
ATP1A1 Mutant in Aldosterone-Producing Adenoma Leads to Cell Proliferation

 , ,
, ,
Abstract
:1. Introduction
2. Results
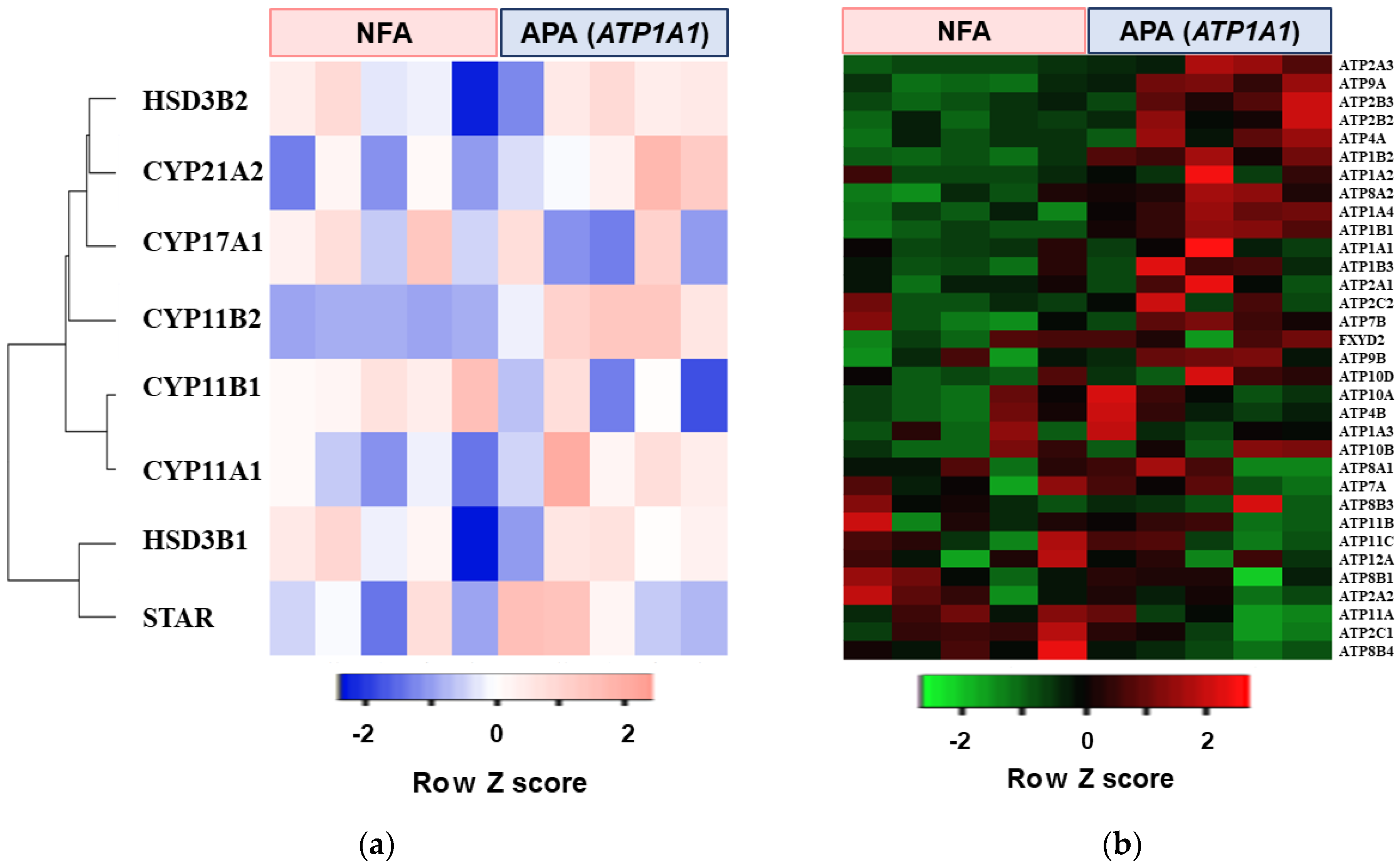
2.1. Transcriptome Analysis in APA with ATP1A1 Mutation
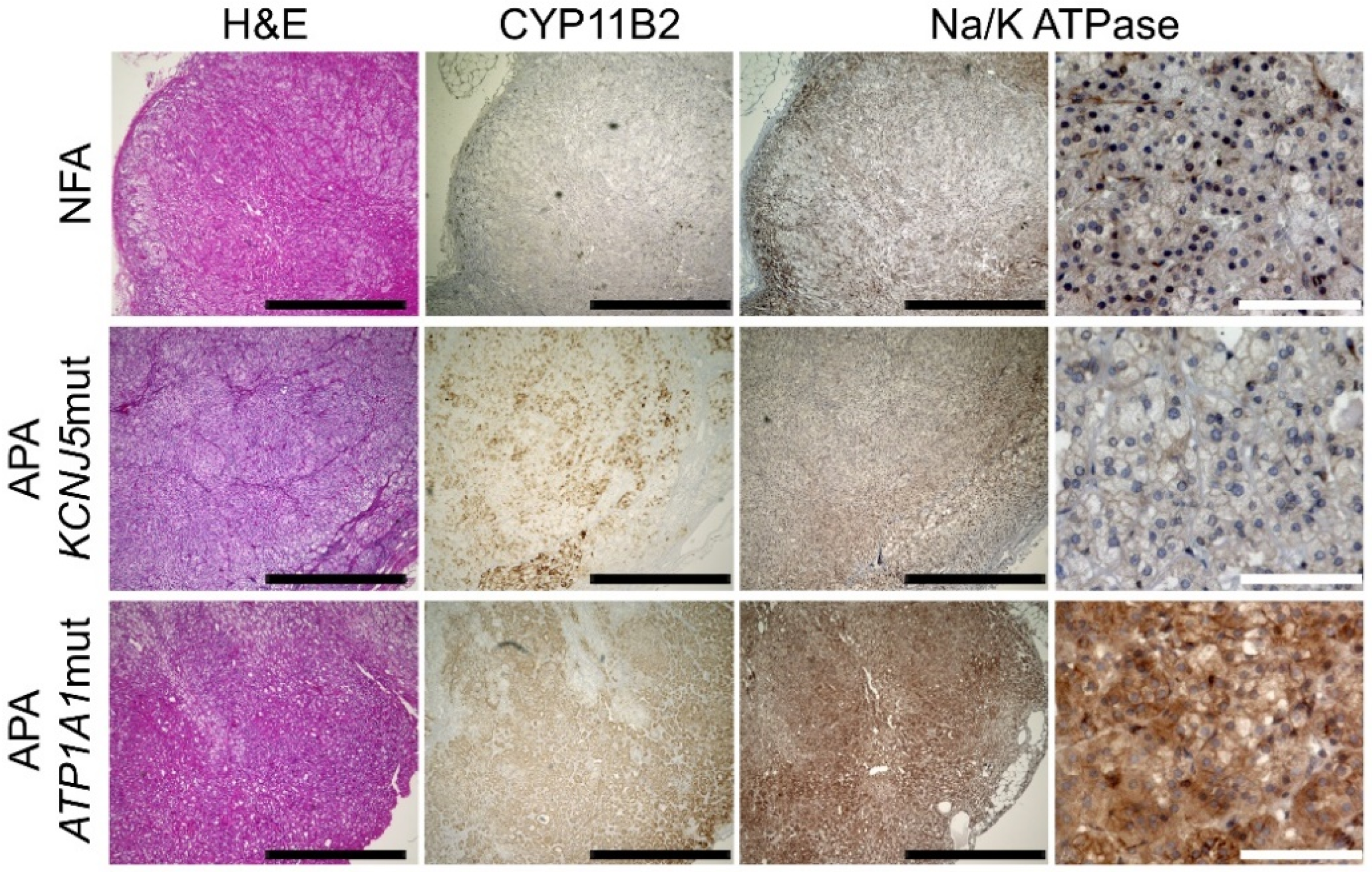
2.2. Immunohistochemical Analysis of NKA in APAs and NFAs
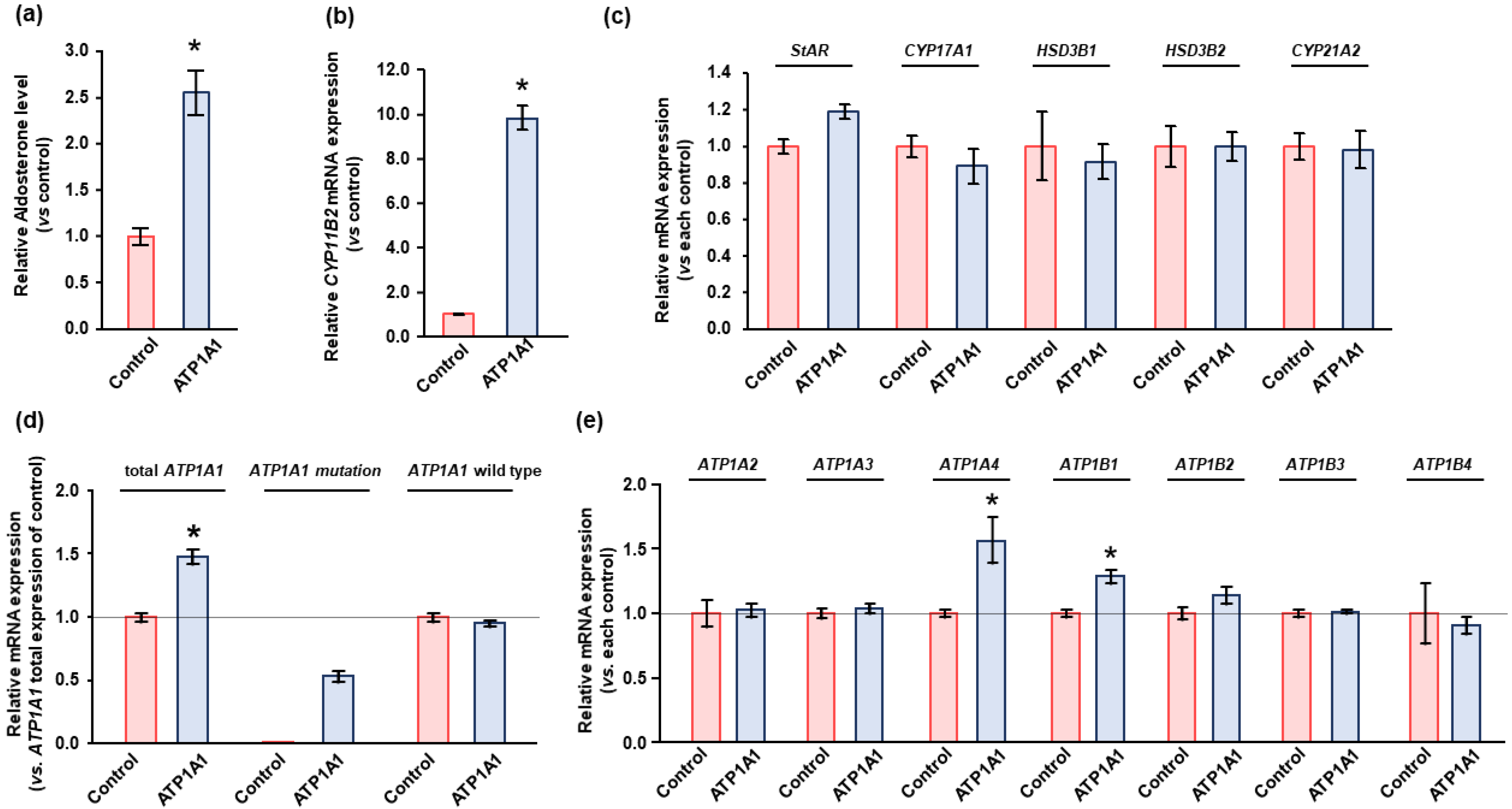
2.3. Effects of ATP1A1 Mutation on NKA Expression in Human Adrenocortical Carcinoma (HAC15) Cells
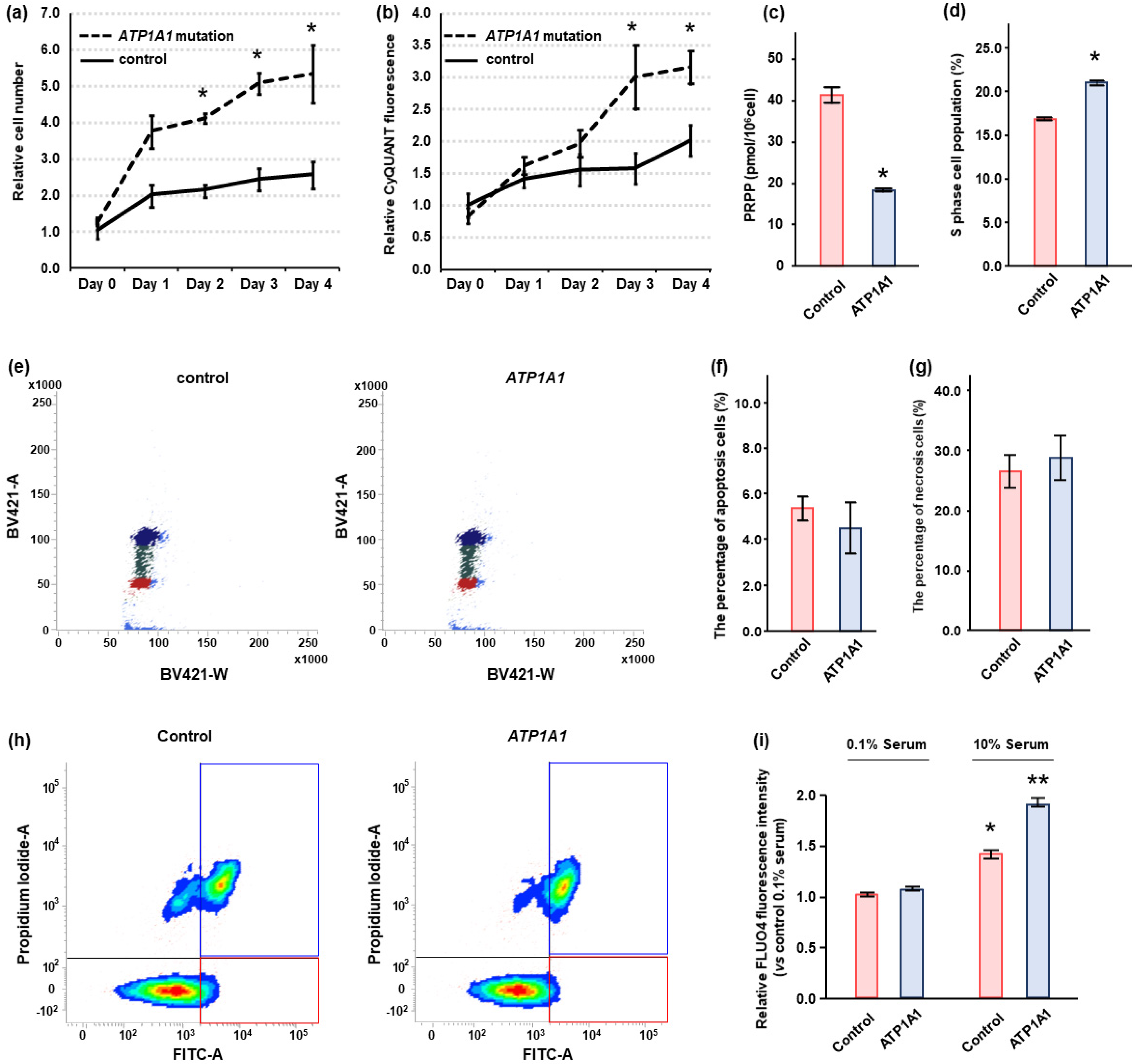
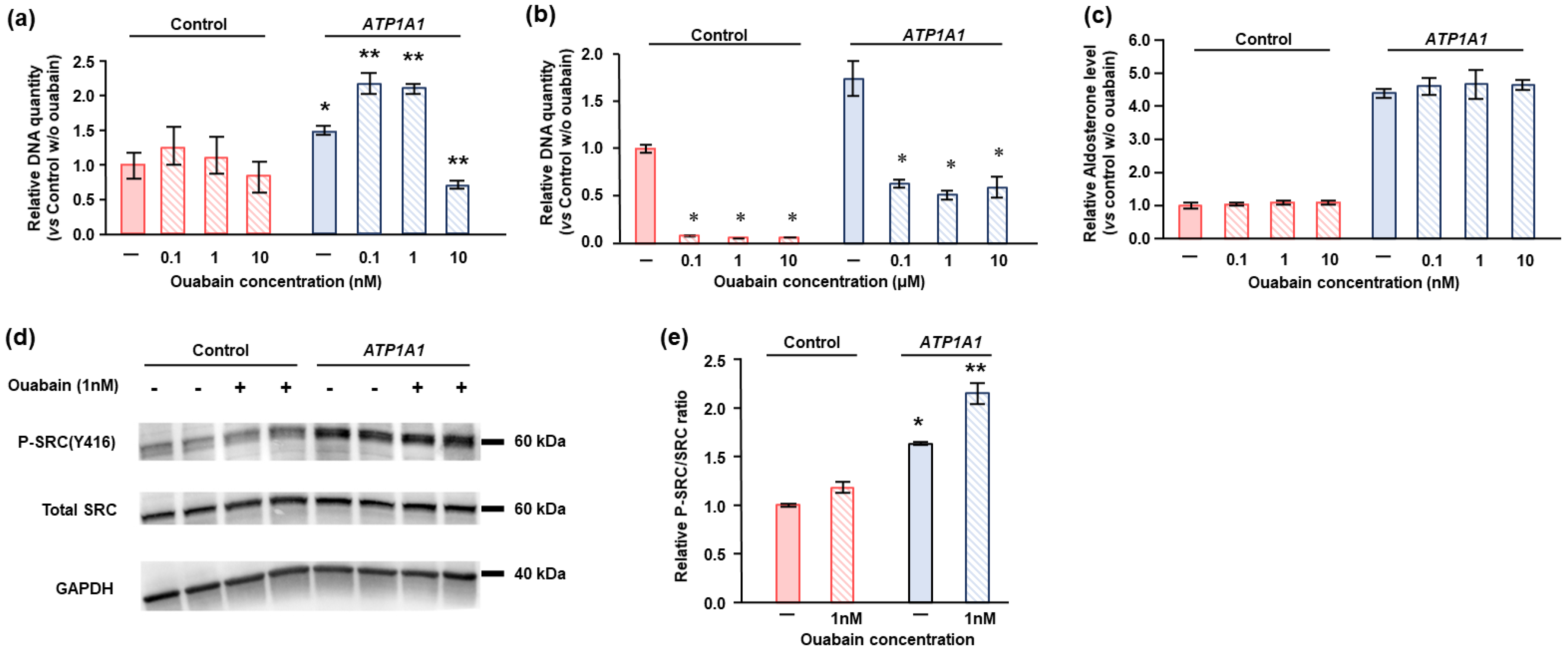
2.4. Effects of ATP1A1 Mutation on Cell Proliferation in HAC15 Cells
2.5. Effects of Ouabain in HAC15 Cells with ATP1A1 Mutation
3. Discussion
4. Materials and Methods
4.1. Adrenal Tissues
4.2. RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR) Assays
4.3. Cell Culture and Reagents
4.4. Lentiviral Production and Infection
4.5. Transcriptome Analysis
4.6. Immunohistochemical Analysis
4.7. Cell Proliferation Assay
4.8. Cell Cycle Assay
4.9. Metabolite Analysis
4.10. Intracellular Ca2+ Detection
4.11. Western Blotting
4.12. Aldosterone Measurement
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funder, J.W.; Carey, R.M.; Fardella, C.; Gomez-Sanchez, C.E.; Mantero, F.; Stowasser, M.; Young, W.F.; Montori, V.M.; Society, E. Case detection, diagnosis, and treatment of patients with primary aldosteronism: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2008, 93, 3266–3281. [Google Scholar] [CrossRef] [Green Version]
- Reincke, M.; Fischer, E.; Gerum, S.; Merkle, K.; Schulz, S.; Pallauf, A.; Quinkler, M.; Hanslik, G.; Lang, K.; Hahner, S.; et al. Observational study mortality in treated primary aldosteronism: The German Conn’s registry. Hypertension 2012, 60, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Savard, S.; Amar, L.; Plouin, P.F.; Steichen, O. Cardiovascular complications associated with primary aldosteronism: A controlled cross-sectional study. Hypertension 2013, 62, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F., Jr. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stölting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, T.; Maharjan, R.; Sven Willenberg, H.; Cupisti, K.; Ip, J.; Moser, A.; Stalberg, P.; Robinson, B.; Alexander Iwen, K.; Dralle, H.; et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 2016, 6, 19546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, K.; Gomez-Sanchez, C.E. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr. J. 2020, 67, 989–995. [Google Scholar] [CrossRef]
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Soderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41. [Google Scholar] [CrossRef]
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 2020, 75, 645–649. [Google Scholar] [CrossRef]
- Shimada, H.; Yamazaki, Y.; Sugawara, A.; Sasano, H.; Nakamura, Y. Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway. Biomedicines 2021, 9, 892. [Google Scholar] [CrossRef]
- Gong, S.; Tetti, M.; Reincke, M.; Williams, T.A. Primary Aldosteronism: Metabolic Reprogramming and the Pathogenesis of Aldosterone-Producing Adenomas. Cancers 2021, 13, 3716. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Nakagawa, K.; Li, D.; Kosaka, T.; Oya, M.; Mikami, S.; Shibata, H.; Itoh, H.; Mitani, F.; Yamazaki, T.; et al. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 2010, 95, 2296–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.A.; Gomez-Sanchez, C.E.; Rainey, W.E.; Giordano, T.J.; Lam, A.K.; Marker, A.; Mete, O.; Yamazaki, Y.; Zerbini, M.C.N.; Beuschlein, F.; et al. International Histopathology Consensus for Unilateral Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2021, 106, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Tomlins, S.A.; Kuick, R.; Cani, A.K.; Giordano, T.J.; Hovelson, D.H.; Liu, C.J.; Sanjanwala, A.R.; Edwards, M.A.; Gomez-Sanchez, C.E.; et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. USA 2015, 112, E4591–E4599. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.; Koga, M.; Seki, T.; Oki, K.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; Naruse, M.; Sakaguchi, T.; Morita, S.; Kosaka, T.; et al. Immunohistochemistry o.of aldosterone synthase leads the way to the pathogenesis of primary aldosteronism. Mol. Cell Endocrinol. 2017, 441, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Lalli, E.; Barhanin, J.; Zennaro, M.C.; Warth, R. Local Control of Aldosterone Production and Primary Aldosteronism. Trends Endocrinol. Metab. 2016, 27, 123–131. [Google Scholar] [CrossRef]
- Seccia, T.M.; Caroccia, B.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; Rossi, G.P. The Biology of Normal Zona Glomerulosa and Aldosterone-Producing Adenoma: Pathological Implications. Endocr. Rev. 2018, 39, 1029–1056. [Google Scholar] [CrossRef] [Green Version]
- Vouillarmet, J.; Fernandes-Rosa, F.; Graeppi-Dulac, J.; Lantelme, P.; Decaussin-Petrucci, M.; Thivolet, C.; Peix, J.L.; Boulkroun, S.; Clauser, E.; Zennaro, M.C. Aldosterone-Producing Adenoma With a Somatic KCNJ5 Mutation Revealing APC-Dependent Familial Adenomatous Polyposis. J. Clin. Endocrinol. Metab. 2016, 101, 3874–3878. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Chen, A.X.; Omata, K.; Vinco, M.; Giordano, T.J.; Else, T.; Hammer, G.D.; Tomlins, S.A.; Rainey, W.E. Molecular Heterogeneity in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. Mutations of the potassium channel KCNJ5 causing aldosterone-producing adenomas: One or two hits? Hypertension 2012, 59, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.J.; Anderson, H.C.; Palade, G.E.; Jamieson, J.D. Intracellular sorting and polarized cell surface delivery of (Na+,K+)ATPase, an endogenous component of MDCK cell basolateral plasma membranes. Cell 1986, 46, 623–631. [Google Scholar] [CrossRef]
- Bejcek, J.; Spiwok, V.; Kmonickova, E.; Rimpelova, S. Na(+)/K(+)-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules 2021, 26, 1905. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 Mutations in Aldosterone-Producing Adenomas. Hypertension 2014, 63, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Itcho, K.; Oki, K.; Ohno, H.; Yoneda, M. Update on Genetics of Primary Aldosteronism. Biomedicines 2021, 9, 409. [Google Scholar] [CrossRef]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef]
- Yudowski, G.A.; Efendiev, R.; Pedemonte, C.H.; Katz, A.I.; Berggren, P.O.; Bertorello, A.M. Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+, K+-ATPase alpha subunit and regulates its trafficking. Proc. Natl. Acad. Sci. USA 2000, 97, 6556–6561. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [Green Version]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticone, S.; Castellano, I.; Versace, K.; Lucatello, B.; Veglio, F.; Gomez-Sanchez, C.E.; Williams, T.A.; Mulatero, P. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol. Cell Endocrinol. 2015, 411, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. Immunohistochemistry of the adrenal in primary aldosteronism. Curr. Opin Endocrinol. Diabetes Obes. 2016, 23, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, T.; ter Meer, M.; Lenders, J.W.; Hermus, A.R.; Schultze Kool, L.; Langenhuijsen, J.F.; Nishimoto, K.; Ogishima, T.; Mukai, K.; Azizan, E.A.; et al. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J. Clin. Endocrinol. Metab. 2014, 99, E1341–E1351. [Google Scholar] [CrossRef] [Green Version]
- Omata, K.; Satoh, F.; Morimoto, R.; Ito, S.; Yamazaki, Y.; Nakamura, Y.; Anand, S.K.; Guo, Z.; Stowasser, M.; Sasano, H.; et al. Cellular and Genetic Causes of Idiopathic Hyperaldosteronism. Hypertension 2018, 72, 874–880. [Google Scholar] [CrossRef]
- Azizan, E.A.; Lam, B.Y.; Newhouse, S.J.; Zhou, J.; Kuc, R.E.; Clarke, J.; Happerfield, L.; Marker, A.; Hoffman, G.J.; Brown, M.J. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J. Clin. Endocrinol. Metab. 2012, 97, E819–E829. [Google Scholar] [CrossRef] [Green Version]
- Oki, K.; Plonczynski, M.W.; Luis Lam, M.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 2012, 153, 1774–1782. [Google Scholar] [CrossRef]
- Sanchez, G.; Nguyen, A.N.; Timmerberg, B.; Tash, J.S.; Blanco, G. The Na,K-ATPase alpha4 isoform from humans has distinct enzymatic properties and is important for sperm motility. Mol. Hum. Reprod. 2006, 12, 565–576. [Google Scholar] [CrossRef]
- Hammann, J.; Bassetti, D.; White, R.; Luhmann, H.J.; Kirischuk, S. alpha2 isoform of Na(+),K(+)-ATPase via Na(+),Ca(2+) exchanger modulates myelin basic protein synthesis in oligodendrocyte lineage cells in vitro. Cell Calcium 2018, 73, 1–10. [Google Scholar] [CrossRef]
- Pezzani, R.; Rubin, B.; Redaelli, M.; Radu, C.; Barollo, S.; Cicala, M.V.; Salvà, M.; Mian, C.; Mucignat-Caretta, C.; Simioni, P.; et al. The antiproliferative effects of ouabain and everolimus on adrenocortical tumor cells. Endocr. J. 2014, 61, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.F.; Amaral, L.S.; Porto, C.S.; Quintas, L.E. Na+/K+-ATPase α1 isoform mediates ouabain-induced expression of cyclin D1 and proliferation of rat sertoli cells. Reproduction 2012, 144, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Tverskoi, A.M.; Sidorenko, S.V.; Klimanova, E.A.; Akimova, O.A.; Smolyaninova, L.V.; Lopina, O.D.; Orlov, S.N. Effects of Ouabain on Proliferation of Human Endothelial Cells Correlate with Na+,K+-ATPase Activity and Intracellular Ratio of Na+ and K. Biochemistry 2016, 81, 876–883. [Google Scholar] [CrossRef]
- Ren, Y.P.; Zhang, M.J.; Zhang, T.; Huang, R.W. Dual effects of ouabain on the regulation of proliferation and apoptosis in human umbilical vein endothelial cells: Involvement of Na(+)-K(+)-ATPase alpha-subunits and NF-kappaB. Int. J. Clin. Exp. Med. 2014, 7, 1214–1222. [Google Scholar]
- Botelho, A.F.M.; Pierezan, F.; Soto-Blanco, B.; Melo, M.M. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon 2019, 158, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.A.; Whitaker, J.M.; Buckalew, V.M., Jr. Endogenous digitalis-like substance in plasma of volume-expanded dogs. Nature 1980, 287, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, F.K.; Dube, P.; Mohamed, A.; Tian, J.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na(+)/K(+)-ATPase. Int. J. Mol. Sci. 2018, 19, 2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baecher, S.; Kroiss, M.; Fassnacht, M.; Vogeser, M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin. Chim Acta 2014, 431, 87–92. [Google Scholar] [CrossRef]
- Lewis, L.K.; Yandle, T.G.; Hilton, P.J.; Jensen, B.P.; Begg, E.J.; Nicholls, M.G. Endogenous ouabain is not ouabain. Hypertension 2014, 64, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Manunta, P.; Rogowski, A.C.; Hamilton, B.P.; Hamlyn, J.M. Ouabain-induced hypertension in the rat: Relationships among plasma and tissue ouabain and blood pressure. J. Hypertens 1994, 12, 549–560. [Google Scholar] [CrossRef]
- Neri, G.; De Toni, R.; Tortorella, C.; Rebuffat, P.; Bova, S.; Cargnelli, G.; Petrelli, L.; Spinazzi, R.; Nussdorfer, G.G. Ouabain chronic infusion enhances the growth and steroidogenic capacity of rat adrenal zona glomerulosa: The possible involvement of the endothelin system. Int. J. Mol. Med. 2006, 18, 315–319. [Google Scholar] [PubMed] [Green Version]
- Nanba, K.; Vaidya, A.; Williams, G.H.; Zheng, I.; Else, T.; Rainey, W.E. Age-Related Autonomous Aldosteronism. Circulation 2017, 136, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Zwermann, O.; Suttmann, Y.; Bidlingmaier, M.; Beuschlein, F.; Reincke, M. Screening for membrane hormone receptor expression in primary aldosteronism. Eur. J. Endocrinol. 2009, 160, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Itcho, K.; Oki, K.; Kobuke, K.; Yoshii, Y.; Ohno, H.; Yoneda, M.; Hattori, N. Aberrant G protein-receptor expression is associated with DNA methylation in aldosterone-producing adenoma. Mol. Cell Endocrinol. 2018, 461, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stindl, J.; Tauber, P.; Sterner, C.; Tegtmeier, I.; Warth, R.; Bandulik, S. Pathogenesis of Adrenal Aldosterone-Producing Adenomas Carrying Mutations of the Na(+)/K(+)-ATPase. Endocrinology 2015, 156, 4582–4591. [Google Scholar] [CrossRef]
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATPase-activated Wnt/beta-catenin signalling. Nat. Cell Biol. 2018, 20, 1421–1433. [Google Scholar] [CrossRef]
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Calneuron 1 Increased Ca(2+) in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma. Hypertension 2018, 71, 125–133. [Google Scholar] [CrossRef]
- Gomez-Sanchez, C.E.; Qi, X.; Velarde-Miranda, C.; Plonczynski, M.W.; Parker, C.R.; Rainey, W.; Satoh, F.; Maekawa, T.; Nakamura, Y.; Sasano, H.; et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol. Cell Endocrinol. 2014, 383, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, R.; Oki, K.; Yoneda, M.; Gomez-Sanchez, C.E.; Ohno, H.; Kobuke, K.; Itcho, K.; Kohno, N. Gonadotropin-Releasing Hormone Stimulate Aldosterone Production in a Subset of Aldosterone-Producing Adenoma. Medicine 2016, 95, e3659. [Google Scholar] [CrossRef] [PubMed]
- Itcho, K.; Oki, K.; Kobuke, K.; Ohno, H.; Yoneda, M.; Hattori, N. Angiotensin 1-7 suppresses angiotensin II mediated aldosterone production via JAK/STAT signaling inhibition. J. Steroid. Biochem. Mol. Biol. 2019, 185, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Itcho, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Kobuke, K.; Nagano, G.; Yoshii, Y.; Baba, R.; Hattori, N.; et al. Endoplasmic Reticulum Chaperone Calmegin Is Upregulated in Aldosterone-Producing Adenoma and Associates With Aldosterone Production. Hypertension 2020, 75, 492–499. [Google Scholar] [CrossRef]
- Hasan, A.; Pokeza, N.; Shaw, L.; Lee, H.S.; Lazzaro, D.; Chintala, H.; Rosenbaum, D.; Grant, M.B.; Chaqour, B. The matricellular protein cysteine-rich protein 61 (CCN1/Cyr61) enhances physiological adaptation of retinal vessels and reduces pathological neovascularization associated with ischemic retinopathy. J. Biol. Chem. 2011, 286, 9542–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2010, 6, 78–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, K.; Plonczynski, M.W.; Lam, M.L.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. The potassium channel, Kir3.4 participates in angiotensin II-stimulated aldosterone production by a human adrenocortical cell line. Endocrinology 2012, 153, 4328–4335. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ranking | Pathway Increased in APA with ATP1A1 Mutation |
|---|---|
| 1 | Cancer Head and Neck vs. Cervical Down |
| 2 | Ion Transport by P Type ATPase |
| 3 | Proximal Tubule Bicarbonate Reclamation |
| 4 | Kidney |
| 5 | LG1 Targets Up |
| Gene Symbol | Fold Increase (vs. NFA) | p Value |
|---|---|---|
| ATP2A3 | 5.12 | 0.036 |
| ATP9A | 4.34 | 0.004 |
| ATP2B3 | 3.84 | 0.040 |
| ATP2B2 | 3.71 | 0.049 |
| ATP4A | 3.43 | 0.059 |
| ATP1B2 | 3.17 | 0.001 |
| ATP1A2 | 2.90 | n.s |
| ATP8A2 | 2.82 | 0.012 |
| ATP1A4 | 2.13 | 0.001 |
| ATP1B1 | 2.01 | 0.001 |
| ATP1A1 | 1.86 | n.s |
| ATP1B3 | 1.67 | n.s |
| ATP2A1 | 1.43 | n.s |
| ATP2C2 | 1.35 | n.s |
| ATP7B | 1.33 | n.s |
| FXYD2 | 1.32 | n.s |
| ATP9B | 1.24 | n.s |
| ATP10D | 1.22 | n.s |
| ATP10A | 1.10 | n.s |
| ATP4B | 1.10 | n.s |
| ATP1A3 | 1.10 | n.s |
| ATP10B | 1.05 | n.s |
| ATP8A1 | 1.03 | n.s |
| ATP7A | 0.98 | n.s |
| ATP8B3 | 0.93 | n.s |
| ATP11B | 0.90 | n.s |
| ATP11C | 0.90 | n.s |
| ATP12A | 0.87 | n.s |
| ATP8B1 | 0.87 | n.s |
| ATP2A2 | 0.79 | n.s |
| ATP11A | 0.79 | n.s |
| ATP2C1 | 0.75 | n.s |
| ATP8B4 | 0.41 | n.s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Itcho, K.; Ohno, H.; Nagano, G.; Yoshii, Y.; Baba, R.; Kodama, T.; et al. ATP1A1 Mutant in Aldosterone-Producing Adenoma Leads to Cell Proliferation. Int. J. Mol. Sci. 2021, 22, 10981. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010981
Kobuke K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Itcho K, Ohno H, Nagano G, Yoshii Y, Baba R, Kodama T, et al. ATP1A1 Mutant in Aldosterone-Producing Adenoma Leads to Cell Proliferation. International Journal of Molecular Sciences. 2021; 22(20):10981. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010981
Chicago/Turabian StyleKobuke, Kazuhiro, Kenji Oki, Celso E. Gomez-Sanchez, Elise P. Gomez-Sanchez, Kiyotaka Itcho, Haruya Ohno, Gaku Nagano, Yoko Yoshii, Ryuta Baba, Takaya Kodama, and et al. 2021. "ATP1A1 Mutant in Aldosterone-Producing Adenoma Leads to Cell Proliferation" International Journal of Molecular Sciences 22, no. 20: 10981. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010981