
The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells

 , and
, and
Abstract
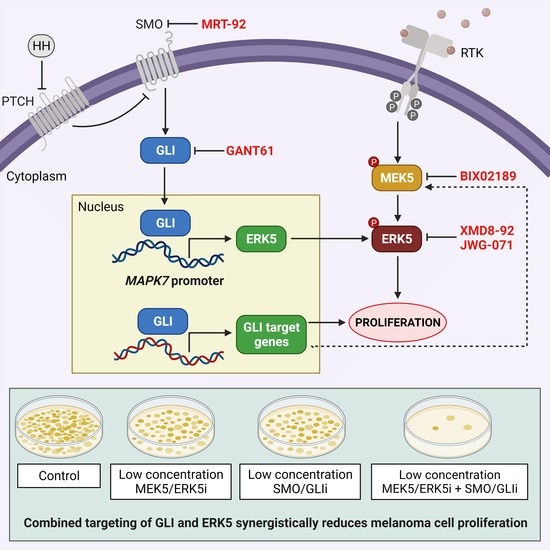
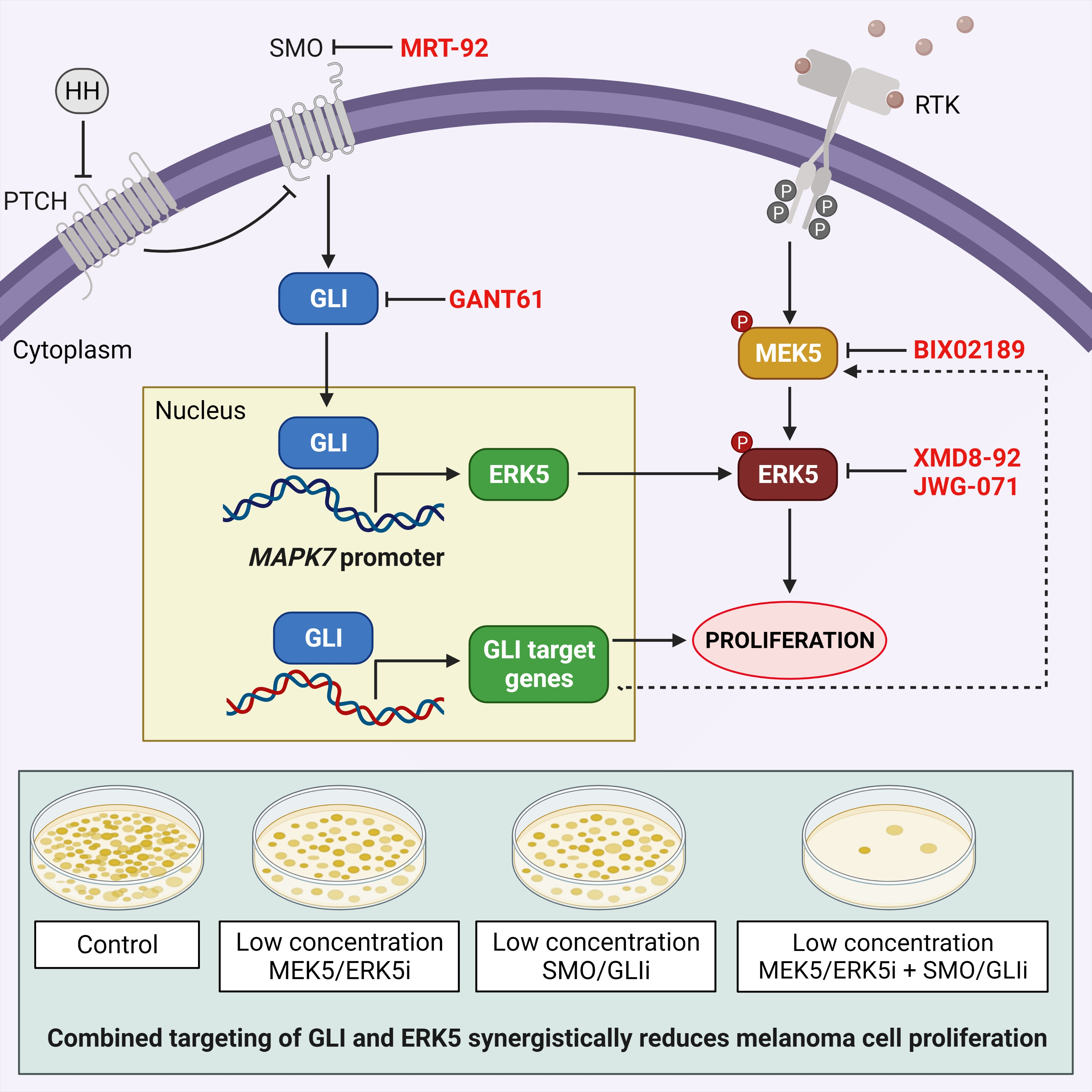
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
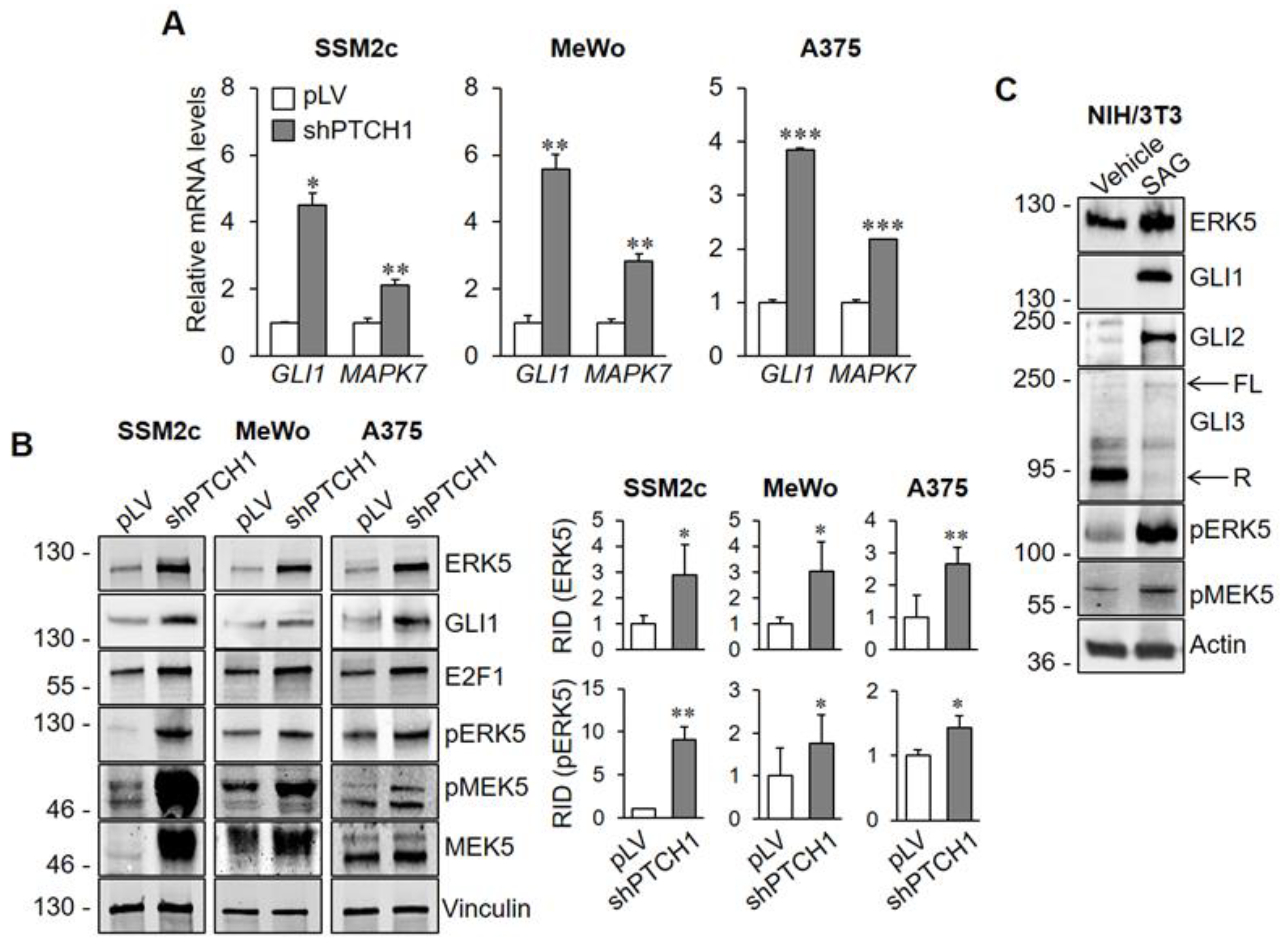
2.1. HH-GLI Signalling Regulates the Amount of ERK5 Protein and mRNA
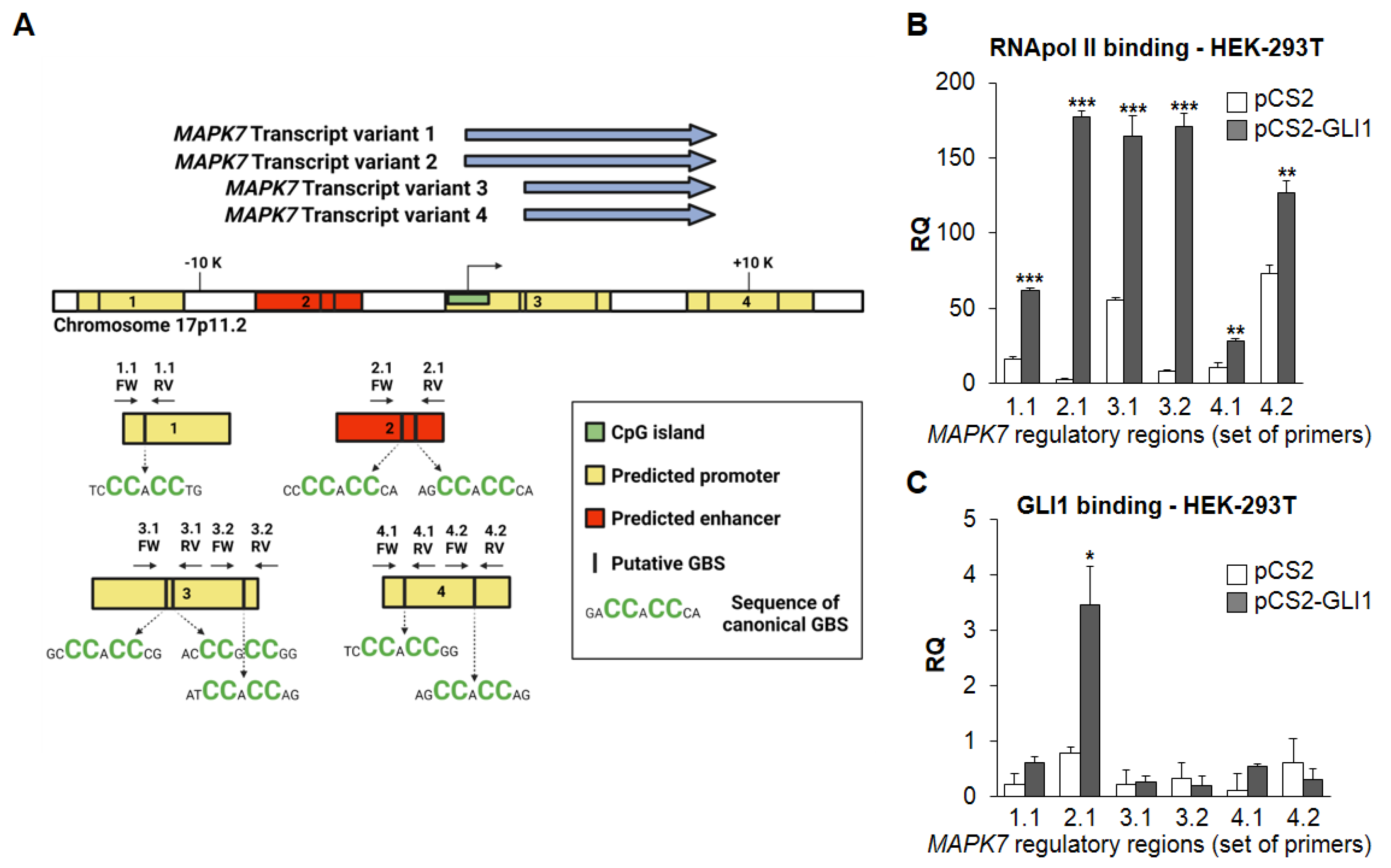
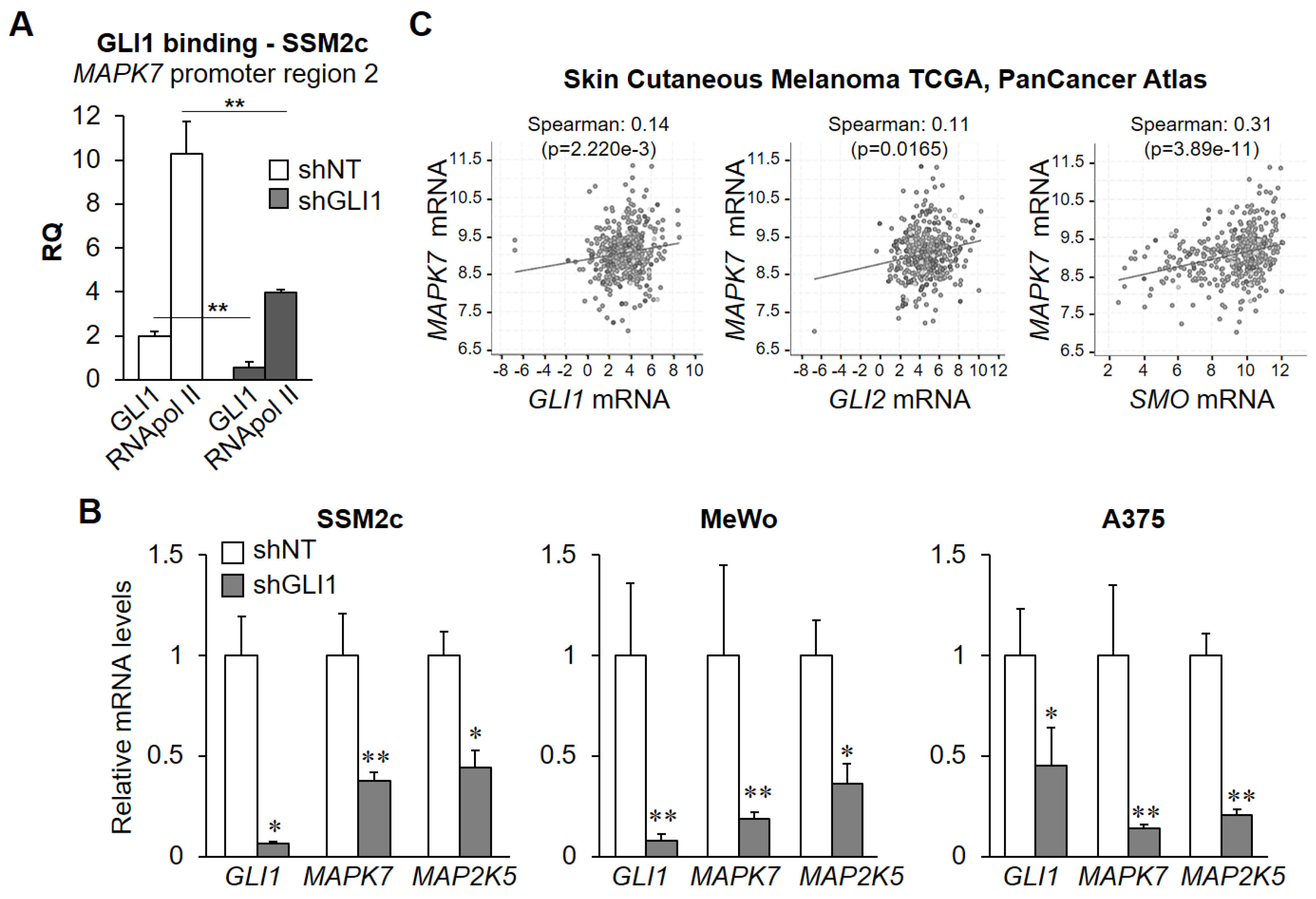
2.2. GLI1 Overexpression Increases RNA Polymerase Type II (RNApol II) Recruitment at MAPK7 Promoter and Results in Increased Binding of GLI1 to MAPK7-Regulatory Regions
2.3. ERK5 Supports HH-GLI-Dependent Proliferation of Melanoma Cells
2.4. Combined Targeting of HH-GLI and ERK5 Pathways Synergistically Reduces Melanoma Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Treatments
4.2. Cell Lysis and Western Blotting
4.3. Measurement of Cell Viability and Cell Cycle Phase Distribution
4.4. RNA Interference
4.5. Plasmids and Transfection
4.6. Quantitative Real-Time (Q-PCR)
4.7. ChIP
4.8. Clonogenic Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.D.; Ulevitch, R.J.; Han, J. Primary structure of BMK1: A new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Dello Sbarba, P.; Rovida, E. Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 2020, 21, 938. [Google Scholar] [CrossRef] [Green Version]
- Erazo, T.; Espinosa-Gil, S.; Diéguez-Martínez, N.; Gómez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef] [Green Version]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA. 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; Stecca, B. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cell. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI1 transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Winklmayr, M.; Schmid, C.; Laner-Plamberger, S.; Kaser, A.; Aberger, F.; Eichberger, T.; Frischauf, A.M. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC. Mol. Biol. 2010, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Nishi, Y.; Ma, W.; Vedenko, A.; Shokri, L.; Zhang, X.; McFarlane, M.; Baizabal, J.M.; Junker, J.P.; van Oudenaarden, A.; et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes. Dev. 2012, 26, 2802–2816. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, S.; Montagnani, V.; Lapucci, A.; Stecca, B. HEDGEHOG/GLI-E2F1 axis modulates iASPP expression and function and regulates melanoma cell growth. Cell Death Differ. 2015, 22, 2006–2019. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Montagnani, V.; Penachioni, J.Y.; Vinci, M.C.; Olivito, B.; Borgognoni, L.; Stecca, B. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 2013, 32, 4737–4747. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.J.; Potter, C.J.; Gohl, D.M.; Silies, M.; Katsov, A.Y.; Clandinin, T.R.; Luo, L. Specific kinematics and motor-related neurons for aversive chemotaxis in Drosophila. Curr. Biol. 2013, 23, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; de Miera, E.V.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel) 2013, 6, 1429–1450. [Google Scholar] [CrossRef]
- Sabol, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2562. [Google Scholar] [CrossRef] [Green Version]
- Gentilini, A.; Lori, G.; Caligiuri, A.; Raggi, C.; Di Maira, G.; Pastore, M.; Piombanti, B.; Lottini, T.; Arcangeli, A.; Madiai, S.; et al. Extracellular Signal-Regulated Kinase 5 Regulates the Malignant Phenotype of Cholangiocarcinoma Cells. Hepatology 2021, 74, 2007–2020. [Google Scholar] [CrossRef]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparís-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tusa, I.; Cheloni, G.; Poteti, M.; Gozzini, A.; DeSouza, N.H.; Shan, Y.; Deng, X.; Gray, N.S.; Li, S.; Rovida, E.; et al. Targeting the Extracellular Signal-Regulated Kinase 5 Pathway to Suppress Human Chronic Myeloid Leukemia Stem Cells. Stem Cell Rep. 2018, 11, 929–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Erazo, T.; Ferguson, F.M.; Buckley, D.L.; Gomez, N.; Muñoz-Guardiola, P.; Diéguez-Martínez, N.; Deng, X.; Hao, M.; Massefski, W.; et al. Structural and Atropisomeric Factors Governing the Selectivity of Pyrimido-benzodiazipinones as Inhibitors of Kinases and Bromodomains. ACS Chem. Biol. 2018, 13, 2438–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatake, R.J.; O’Neill, M.M.; Kennedy, C.A.; Wayne, A.L.; Jakes, S.; Wu, D.; Kugler, S.Z.; Kashem, M.A., Jr.; Kaplita, P.; Snow, R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, L.; Faure, H.; Roudaut, H.; Schoenfelder, A.; Mann, A.; Girard, N.; Bihannic, L.; Ayrault, O.; Petricci, E.; Taddei, M.; et al. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor. FASEB J. 2015, 29, 1817–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Santini, R.; Gagliardi, S.; Dapporto, F.; Colecchia, D.; Chiariello, M.; Leone, C.; Valoti, M.; Manetti, F.; Petricci, E.; et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Poggetto, E.; Tanturli, M.; Ben-Califa, N.; Gozzini, A.; Tusa, I.; Cheloni, G.; Marzi, I.; Cipolleschi, M.G.; Kashman, Y.; Neumann, D.; et al. Salarin C inhibits the maintenance of chronic myeloid leukemia progenitor cells. Cell Cycle 2015, 14, 3146–3154. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Barbetti, V.; Tusa, I.; Cipolleschi, M.G.; Rovida, E.; Dello Sbarba, P. AML1/ETO sensitizes via TRAIL acute myeloid leukemia cells to the pro-apoptotic effects of hypoxia. Cell Death Dis. 2013, 4, e536. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Menconi, A.; Lulli, M.; Dello Sbarba, P.; Stecca, B.; Rovida, E. The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 11259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011259
Tusa I, Gagliardi S, Tubita A, Pandolfi S, Menconi A, Lulli M, Dello Sbarba P, Stecca B, Rovida E. The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells. International Journal of Molecular Sciences. 2021; 22(20):11259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011259
Chicago/Turabian StyleTusa, Ignazia, Sinforosa Gagliardi, Alessandro Tubita, Silvia Pandolfi, Alessio Menconi, Matteo Lulli, Persio Dello Sbarba, Barbara Stecca, and Elisabetta Rovida. 2021. "The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells" International Journal of Molecular Sciences 22, no. 20: 11259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011259