019, 34, 1984–1998. [Google Scholar] [CrossRef]
- Khatun, H.; Ihara, Y.; Takakura, K.; Egashira, J.; Wada, Y.; Konno, T.; Tatemoto, H.; Yamanaka, K.I. Role of endoplasmic reticulum stress on developmental competency and cryo-tolerance in bovine embryos. Theriogenology 2020, 142, 131–137. [Google Scholar] [CrossRef]
- Liu, L.; Trimarchi, J.R.; Keefe, D.L. Involvement of mitochondria in oxidative stress-induced cell death in mouse zygotes. Biol. Reprod. 2000, 62, 1745–1753. [Google Scholar] [CrossRef] [Green Version]
- Binelli, M.; Gonella-Diaza, A.M.; Mesquita, F.S.; Membrive, C.M.B. Sex steroid-mediated control of oviductal function in cattle. Biology 2018, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Ramos, R.S.; Oliveira, M.L.; Izaguirry, A.P.; Vargas, L.M.; Soares, M.B.; Mesquita, F.S.; Santos, F.W.; Binelli, M. The periovulatory endocrine milieu affects the uterine redox environment in beef cows. Reprod. Biol. Endocrinol. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wysoker, A. Durbin R; 1000 Genome project data processing subgroup. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Precedings 2010. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: http://www.repeatmasker.org (accessed on 20 July 2015).
- Da Silveira, J.C.; Andrade, G.M.; del Collado, M.; Sampaio, R.V.; Sangalli, J.R.; Silva, L.A.; Pinaffi, F.V.; Jardim, I.B.; Cesar, M.C.; Nogueira, M.F.; et al. Supplementation with small-extracellular vesicles from ovarian follicular fluid during in vitro production modulates bovine embryo development. PLoS ONE 2017, 12, e0179451. [Google Scholar] [CrossRef]
- Russo, P.S.; Ferreira, G.R.; Cardozo, L.E.; Bürger, M.C.; Arias-Carrasco, R.; Maruyama, S.R.; Hirata, T.D.; Lima, D.S.; Passos, F.M.; Fukutani, K.F.; et al. CEMiTool: A Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- De Mendiburu, F. Statistical procedures for agricultural research. In Package “Agricolae” Version 1.44. Comprehensive R Archive Network; Institute for Statistics and Mathematics: Vienna, Austria, 2013. [Google Scholar]
- De Mendiburu, F.; Simon, R. Agricolae-Ten years of an open source statistical tool for experiments in breeding, agriculture and biology. Peer. J. Prepr. 2015, e1748. [Google Scholar] [CrossRef]
- Kolde, E.; Kolde, M.R. Package ‘pheatmap. R Package 2015, 1, 7. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html. (accessed on 15 May 2019).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | LF-LCL | SF-SCL | p Value | Log2 (Fold Change) |
|---|---|---|---|---|
| let-7b | 0.271 ± 0.046 | 1.052 ± 0.634 | ≤0.01 | 1.955 |
| miR-106a | 0.008 ± 0.002 | 0.020 ± 0.009 | 0.01 | 1.362 |
| miR-106b | 0.047 ± 0.038 | 0.015 ± 0.009 | 0.02 | −1.692 |
| miR-181d | 0.028 ± 0.010 | 0.088 ± 0.058 | ≤0.01 | 1.668 |
| miR-200b | 0.193 ± 0.078 | 0.159 ± 0.025 | 0.04 | −0.273 |
| miR-30c | 0.026 ± 0.005 | 0.056 ± 0.034 | ≤0.01 | 1.114 |
| miR-339b | 0.164 ± 0.053 | 0.301 ± 0.261 | 0.02 | 0.870 |
| miR-375 | 0.241 ± 0.085 | 0.036 ± 0.013 | 0.01 | −2.732 |
| miR-378 | 0.046 ± 0.015 | 0.125 ± 0.107 | 0.03 | 1.448 |
| miR-631 | 5.740 ± 2.225 | 15.586 ± 8.933 | 0.04 | 1.441 |
| miR-92a | 0.142 ± 0.068 | 0.032 ± 0.003 | 0.04 | −2.150 |
| miR-92b | 0.521 ± 0.250 | 2.785 ± 2.683 | ≤0.01 | 2.417 |
| miR-940 | 0.928 ± 0.414 | 4.714 ± 4.399 | ≤0.01 | 2.346 |
| miR-99a-5p | 0.200 ± 0.079 | 0.130 ± 0.020 | 0.02 | −0.623 |
| MicroRNA | LF-LCL | SF-SCL | p Value | Log2 (Fold Change) |
|---|---|---|---|---|
| miR-106a | 0.038 ± 0.018 | 0.011 ± 0.004 | 0.02 | −1.818 |
| miR-122 | 1.838 ± 1.363 | 0.244 ± 0.153 | ≤0.01 | −2.910 |
| miR-125b | 0.036 ± 0.007 | 0.239 ± 0.202 | ≤0.01 | 2.736 |
| miR-132 | 0.393 ± 0.126 | 9.506 ± 9.013 | ≤0.01 | 4.598 |
| miR-138 | 0.406 ± 0.144 | 13.037 ± 11.970 | ≤0.01 | 5.005 |
| miR-143 | 0.317 ± 0.174 | 15.701 ± 15.104 | ≤0.01 | 5.628 |
| miR-154b | 0.297 ± 0.097 | 5.649 ± 5.003 | ≤0.01 | 4.247 |
| miR-17-3p | 0.022 ± 0.009 | 0.119 ± 0.096 | ≤0.01 | 2.454 |
| miR-17-5p | 0.147 ± 0.101 | 0.466 ± 0.425 | 0.02 | 1.661 |
| miR-181b | 0.017 ± 0.005 | 0.272 ± 0.215 | ≤0.01 | 3.961 |
| miR-188 | 0.186 ± 0.049 | 0.372 ± 0.273 | 0.01 | 0.999 |
| miR-192 | 0.230 ± 0.037 | 0.084 ± 0.002 | ≤0.01 | −1.457 |
| miR-193a-5p | 0.111 ± 0.049 | 0.289 ± 0.192 | 0.02 | 1.375 |
| miR-193b | 0.050 ± 0.022 | 0.004 ± 0.002 | ≤0.01 | −3.686 |
| miR-196b | 0.032 ± 0.011 | 0.296 ±0.253 | ≤0.01 | 3.222 |
| miR-199a-3p | 0.007 ± 0.002 | 0.822 ± 0.818 | ≤0.01 | 6.885 |
| miR-200b | 0.019 ± 0.009 | 0.307 ± 0.256 | ≤0.01 | 4.032 |
| miR-211 | 0.048 ± 0.028 | 0.168 ± 0.152 | ≤0.01 | 1.818 |
| miR-219 | 0.060 ± 0.019 | 0.590 ± 0.555 | ≤0.01 | 3.295 |
| miR-30b-5p | 0.028 ± 0.012 | 0.374 ± 0.365 | ≤0.01 | 3.754 |
| miR-30d | 0.119 ± 0.031 | 0.734 ± 0.587 | ≤0.01 | 2.620 |
| miR-345-5p | 0.597 ± 0.257 | 11.439 ± 11.292 | ≤0.01 | 4.260 |
| miR-378 | 0.184 ± 0.068 | 0.025 ± 0.010 | ≤0.01 | −2.892 |
| miR-383 | 0.324 ± 0.151 | 0.130 ± 0.027 | 0.04 | −1.318 |
| miR-409b | 0.021 ± 0.006 | 0.039 ± 0.032 | 0.02 | 0.925 |
| miR-423-5p | 0.210 ± 0.052 | 0.024 ± 0.013 | 0.04 | −3.149 |
| miR-425-3p | 0.639 ± 0.180 | 0.109 ± 0.053 | 0.03 | −2.556 |
| miR-431 | 0.086 ± 0.036 | 0.026 ± 0.008 | 0.02 | −1.743 |
| miR-432 | 0.153 ± 0.038 | 0.327 ± 0.301 | 0.02 | 1.099 |
| miR-532 | 0.352 ± 0.086 | 0.647 ± 0.593 | 0.02 | 0.877 |
| miR-631 | 105.937 ± 28.771 | 316.714 ± 131.871 | 0.01 | 1.580 |
| miR-654 | 0.386 ± 0.220 | 0.034 ± 0.019 | 0.01 | −3.509 |
| miR-671 | 0.023 ± 0.022 | 0.000 ± 0.000 | ≤0.01 | −8.618 |
| miR-769 | 1.896 ± 1.746 | 0.134 ± 0.112 | ≤0.01 | −3.824 |
| GenBank ID | Gene | Primer Sequence (5′–3′) Forward/Reverse | Amplicon Length (bp) | PCR Efficiency |
|---|---|---|---|---|
| XM_005196186.2 | Drosha ribonuclease type III (DROSHA) | AAGGCAGTGCATGTCACAGAA GCTGGGAGGTTCGTATTGGT | 170 | 94.35 |
| NM_203359.1 | Dicer ribonuclease type III (DICER1) | TCACGATCAACACGGCCATT TTGGGGGACCAACAATGGAG | 177 | 101.11 |
| NM_001205899.1 | Argonaute RISC catalytic component 1 (AGO1) | ATTGATGTCTCAGCCACTGCCT CTTGATCTCCTTGGTGAAGCGTAC | 140 | 91.92 |
| NM_205794.1 | Argonaute RISC catalytic component 2 (AGO2) | TTACAAGTCGGACAGGAGCAGA AGTCGCTCTGATCATGGTTGAG | 121 | 103.09 |
| NM_001001133.1 | Argonaute RISC catalytic component 3 (AGO3) | GGGCAGTTCAGGCAGGTATTA GTCTGTGTCCACCGTCGTT | 197 | 109.25 |
| XM_002686552.4 | Argonaute RISC catalytic component 4 (AGO4) | CATCAGTCTGTGAGACCTGCCAT TTGACACGCTGGGAGTCTGTTAG | 163 | 97.63 |
| XM_002697301.3 | Exportin 5 (XPO5) | AGGCTACATTGACTGGGTGC TCCAACTTGCCTTTCCTGCT | 150 | 94.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonella-Diaza, A.M.; Lopes, E.; Ribeiro da Silva, K.; Perecin Nociti, R.; Mamede Andrade, G.; Atuesta-Bustos, J.E.; Coelho da Silveira, J.; Vieira Meirelles, F.; Binelli, M. Steroidal Regulation of Oviductal microRNAs Is Associated with microRNA-Processing in Beef Cows. Int. J. Mol. Sci. 2021, 22, 953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020953
Gonella-Diaza AM, Lopes E, Ribeiro da Silva K, Perecin Nociti R, Mamede Andrade G, Atuesta-Bustos JE, Coelho da Silveira J, Vieira Meirelles F, Binelli M. Steroidal Regulation of Oviductal microRNAs Is Associated with microRNA-Processing in Beef Cows. International Journal of Molecular Sciences. 2021; 22(2):953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020953
Chicago/Turabian StyleGonella-Diaza, Angela Maria, Everton Lopes, Kauê Ribeiro da Silva, Ricardo Perecin Nociti, Gabriella Mamede Andrade, Jorge Eduardo Atuesta-Bustos, Juliano Coelho da Silveira, Flávio Vieira Meirelles, and Mario Binelli. 2021. "Steroidal Regulation of Oviductal microRNAs Is Associated with microRNA-Processing in Beef Cows" International Journal of Molecular Sciences 22, no. 2: 953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020953