
Development of a Multiplex Immunohistochemistry Workflow to Investigate the Immune Microenvironment in Mouse Models of Inflammatory Bowel Disease and Colon Cancer


Abstract
:1. Introduction
2. Results
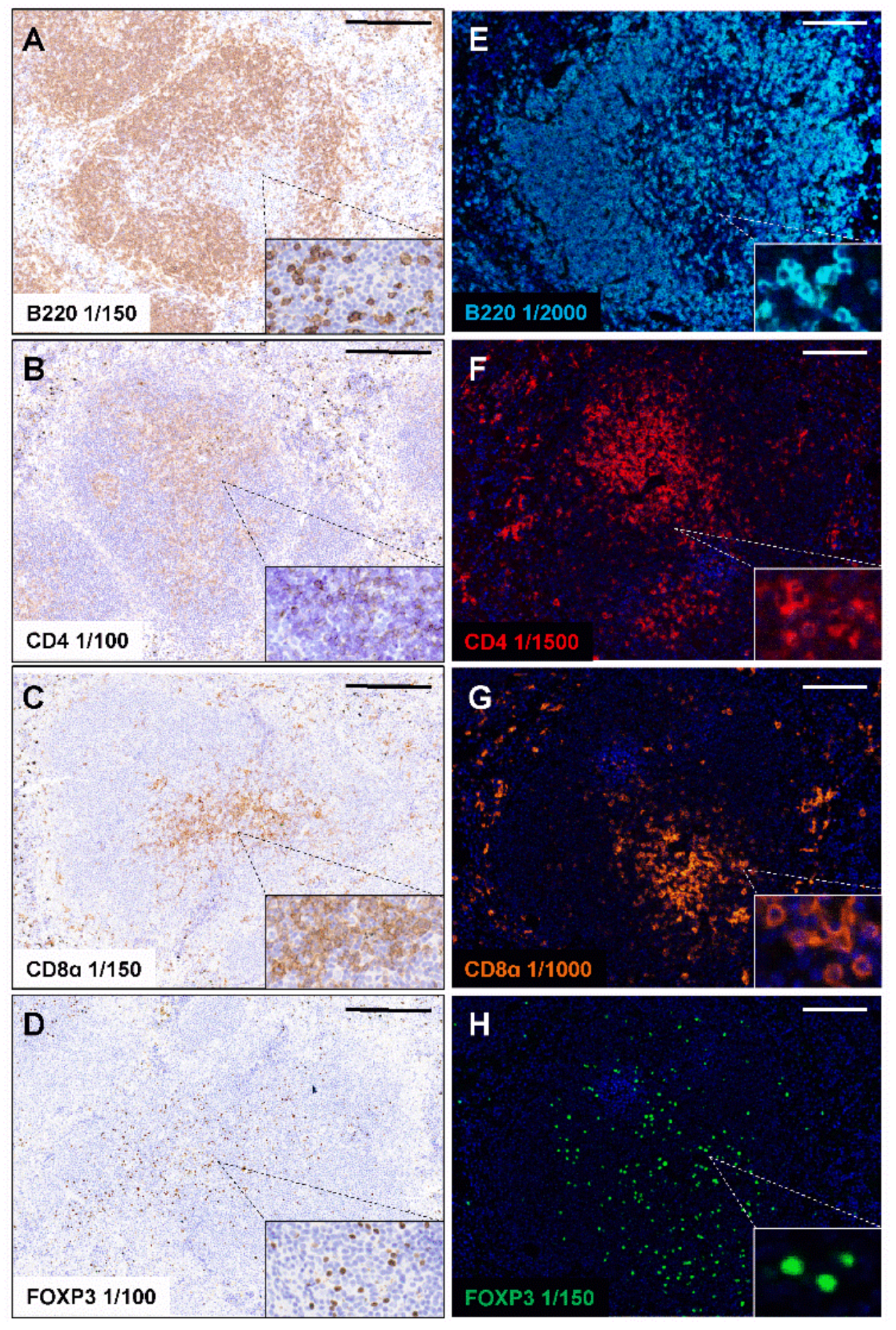
2.1. Standard Chromogenic IHC and Opal Monoplex Assay Development
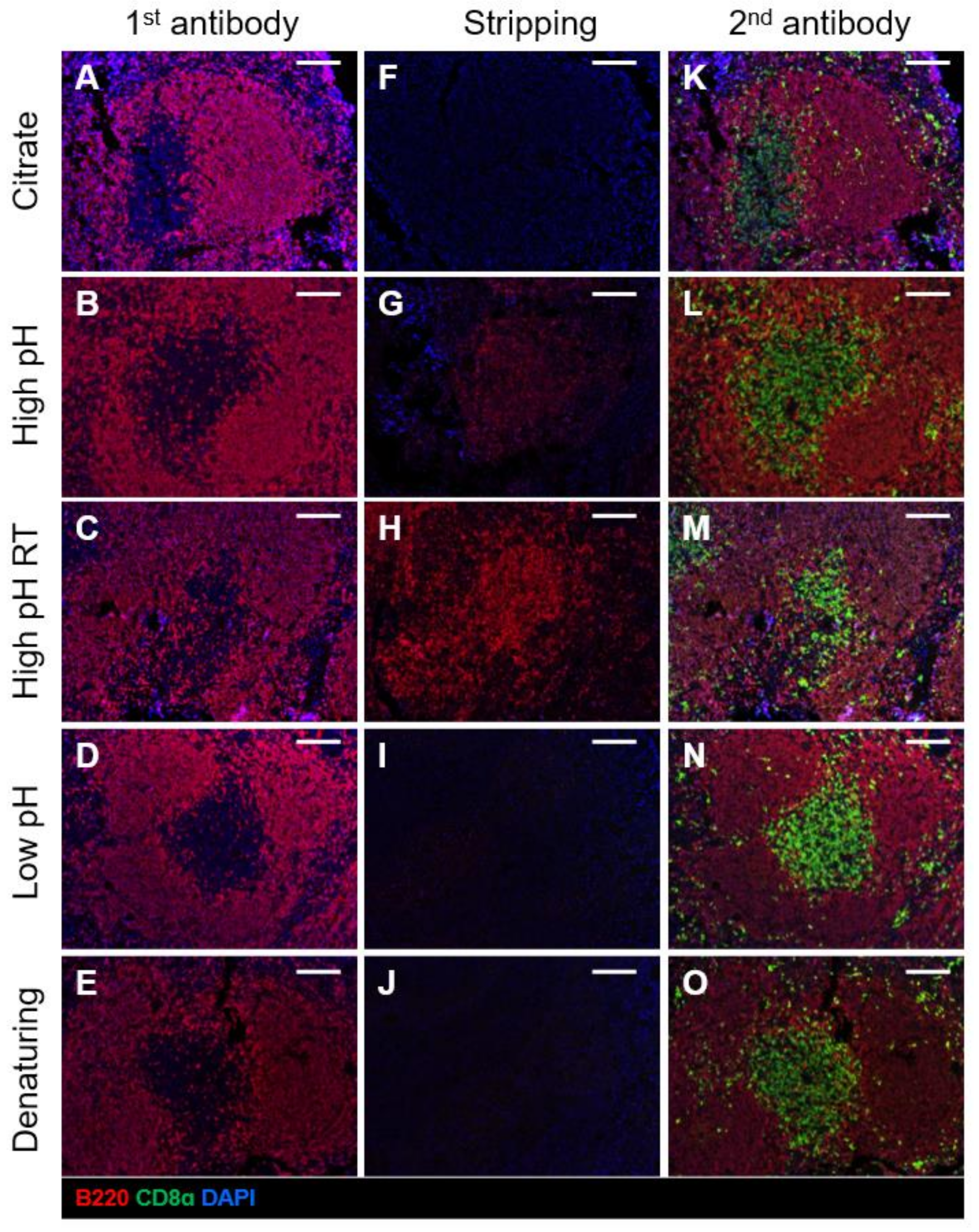
2.2. Comparison of Different Antibody Stripping Methods
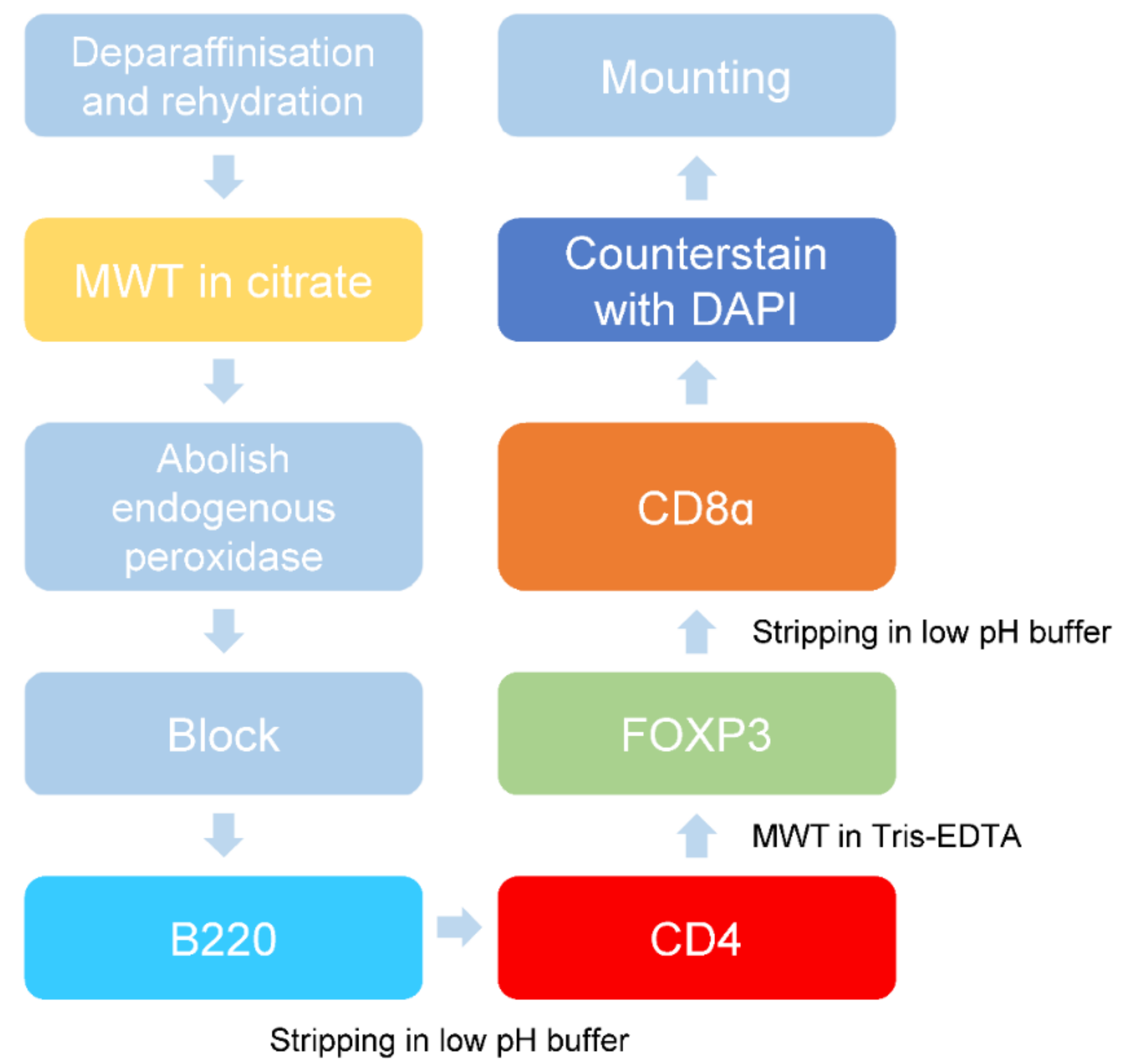
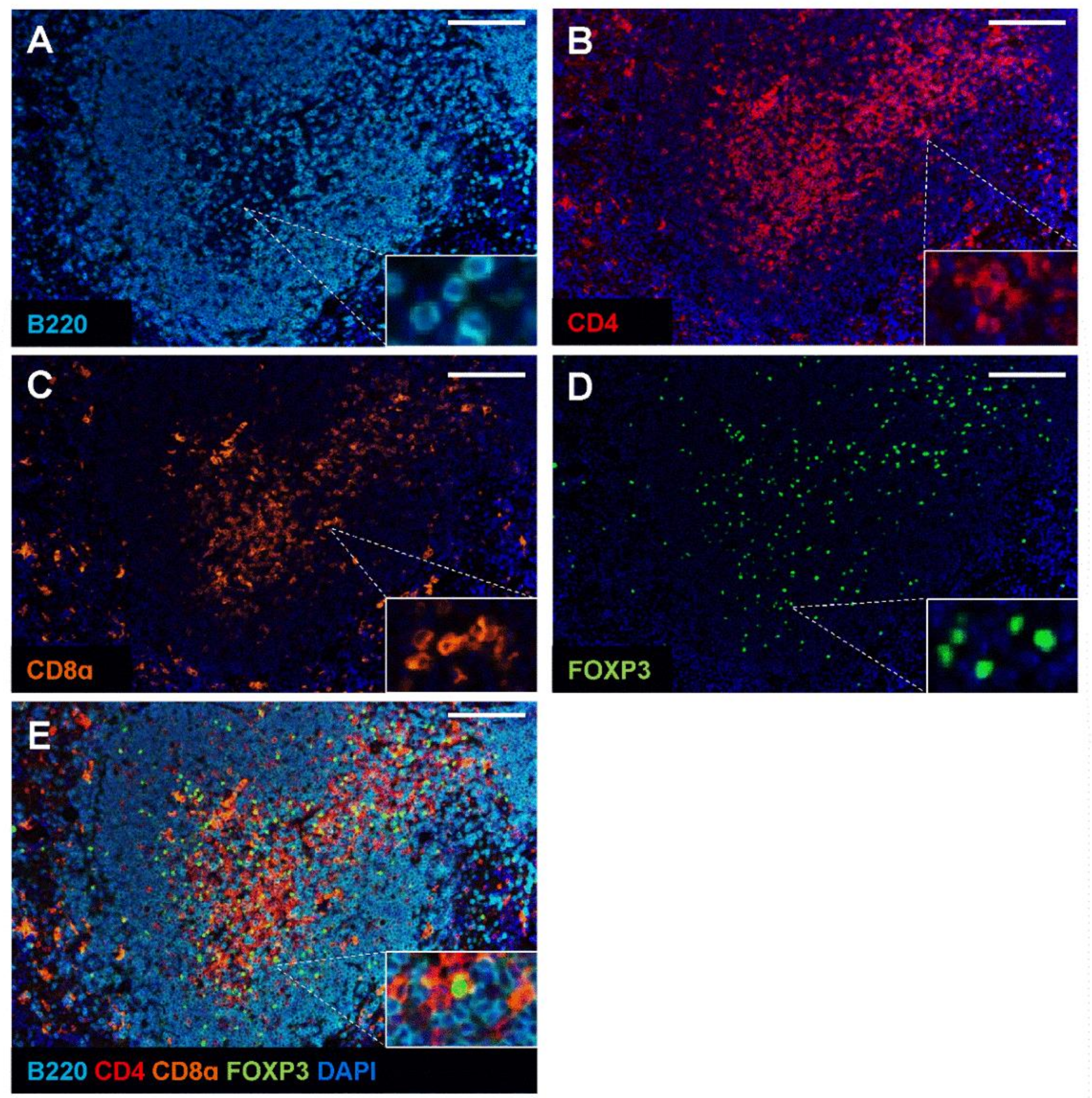
2.3. Validation of Multiplex Staining Protocol on Mouse Spleen and Different Disease Models
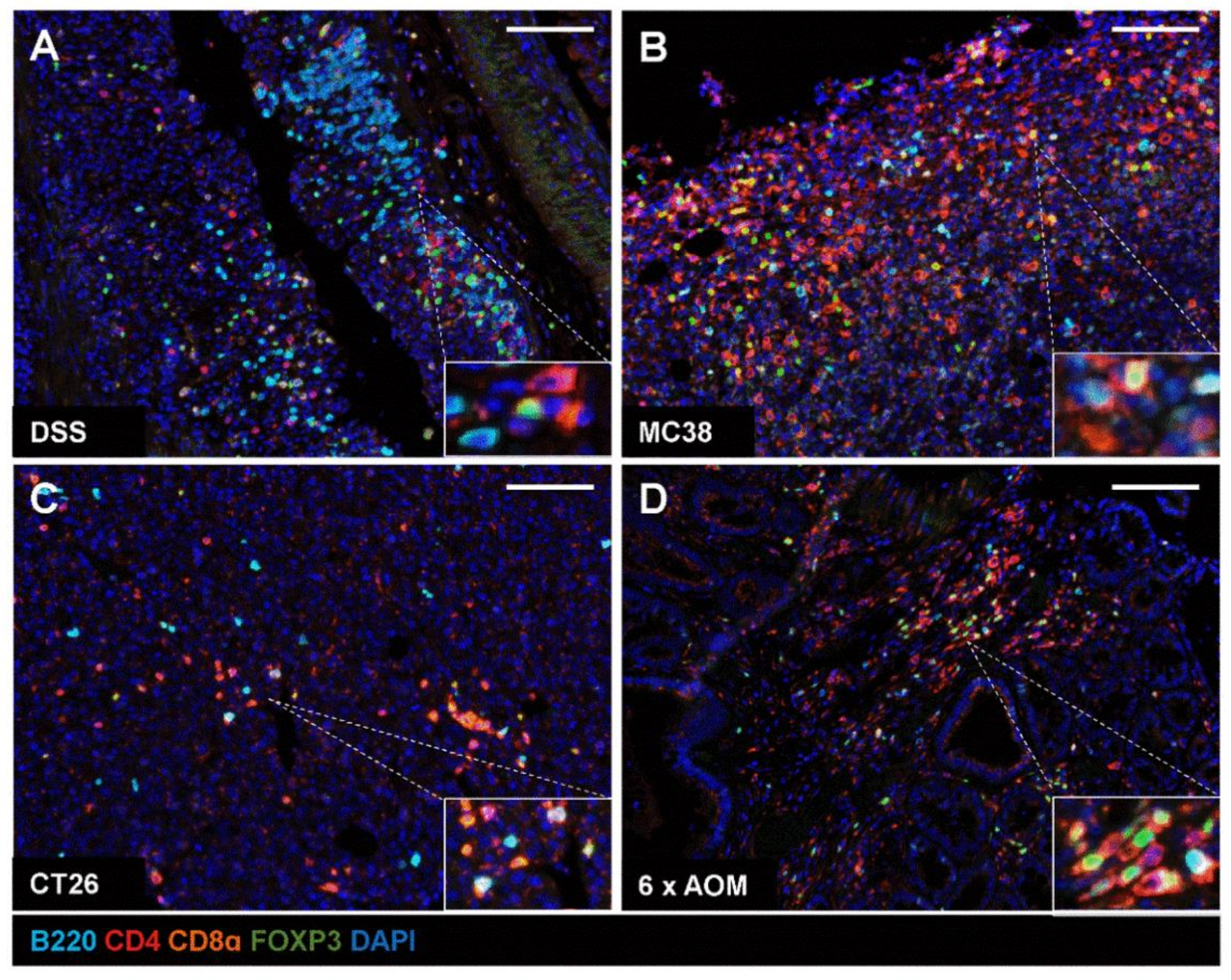
2.4. Application of mIHC Panel in Mouse Models of IBD and Colon Tumour Models
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. DSS-Induced Colitis
4.3. Tumour Cell Lines
4.4. Tissue Fixation
4.5. Chromogenic Immunohistochemistry and Opal Monoplex Assay Development
4.6. Antibody Stripping
4.7. mIHC Staining
4.8. Image Acquisition and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef]
- Pivetta, E.; Capuano, A.; Scanziani, E.; Minoli, L.; Andreuzzi, E.; Mongiat, M.; Baldassarre, G.; Doliana, R.; Spessotto, P. Multiplex staining depicts the immune infiltrate in colitis-induced colon cancer model. Sci. Rep. 2019, 9, 12645. [Google Scholar] [CrossRef]
- Tóth, Z.E.; Mezey, É. Simultaneous Visualization of Multiple Antigens with Tyramide Signal Amplification using Antibodies from the same Species. J. Histochem. Cytochem. 2007, 55, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, J.; Akiska, Y.; Perusina Lanfranca, M.; Delrosario, L.; Sun, L.; Long, D.; Shi, J.; Crawford, H.; Di Magliano, M.P.; Zou, W.; et al. Optimization, Design and Avoiding Pitfalls in Manual Multiplex Fluorescent Immunohistochemistry. J. Vis. Exp. 2019, 10, e59915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, E.R.; Uraoka, N.; Jiang, M.; Cook, P.; Gibbons, D.; Forget, M.-A.; Bernatchez, C.; Haymaker, C.; Wistuba, I.I.; Rodriguez-Canales, J. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci. Rep. 2017, 7, 13380. [Google Scholar] [CrossRef] [Green Version]
- Gorris, M.A.J.; Halilovic, A.; Rabold, K.; van Duffelen, A.; Wickramasinghe, I.N.; Verweij, D.; Wortel, I.M.N.; Textor, J.C.; de Vries, I.J.M.; Figdor, C.G. Eight-Color Multiplex Immunohistochemistry for Simultaneous Detection of Multiple Immune Checkpoint Molecules within the Tumor Microenvironment. J. Immunol. 2018, 200, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Bolen, J.; Schuetter, L.; Massion, P.; Hoyt, C.C.; VandenBerg, S.; Esserman, L.; Borowsky, A.D.; Campbell, M.J. Characterizing the Tumor Immune Microenvironment with Tyramide-Based Multiplex Immunofluorescence. J. Mammary Gland. Biol. Neoplasia 2020, 25, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.T.; Yeong, J.P.S.; Lim, C.J.; Ong, C.C.H.; Wong, S.C.; Chew, V.S.P.; Ahmed, S.S.; Tan, P.H.; Iqbal, J. An automated staining protocol for seven-colour immunofluorescence of human tissue sections for diagnostic and prognostic use. Pathology 2018, 50, 333–341. [Google Scholar] [CrossRef]
- Mauldin, I.S.; Sheybani, N.D.; Young, S.J.; Price, R.J.; Slingluff, C.L. Deconvolution of the immunological contexture of mouse tumors with multiplexed immunohistochemistry. Methods Enzymol. 2020, 635, 81–93. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- McIntyre, R.E.; Buczacki, S.J.A.; Arends, M.J.; Adams, D.J. Mouse models of colorectal cancer as preclinical models. Bioessays 2015, 37, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, A.J.; Bialkowska, A.B.; Ghaleb, A.M.; Yang, V.W.; Obeid, L.M.; Hannun, Y.A. Murine Model for Colitis-Associated Cancer of the Colon. In Mouse Models for Drug Discovery: Methods and Protocols; Proetzel, G., Wiles, M.V., Eds.; Springer: New York, NY, USA, 2016; pp. 245–254. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirilovsky, A.; Marliot, F.; El Sissy, C.; Haicheur, N.; Galon, J.; Pagès, F. Rational bases for the use of the Immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. Int. Immunol. 2016, 28, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Jensen, S.M.; Messenheimer, D.J.; Farhad, M.; Neuberger, M.; Bifulco, C.B.; Fox, B.A. Multispectral Imaging of T and B Cells in Murine Spleen and Tumor. J. Immunol. 2016, 196, 3943–3950. [Google Scholar] [CrossRef] [Green Version]
- Beckstead, J.H. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J. Histochem. Cytochem. 1994, 42, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, M.; Krebbers, G.; Bekkenk, M.W.; Teunissen, M.B.M.; Luiten, R.M. Improvement of Opal Multiplex Immunofluorescence Workflow for Human Tissue Sections. J. Histochem. Cytochem. 2021, 69, 339–346. [Google Scholar] [CrossRef]
- Eckhard, A.H.; O’Malley, J.T.; Nadol, J.B., Jr.; Adams, J.C. Mechanical Compression of Coverslipped Tissue Sections during Heat-Induced Antigen Retrieval Prevents Section Detachment and Preserves Tissue Morphology. J. Histochem. Cytochem. 2019, 67, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Wester, K.; Asplund, A.; Bäckvall, H.; Micke, P.; Derveniece, A.; Hartmane, I.; Malmström, P.-U.; Pontén, F. Zinc-Based Fixative Improves Preservation of Genomic DNA and Proteins in Histoprocessing of Human Tissues. Lab. Investig. 2003, 83, 889–899. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Soonsawad, P.; Schuetter, L.; Chen, Q.; Hubbard, N.E.; Cardiff, R.D.; Borowsky, A.D. Introduction of Zinc-salt Fixation for Effective Detection of Immune Cell-related Markers by Immunohistochemistry. Toxicol. Pathol. 2015, 43, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Sorrelle, N.; Ganguly, D.; Dominguez, A.T.A.; Zhang, Y.; Huang, H.; Dahal, L.N.; Burton, N.; Ziemys, A.; Brekken, R.A. Improved Multiplex Immunohistochemistry for Immune Microenvironment Evaluation of Mouse Formalin-Fixed, Paraffin-Embedded Tissues. J. Immunol. 2019, 202, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Paavilainen, L.; Edvinsson, A.; Asplund, A.; Hober, S.; Kampf, C.; Pontén, F.; Wester, K. The impact of tissue fixatives on morphology and antibody-based protein profiling in tissues and cells. J. Histochem. Cytochem. 2010, 58, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.Y.; Song, J.S.; Ylaya, K.; Sears, J.D.; Choi, L.; Cho, H.; Rosenberg, A.Z.; Hewitt, S.M. Histomorphological and Molecular Assessments of the Fixation Times Comparing Formalin and Ethanol-Based Fixatives. J. Histochem. Cytochem. 2018, 66, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, L.; Huynh, J.; Alorro, M.G.; Li, X.; Ernst, M.; Chand, A.L. STAT3 Signalling via the IL-6ST/gp130 Cytokine Receptor Promotes Epithelial Integrity and Intestinal Barrier Function during DSS-Induced Colitis. Biomedicines 2021, 9, 187. [Google Scholar] [CrossRef]
- Shi, S.R.; Imam, S.A.; Young, L.; Cote, R.J.; Taylor, C.R. Antigen retrieval immunohistochemistry under the influence of pH using monoclonal antibodies. J. Histochem. Cytochem. 1995, 43, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Faget, L.; Hnasko, T.S. Tyramide Signal Amplification for Immunofluorescent Enhancement. In ELISA: Methods and Protocols; Hnasko, R., Ed.; Springer: New York, NY, USA, 2015; pp. 161–172. [Google Scholar] [CrossRef]
- Pirici, D.; Mogoanta, L.; Kumar-Singh, S.; Pirici, I.; Margaritescu, C.; Simionescu, C.; Stanescu, R. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J. Histochem. Cytochem. 2009, 57, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenberg, A.J.; Morales, D.O.; Piergies, A.M.H.; Li, S.H.; Tejedor, J.S.; Mladinov, M.; Mulder, J.; Grinberg, L.T. A manual multiplex immunofluorescence method for investigating neurodegenerative diseases. J. Neurosci. Methods 2020, 339, 108708. [Google Scholar] [CrossRef]
- Syed, J.; Ashton, J.; Joseph, J.; Jones, G.N.; Slater, C.; Sharpe, A.; Ashton, G.; Howat, W.; Byers, R.; Angell, H.K. Multiplex immunohistochemistry: The importance of staining order when producing a validated protocol. Immunotherapy 2019, 5, 157. [Google Scholar]
- Halse, H.; Colebatch, A.J.; Petrone, P.; Henderson, M.A.; Mills, J.K.; Snow, H.; Westwood, J.A.; Sandhu, S.; Raleigh, J.M.; Behren, A.; et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci. Rep. 2018, 8, 11158. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Nyberg, R.; Wu, Y.; Bernard, B.; Redmond, W.L. Developing an enhanced 7-color multiplex IHC protocol to dissect immune infiltration in human cancers. PLoS ONE 2021, 16, e0247238. [Google Scholar] [CrossRef]
- Rehg, J.E.; Bush, D.; Ward, J.M. The Utility of Immunohistochemistry for the Identification of Hematopoietic and Lymphoid Cells in Normal Tissues and Interpretation of Proliferative and Inflammatory Lesions of Mice and Rats. Toxicol. Pathol. 2012, 40, 345–374. [Google Scholar] [CrossRef]
- Steinert, E.M.; Schenkel, J.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyártó, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Hubbard, A.; Jones, T.; Racolta, A.; Bhaumik, S.; Cummins, N.; Zhang, L.; Garsha, K.; Ventura, F.; Lefever, M.R.; et al. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal amplification and same species antibodies. Lab. Invest. 2017, 97, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Viratham Pulsawatdi, A.; Craig, S.G.; Bingham, V.; McCombe, K.; Humphries, M.P.; Senevirathne, S.; Richman, S.D.; Quirke, P.; Campo, L.; Domingo, E.; et al. A robust multiplex immunofluorescence and digital pathology workflow for the characterisation of the tumour immune microenvironment. Mol. Oncol. 2020, 14, 2384–2402. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.J.; Wilkinson, D.J.; Rankin, D.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Atherton, P.J. An overview of technical considerations for Western blotting applications to physiological research. Scand. J. Med. Sci. Sports 2017, 27, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Hasui, K.; Takatsuka, T.; Sakamoto, R.; Matsushita, S.; Tsuyama, S.; Izumo, S.; Murata, F. Double autoimmunostaining with glycine treatment. J. Histochem. Cytochem. 2003, 51, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Haughey, C.M.; Mukherjee, D.; Steele, R.E.; Popple, A.; Dura-Perez, L.; Pickard, A.; Patel, M.; Jain, S.; Mullan, P.B.; Williams, R.; et al. Investigating Radiotherapy Response in a Novel Syngeneic Model of Prostate Cancer. Cancers 2020, 12, 2804. [Google Scholar] [CrossRef]
- Huynh, J.; Baloyan, D.; Chisanga, D.; Shi, W.; Brien, M.; Afshar-Sterle, S.; Alorro, M.; Pang, L.; Williams, D.S.; Parslow, A.C.; et al. Host IL11 Signaling Suppresses CD4+ T cell–Mediated Antitumor Responses to Colon Cancer in Mice. Cancer Immunol. Res. 2021, 9, 735. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Buffer | Incubation Time |
|---|---|---|
| Microwave | 10 mM citrate, pH 6.0 | 1 min on 100% power (till boiling point is reached), followed by 7.5 min on 10% power |
| High pH | 100 mM glycine NaOH, pH 10.0 | 50 °C 30 min or RT 15 min |
| Low pH | 50 mM glycine HCl, pH 2.2 | 50 °C 30 min |
| Denaturing | 25 mM glycine HCl, 10% SDS, pH 2.0 | 50 °C 30 min |
| Staining Order | Antigen Retrieval | Primary Antibody | Secondary Antibody | Opal Fluorophore | Stripping |
|---|---|---|---|---|---|
| 1 | Citrate pH 6.0 | B220 (1/2000) 1 h RT | Anti-rat secondary HRP 10 min | 690 10 min | Low pH glycine buffer 30 min, 50 °C |
| 2 | - | CD4 (1/1000) Overnight 4 °C | Anti-rat secondary HRP 10 min | 620 10 min | - |
| 3 | Tris-EDTA pH 9.0 | Foxp3 (1/150) 1 h RT | Anti-rat secondary HRP 10 min | 520 10 min | Low pH glycine buffer 30 min, 50 °C |
| 4 | - | CD8α (1/1000) Overnight 4 °C | Anti-rat secondary HRP 10 min | 570 10 min | - |
| Antibody | Supplier | Catalogue No. | Species | IHC Dilution | mIHC Dilution |
|---|---|---|---|---|---|
| B220 | BD Pharmingen | 550286 | Rat | 1/150 | 1/2000 |
| CD4 | eBioscience | 14-9766-82 | Rat | 1/100 | 1/1500 |
| CD8α | eBioscience | 14-0808 | Rat | 1/150 | 1/1000 |
| FOXP3 | eBioscience | 14-5773-80 | Rat | 1/100 | 1/150 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Ernst, M.; Huynh, J. Development of a Multiplex Immunohistochemistry Workflow to Investigate the Immune Microenvironment in Mouse Models of Inflammatory Bowel Disease and Colon Cancer. Int. J. Mol. Sci. 2021, 22, 11001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011001
Pang L, Ernst M, Huynh J. Development of a Multiplex Immunohistochemistry Workflow to Investigate the Immune Microenvironment in Mouse Models of Inflammatory Bowel Disease and Colon Cancer. International Journal of Molecular Sciences. 2021; 22(20):11001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011001
Chicago/Turabian StylePang, Lokman, Matthias Ernst, and Jennifer Huynh. 2021. "Development of a Multiplex Immunohistochemistry Workflow to Investigate the Immune Microenvironment in Mouse Models of Inflammatory Bowel Disease and Colon Cancer" International Journal of Molecular Sciences 22, no. 20: 11001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011001