
8-Hydroxyquinoline-Amino Acid Hybrids and Their Half-Sandwich Rh and Ru Complexes: Synthesis, Anticancer Activities, Solution Chemistry and Interaction with Biomolecules †

 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
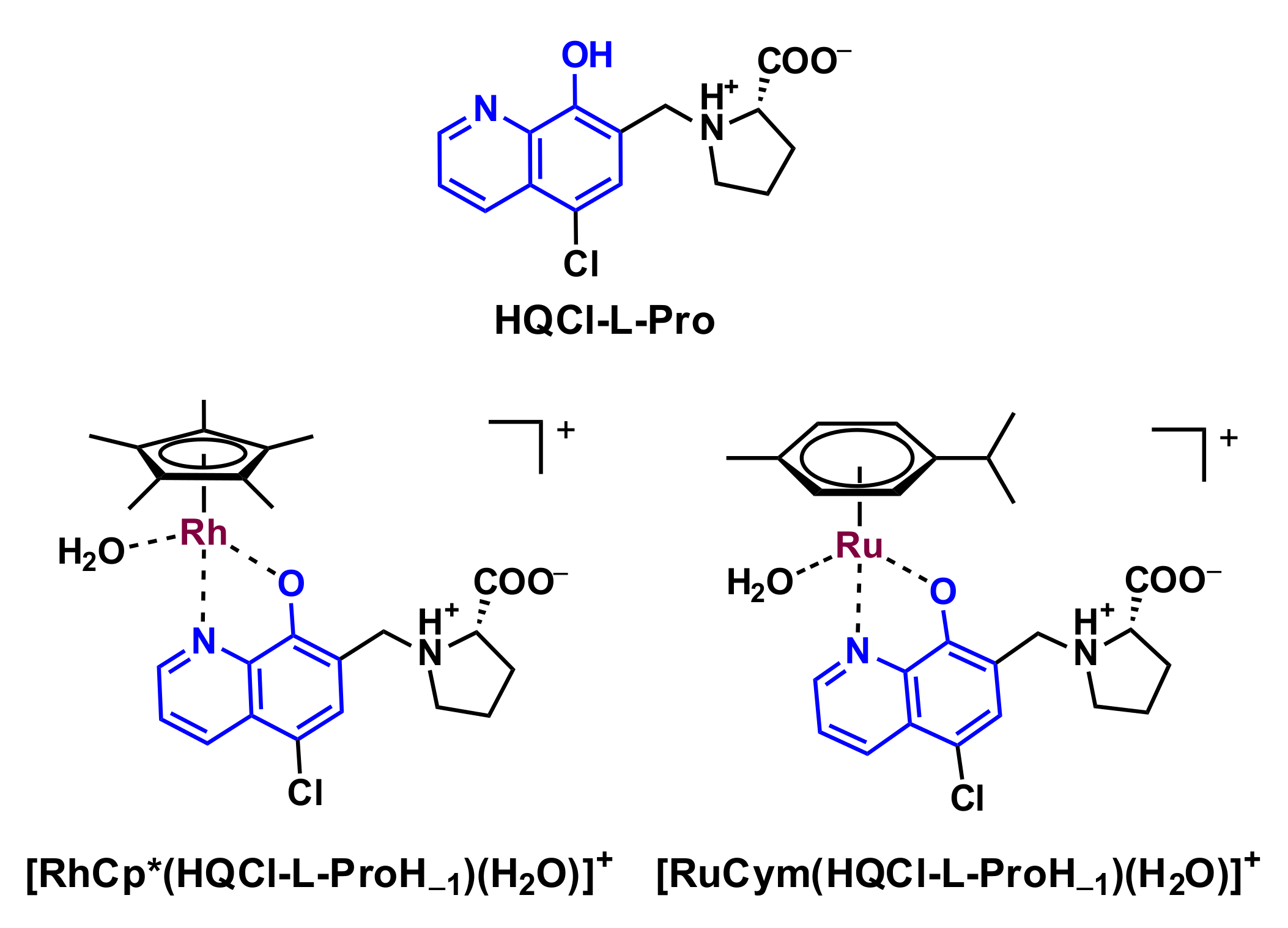
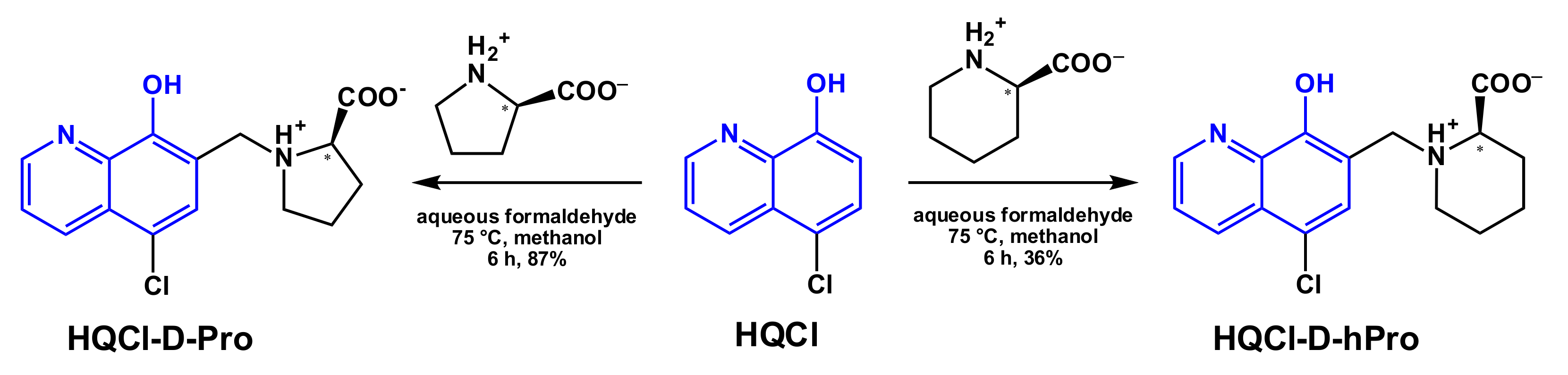
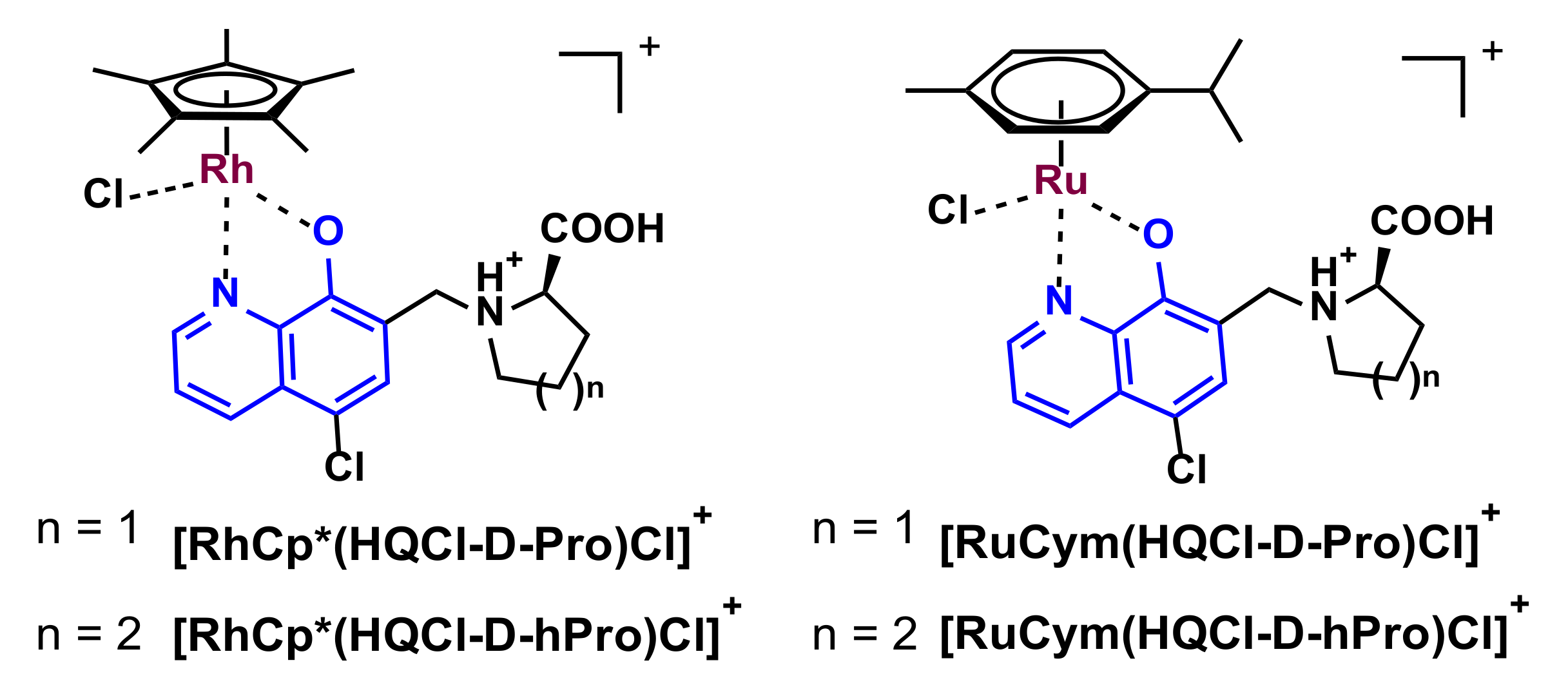
2.1. Synthesis of HQCl-D-Pro and HQCl-D-hPro, Their Ru(η6-p-cymene) and Rh(η5-C5Me5) Complexes
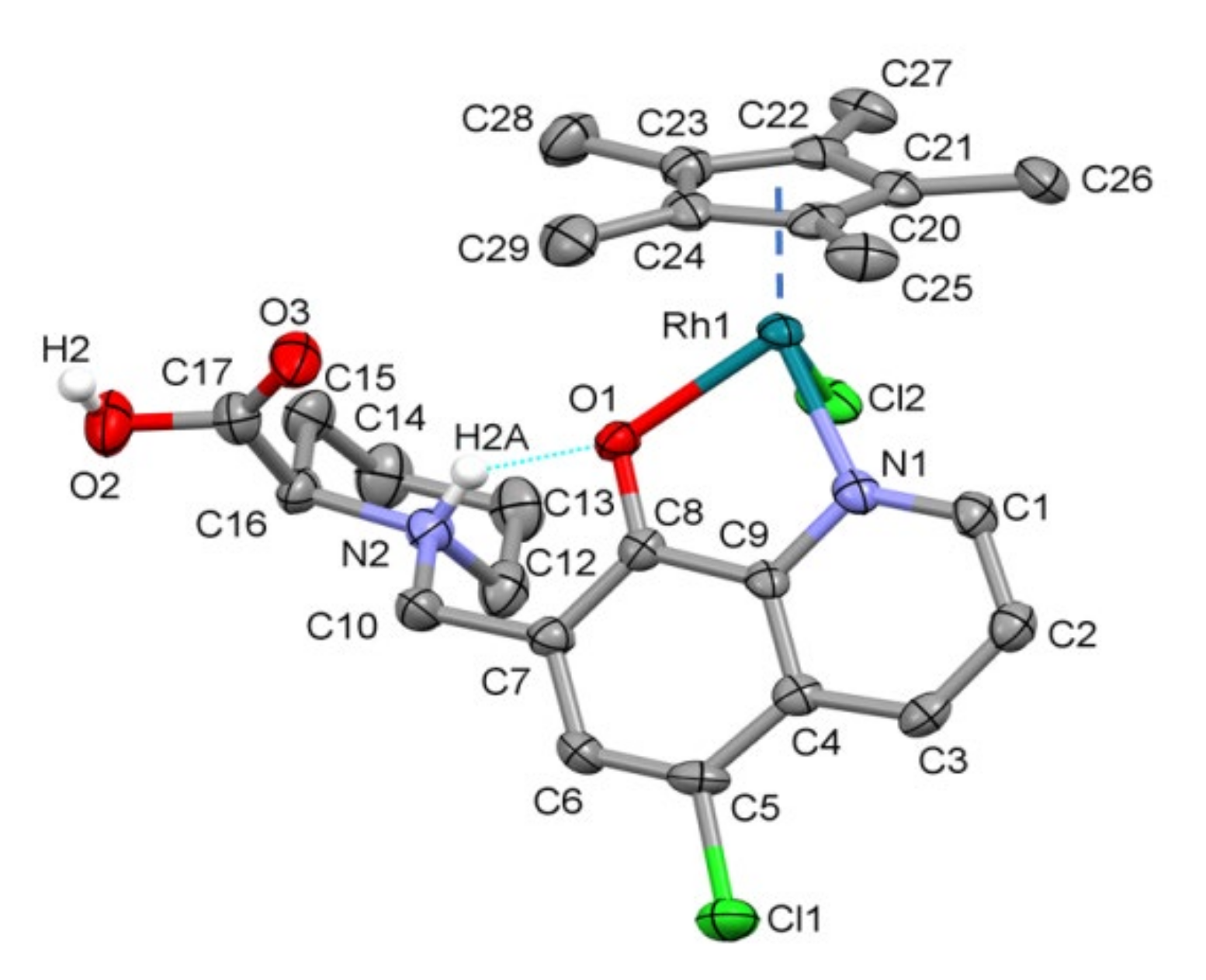
2.2. Structural Studies of [RhCp*(HQCl-D-hPro)Cl]Cl∙H2O∙CH3OH (1) by X-ray Crystallography
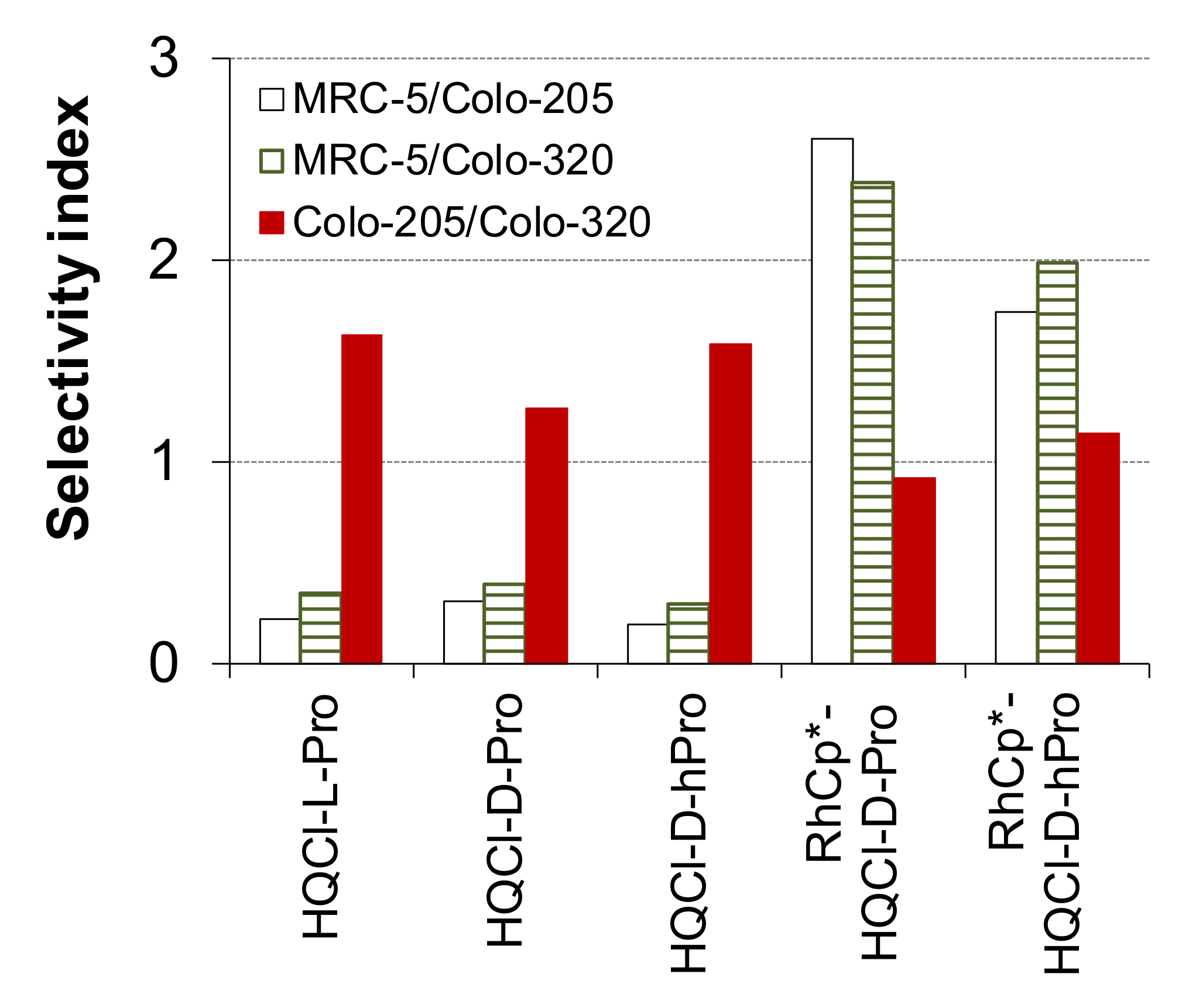
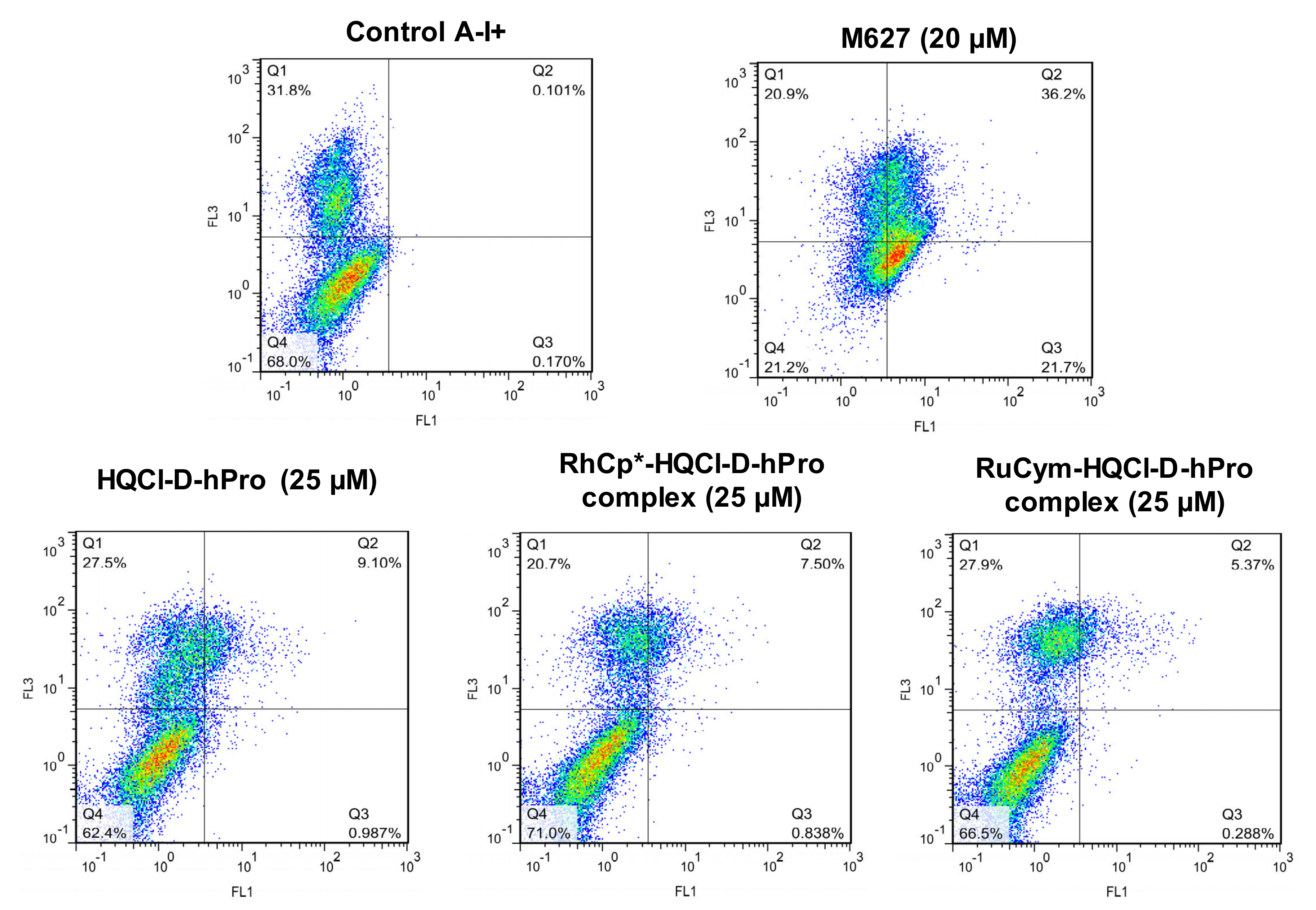
2.3. In Vitro Cytotoxicity and Induction of Apoptosis of the Ligands and Their Complexes
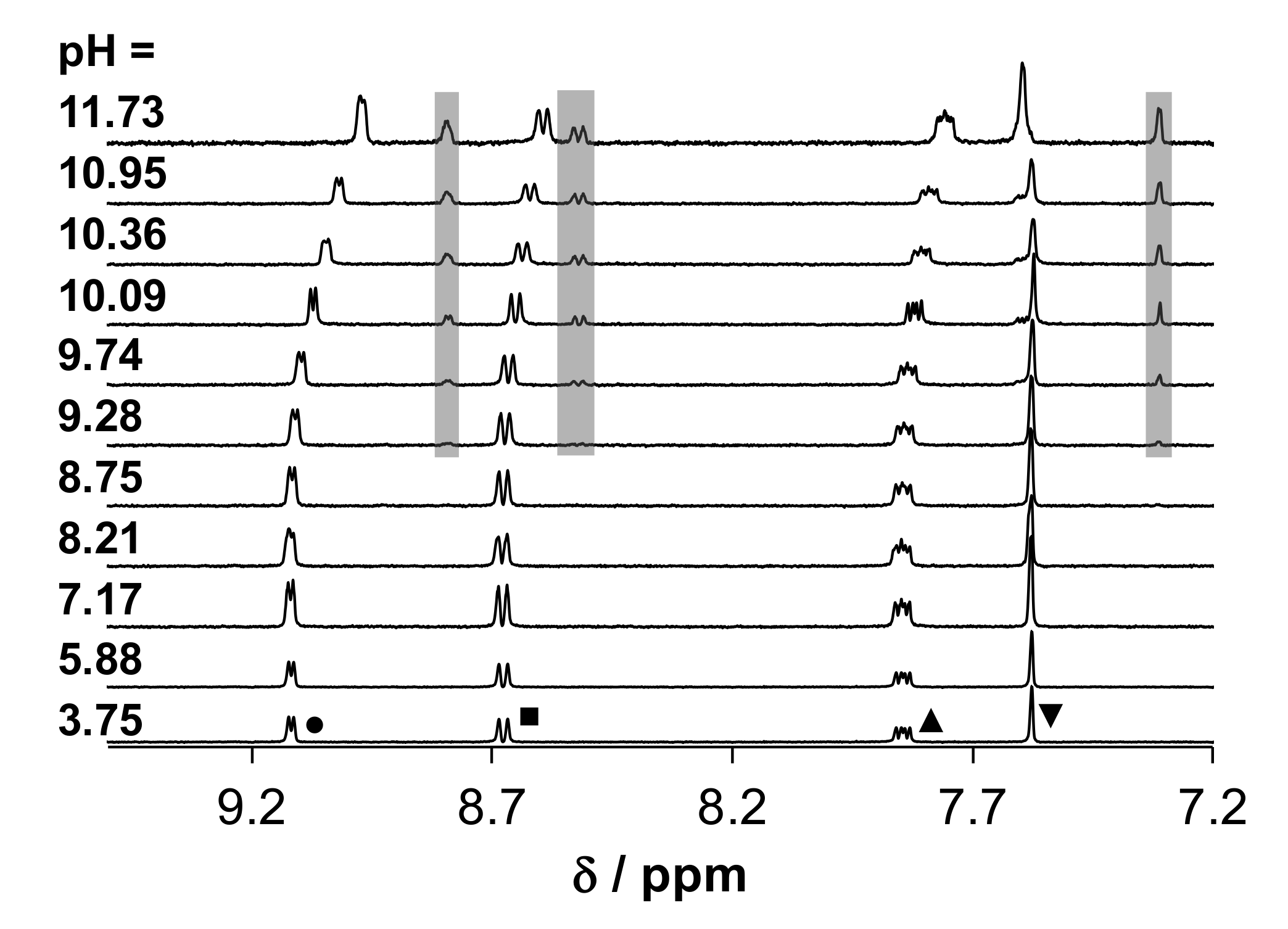
2.4. Solution Chemical Behavior of the Title Ligands and Their Organometallic Complexes
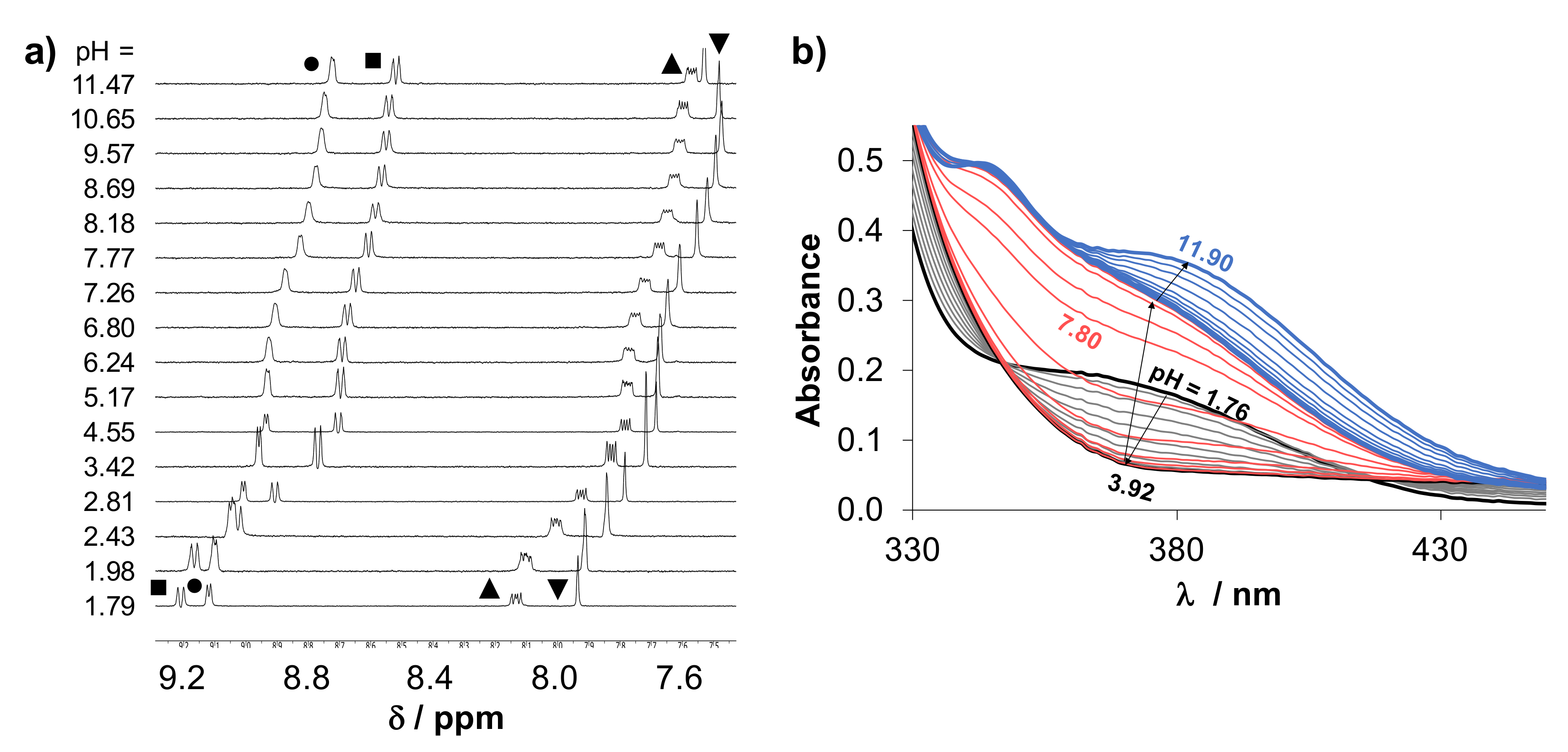
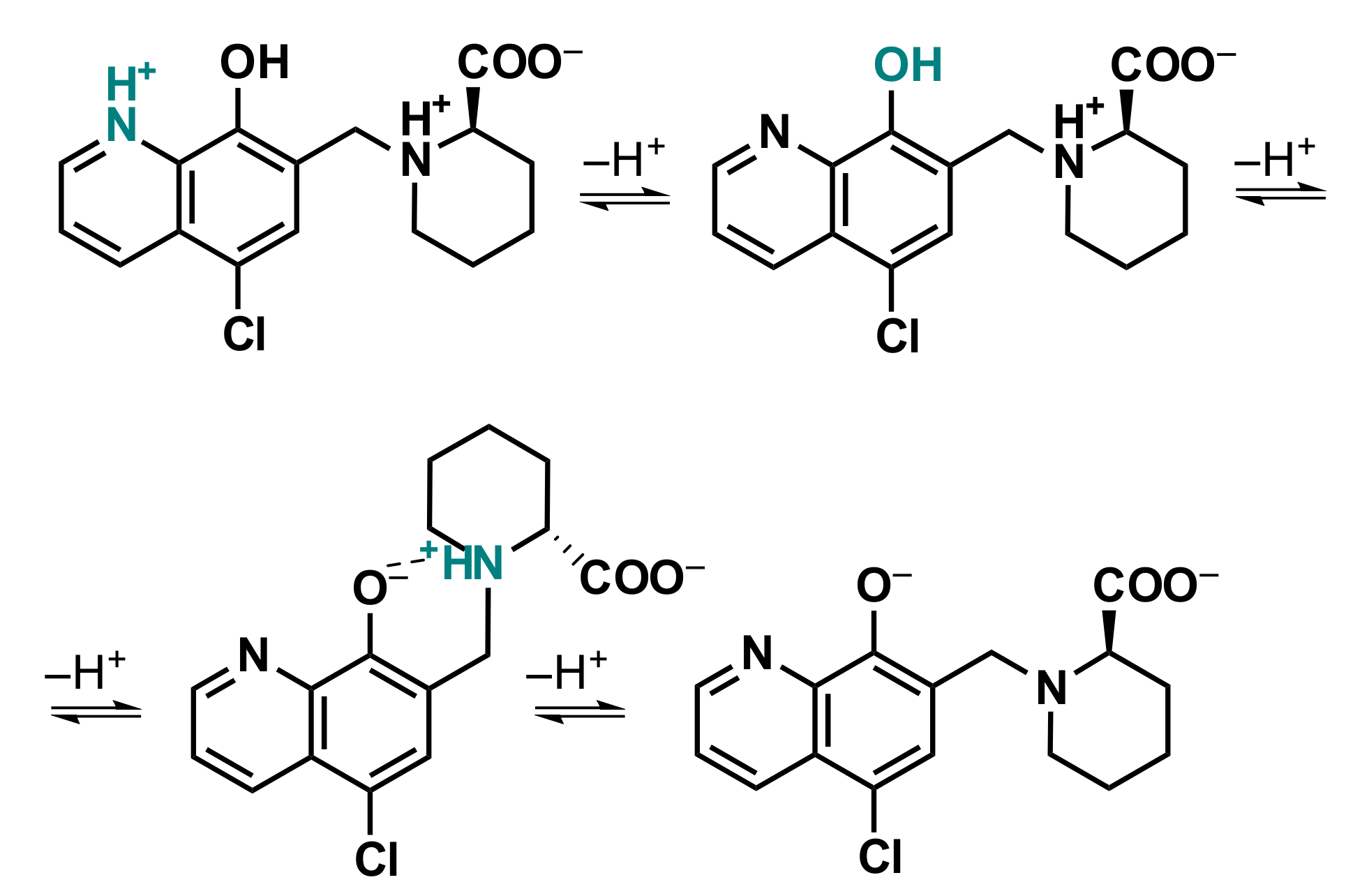
2.4.1. Solution Equilibria and Lipophilicity of HQCl-D-Pro and HQCl-D-hPro
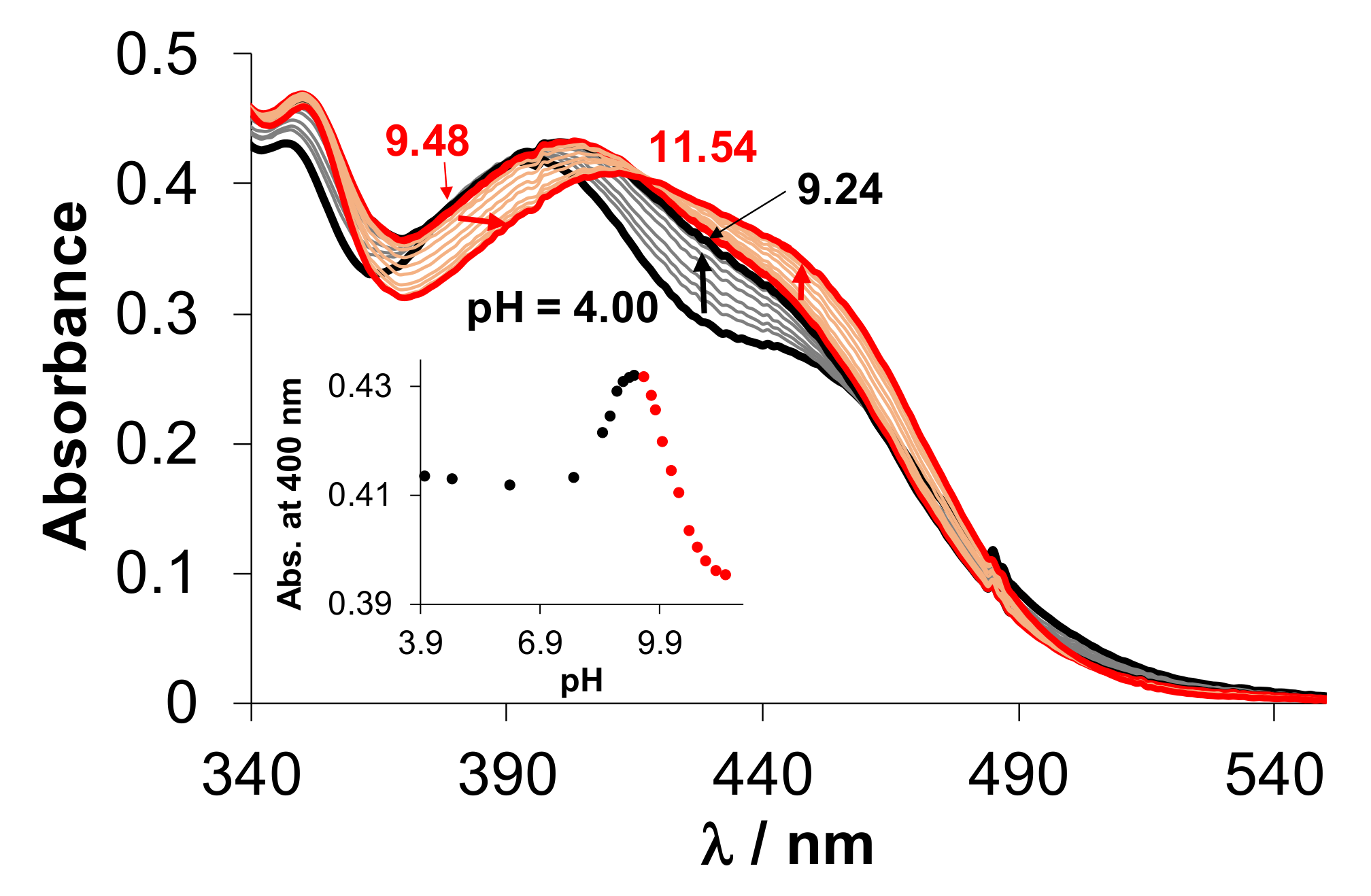
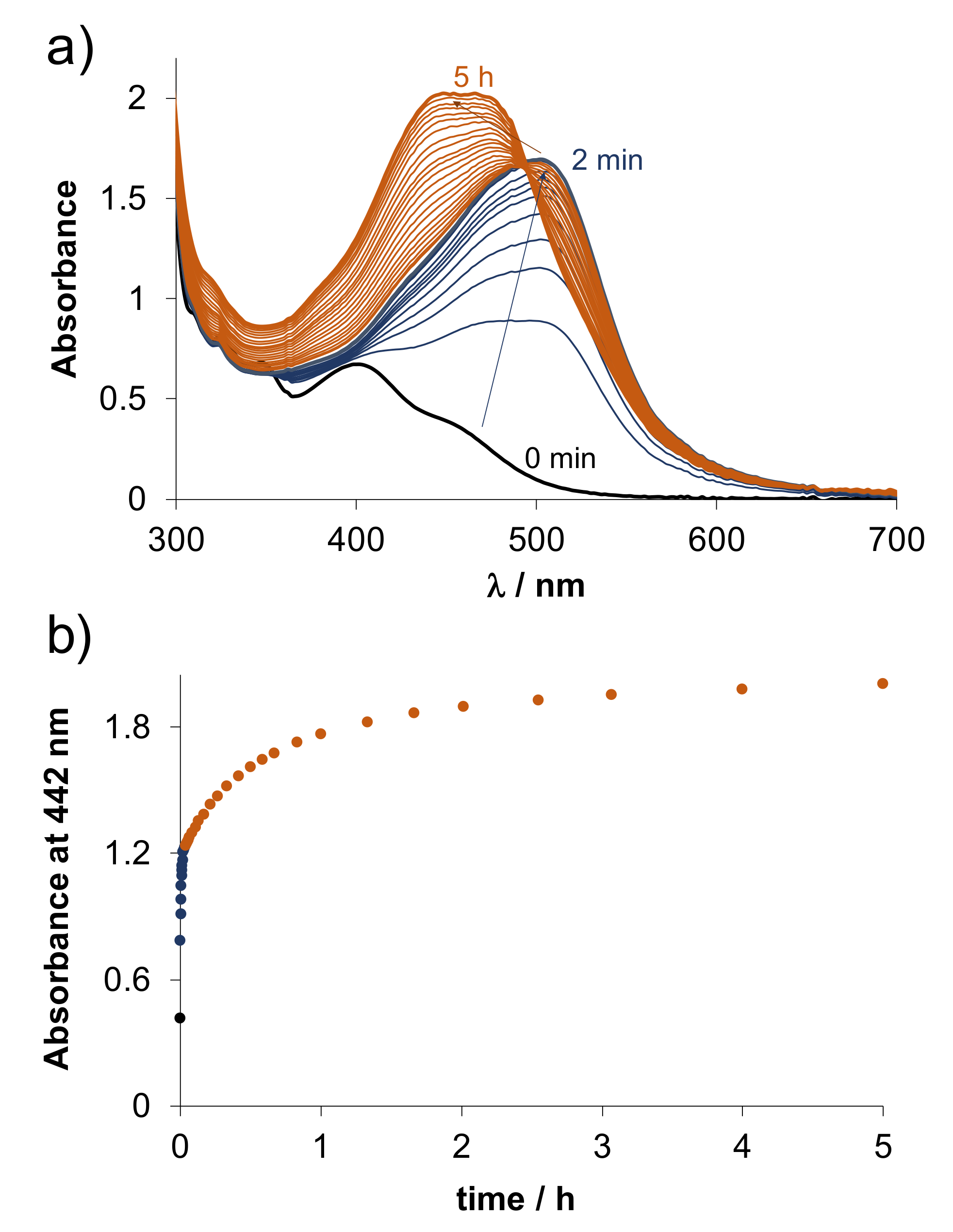
2.4.2. Solution Equilibria and Lipophilicity of the Title Organometallic Complexes
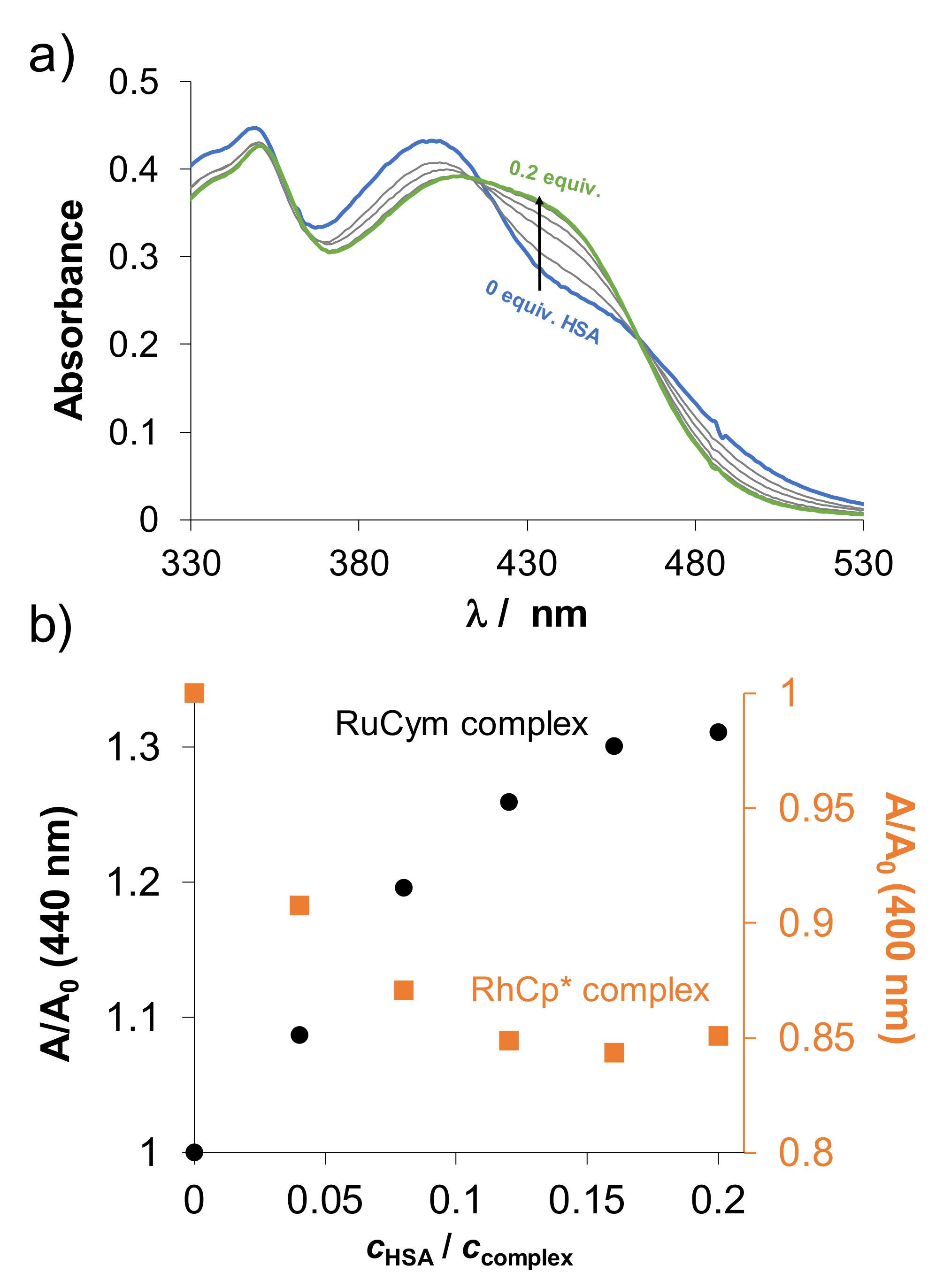
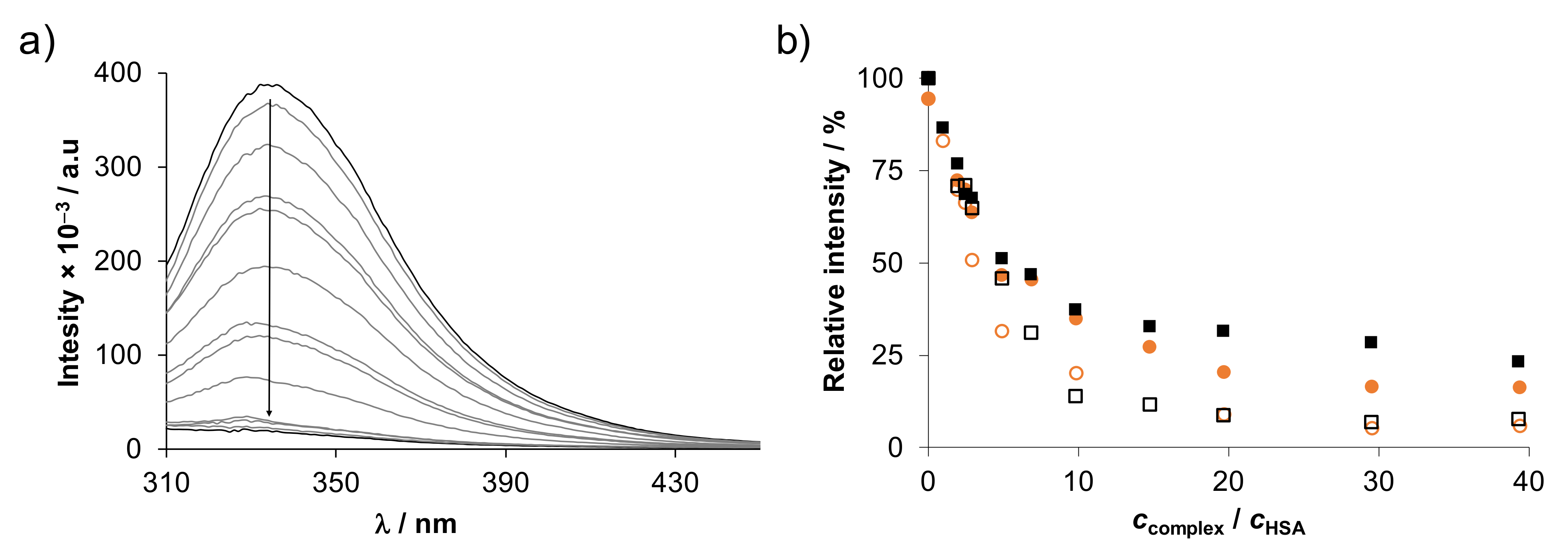
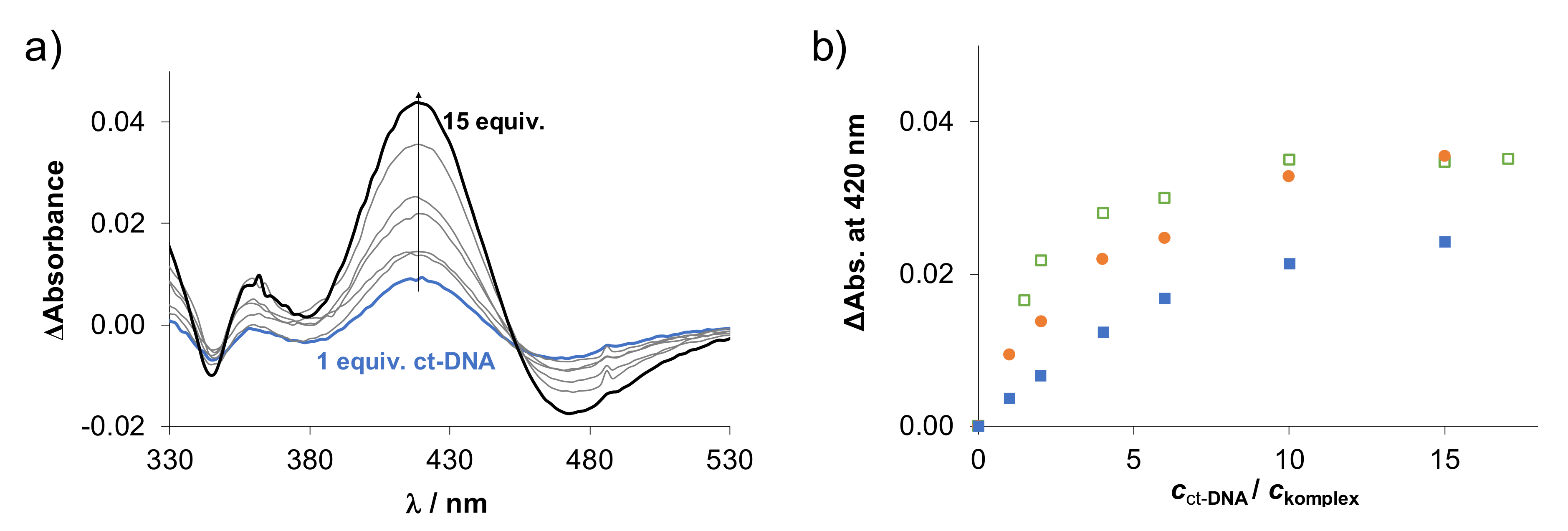
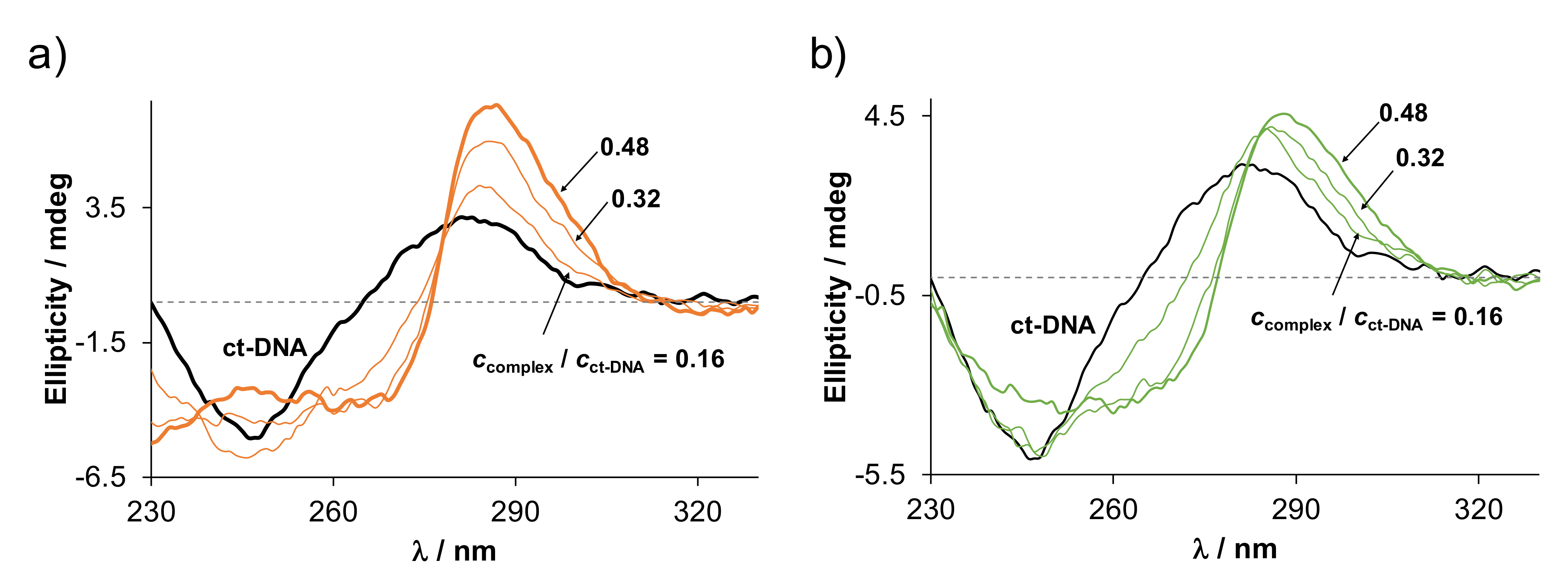
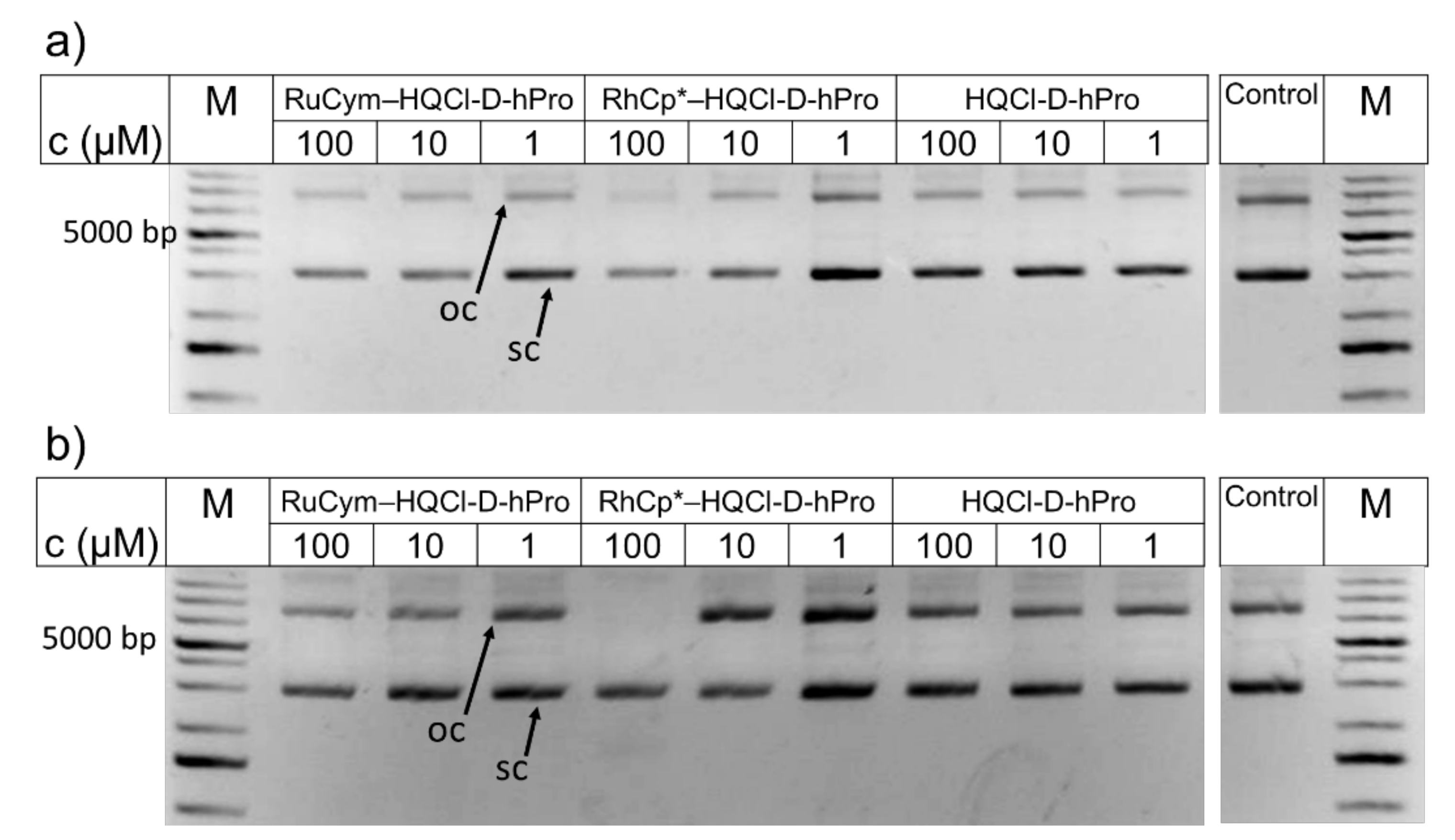
2.5. Binding of the Complexes to Biological Macromolecules
2.5.1. Interaction of the Complexes with Human Serum Albumin
2.5.2. Interaction of the Complexes with DNA
3. Materials and Methods
3.1. Chemicals
3.2. Stock Solutions and Sample Preparation
3.3. Synthesis and Characterization of Ligands and Their Complexes
3.3.1. Synthesis of HQCl-D-Pro
3.3.2. Synthesis of HQCl-D-hPro
3.3.3. Synthesis of [RuCym(HQCl-D-Pro)Cl]Cl
3.3.4. Synthesis of [RhCp*(HQCl-D-Pro)Cl]Cl
3.3.5. Synthesis of [RuCym(HQCl-D-hPro)Cl]Cl
3.3.6. Synthesis of [RhCp*(HQCl-D-hPro)Cl]Cl
3.4. Electrospray Mass Spectrometry
3.5. pH-Potentiometric Measurements
3.6. UV-Visible Spectrophotometry and Determination of Distribution Coefficients
3.7. Circular Dichroism Spectroscopy and Spectrofluorometry
3.8. NMR Spectroscopy
3.9. Capillary Zone Electrophoresis
3.10. X-ray Data Collection, Structure Solution and Refinement for Compound [RhCp*(HQCl-D-hPro)Cl]Cl∙H2O∙CH3OH
3.11. In Vitro Cell Studies
3.11.1. Cell Lines and Culture Conditions
3.11.2. MTT Assay
3.11.3. Assay for Apoptosis Induction
3.12. In Vitro DNA Cleavage Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tremlett, W.D.J.; Goodman, D.M.; Steel, T.R.; Kumar, S.; Wieczorek-Błauż, A.; Walsh, F.P.; Sullivan, M.; Hartinger, C.G. Design concepts of half-sandwich organoruthenium anticancer agents based on bidentate bioactive ligands. Coord. Chem. Rev. 2021, 445, 213950. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent biological and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Noffke, A.L.; Habtemariam, A.; Pizarro, A.M.; Sadler, P.J. Designing organometallic compounds for catalysis and therapy. Chem. Commun. 2012, 48, 5219–5246. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, O.; Aicher, S.; Schmidlehner, M.; Novak, M.S.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Hartinger, C.G.; Keppler, B.K.; Enyedy, É.A. Antitumor pentamethylcyclopentadienyl rhodium complexes of maltol and allomaltol: Synthesis, solution speciation and bioactivity. J. Inorg. Biochem. 2014, 134, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mészáros, J.P.; Pape, V.F.S.; Szakács, G.; Németi, G.; Dénes, M.; Holczbauer, T.; May, N.V.; Enyedy, É.A. Half-sandwich organometallic Ru and Rh complexes of (N,N) donor compounds: Effect of ligand methylation on solution speciation and anticancer activity. Dalton Trans. 2021, 50, 8218–8231. [Google Scholar] [CrossRef] [PubMed]
- Kubanik, M.; Holtkamp, H.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Impact of the Halogen Substitution Pattern on the Biological Activity of Organoruthenium 8-Hydroxyquinoline Anticancer Agents. Organometallics 2015, 34, 5658–5668. [Google Scholar] [CrossRef]
- Zhu, L.-G.; Wang, Z.-F.; Gao, Y.; Qin, Q.-P.; Huang, X.-L.; Tan, M.-X.; Zeng, C.-J.; Zou, B.-Q. New 5-chloro-8-hydroxyquinoline derivatives organometallic Ru(II)-arene complexes as antitumor agents. Inorg. Chem. Commun. 2019, 108, 107537. [Google Scholar] [CrossRef]
- Pivarcsik, T.; Tóth, G.; Szemerédi, N.; Bogdanov, A.; Spengler, G.; Kljun, J.; Kladnik, J.; Turel, I.; Enyedy, É.A. Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands. Pharmaceuticals 2021, 14, 518. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Dev. Ther. 2013, 7, 1157–1178. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A privileged structure with a broad-ranging pharmacological potential. Med. Chem. Commun. 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Oliveri, V.; Vecchio, G. 8-Hydroxyquinolines in medicinal chemistry: A structural perspective. Eur. J. Med. Chem. 2016, 120, 252–274. [Google Scholar] [CrossRef]
- Zhai, S.; Yang, L.; Cui, Q.C.; Sun, Y.; Dou, Q.P.; Yan, B. Tumor cellular proteasome inhibition and growth suppression by 8-hydroxyquinoline and clioquinol requires their capabilities to bind copper and transport copper into cells. J. Biol. Inorg. Chem. 2010, 15, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leanderson, P.; Tagesson, C.; Yan, B. Iron bound to the lipophilic iron chelator, 8-hydroxyquinoline, causes DNA strand breakage in cultured lung cells. Carcinogenesis 1996, 17, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Pape, V.F.S.; May, N.V.; Gál, G.T.; Szatmári, I.; Szeri, F.; Fülöp, F.; Szakács, G.; Enyedy, É.A. Impact of copper and iron binding properties on the anticancer activity of 8-hydroxyquinoline derived Mannich bases. Dalton Trans. 2018, 47, 17032–17045. [Google Scholar] [CrossRef] [Green Version]
- Pape, V.F.S.; Gaál, A.; Szatmári, I.; Kucsma, N.; Szoboszlai, N.; Streli, C.; Fülöp, F.; Enyedy, É.A.; Szakács, G. Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells. Cancers 2021, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Neldner, K.H. The halogenated 8-hydroxyquinolines. Int. J. Dermatol. 1977, 16, 267–273. [Google Scholar] [CrossRef]
- Naber, K.G.; Niggemann, H.; Stein, G.; Stein, G. Review of the literature and individual patients’ data meta-analysis on efficacy and tolerance of nitroxoline in the treatment of uncomplicated urinary tract infections. BMC Infect. Dis. 2014, 14, 628. [Google Scholar] [CrossRef] [Green Version]
- Shaw, Y.A.; Chang, C.-Y.; Hsu, M.-Y.; Lu, P.-J.; Yang, C.-N.; Chen, H.-L.; Lo, C.-W.; Shiau, C.-W.; Chern, M.-K. Synthesis and structure-activity relationship study of 8-hydroxyquinoline-derived Mannich bases as anticancer agents. Eur. J. Med. Chem. 2010, 45, 2860–2867. [Google Scholar] [CrossRef]
- Yang, Q.-Y.; Cao, Q.-Q.; Zhang, Y.-L.; Xu, X.-F.; Deng, C.-X.; Kumar, R.; Zhu, X.-M.; Wang, X.-J.; Liang, H.; Chen, Z.-F. Synthesis, structural characterization and antitumor activity of six rare earth metal complexes with 8-hydroxyquinoline derivatives. J. Inorg. Biochem. 2020, 211, 111175. [Google Scholar] [CrossRef]
- Mohammadi, F.; Torshizi-Mansouri, H. Five novel palladium(II) complexes of 8-hydroxyquinoline and amino acids with hydrophobic side chains: Synthesis, characterization, cytotoxicity, DNA- and BSA-interaction studies. J. Biomol. Struct. Dyn. 2019, 38, 3059–3073. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Tang, S.-F.; Qin, Q.-P.; Liang, Y.-L.; Wu, C.-X.; Wang, C.-Y.; Yan, H.-T.; Dong, J.-X.; Liu, Y.-C. Evaluation of the effect of iodine substitution of 8-hydroxyquinoline on its platinum(ii) complex: Cytotoxicity, cell apoptosis and telomerase inhibition. Med. Chem. Commun. 2016, 7, 1802–1811. [Google Scholar] [CrossRef]
- Gogna, R.; Madan, E.; Keppler, B.; Pati, U. Gallium compound GaQ3-induced Ca2+ signalling triggers p53-dependent and -independent apoptosis in cancer cells. Br. J. Pharmacol. 2011, 166, 617–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dömötör, O.; Pape, V.F.S.; May, N.V.; Szakács, G.; Enyedy, É.A. Comparative solution equilibrium studies of antitumor ruthenium(η6-p-cymene) and rhodium (η5-C5Me5) complexes of 8-hydroxyquinolines. Dalton Trans. 2017, 46, 4382–4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mészáros, J.P.; Poljarevic, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, N.; Spengler, G.; Enyedy, É.A. An 8-hydroxyquinoline–proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef]
- Tremlett, W.D.J.; Tong, K.K.H.; Steel, T.R.; Movassaghi, S.; Hanif, M.; Jamieson, S.M.F.; Söhnel, T.; Hartinger, C.G. Hydroxyquinoline-derived anticancer organometallics: Introduction of amphiphilic PTA as an ancillary ligand increases their aqueous solubility. J. Inorg. Biochem. 2019, 199, 110768–110775. [Google Scholar] [CrossRef]
- Mitrovic, A.; Kljun, J.; Sosic, I.; Ursic, M.; Meden, A.; Gobec, S.; Kos, J.; Turel, I. Organoruthenated Nitroxoline Derivatives Impair Tumor Cell Invasion through Inhibition of Cathepsin B Activity. Inorg. Chem. 2019, 58, 12334–12347. [Google Scholar] [CrossRef] [Green Version]
- Movassaghi, S.; Hanif, M.; Holtkamp, H.U.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Making organoruthenium complexes of 8-hydroxyquinolines more hydrophilic: Impact of a novel l-phenylalanine-derived arene ligand on the biological activity. Dalton Trans. 2018, 47, 2192–2201. [Google Scholar] [CrossRef] [Green Version]
- Gobec, M.; Kljun, J.; Sosic, I.; Mlinarič-Rascan, I.; Ursic, M.; Gobec, S.; Turel, I. Structural characterization and biological evaluation of a clioquinol–ruthenium complex with copper-independent antileukaemic activity. Dalton Trans. 2014, 43, 9045–9051. [Google Scholar] [CrossRef] [Green Version]
- Füredi, A.; Tóth, S.; Szebényi, K.; Pape, V.F.S.; Türk, D.; Kucsma, N.; Cervenak, L.; Tóvári, J.; Szakács, G. Identification and Validation of Compounds Selectively Killing Resistant Cancer: Delineating Cell Line–Specific Effects from P-Glycoprotein–Induced Toxicity. Mol. Cancer Ther. 2016, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Cserepes, M.; Türk, D.; Tóth, S.; Pape, V.F.S.; Gaál, A.; Gera, M.; Szabó, J.E.; Kucsma, N.; Várady, G.; Vértessy, B.G.; et al. Unshielding Multidrug Resistant Cancer through Selective Iron Depletion of P-Glycoprotein–Expressing Cells. Cancer Res. 2020, 80, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, J.P.; Dömötör, O.; Hackl, C.M.; Roller, A.; Keppler, B.K.; Kandioller, W.; Enyedy, É.A. Structural and solution equilibrium studies on half-sandwich organorhodium complexes of (N,N) donor bidentate ligands. New. J. Chem. 2018, 42, 11174–11184. [Google Scholar] [CrossRef] [Green Version]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Controll. Release 2012, 157, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, O.; Pivarcsik, T.; Mészáros, J.P.; Szatmári, I.; Fülöp, F.; Enyedy, É.A. Critical factors affecting the albumin binding of half-sandwich Ru(II) and Rh(III) complexes of 8-hydroxyquinolines and oligopyridines. Dalton Trans. 2021, 50, 11918–11930. [Google Scholar] [CrossRef]
- Dömötör, O.; Enyedy, É.A. Binding mechanisms of half-sandwich Rh(III) and Ru(II) arene complexes on human serum albumin: A comparative study. J. Biol. Inorg. Chem. 2019, 24, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Luo, Q.; Ma, X.; Wu, K.; Liu, J.; Chen, Y.; Xiong, S.; Wang, J.; Sadler, P.J.; Wang, F. Arene Control over Thiolate to Sulfinate Oxidation in Albumin by Organometallic Ruthenium Anticancer Complexes. Chem. Eur. J. 2009, 15, 6586–6594. [Google Scholar] [CrossRef]
- Adhireksan, Z.; Palermo, G.; Riedel, T.; Ma, Z.; Muhammad, R.; Rothlisberger, U.; Dyson, P.J.; Davey, C.A. Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat. Commun. 2017, 8, 14860. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Bella, J.; Parkinson, A.; Sadler, P.J. Competitive reactions of a ruthenium arene anticancer complex with histidine, cytochrome c and an oligonucleotide. J. Biol. Inorg. Chem. 2005, 10, 147–155. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents—Towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Coverdale, J.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.C. Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N., Nakanishi, K., Woody, R.W., Eds.; VCH Publishers: New York, NY, USA, 2000; p. 523. [Google Scholar]
- Geldmacher, Y.; Rubbiani, R.; Wefelmeier, P.; Prokop, A.; Ott, I.; Sheldrick, W.S. Synthesis and DNA-binding properties of apoptosis-inducing cytotoxic half-sandwich rhodium(III) complexes with methyl-substituted polypyridyl ligands. J. Organomet. Chem. 2011, 696, 1023–1031. [Google Scholar] [CrossRef]
- Schäfer, S.; Ott, I.; Gust, R.; Sheldrick, W.S. Influence of the Polypyridyl (pp) Ligand Size on the DNA Binding Properties, Cytotoxicity and Cellular Uptake of Organoruthenium(II) Complexes of the Type [(η6-C6Me6)Ru(L)(pp)]n+ [L = Cl, n = 1; L = (NH2)2CS, n = 2]. Eur. J. Inorg. Chem. 2007, 19, 3034–3046. [Google Scholar] [CrossRef]
- Heller, D.P.; Greenstock, C.L. Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye. Biophys. Chem. 1994, 50, 305–312. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Beaven, G.H.; Chen, S.-H.; D’albis, A.; Gratzer, W.B. A Spectroscopic Study of the Haemin–Human-Serum-Albumin System. Eur. J. Biochem. 1974, 42, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.R. Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; Greene and Wiley-Interscience: New York, NY, USA, 1994; p. A-3D-1-14. [Google Scholar]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- SCQuery. The IUPAC Stability Constants Database, Academic Software, Version 5.5; Royal Society of Chemistry: London, UK, 1993. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Higashi, T. Numerical Absorption Correction, NUMABS; Rigaku/MSC Inc.: Austin, TX, USA, 2002. [Google Scholar]
- CrystalClear SM 1.4.0; Rigaku/MSC Inc.: Austin, TX, USA, 2008.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.H.; Johnson, O.; Shields, G.P.; Smith, B.R.; Shields, G.P.; Towler, M. CIF applications. XV. enCIFer: A program for viewing, editing and visualizing CIFs. J. Appl. Crystallogr. 2004, 37, 335–338. [Google Scholar] [CrossRef] [Green Version]
- GraphPad Prism Version 7.00 for Windows, Graph Pad Software, La Jolla, California, USA. 2018. Available online: http://www.graphpad.com (accessed on 18 October 2021).


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | Colo-205 | Colo-320 | MRC-5 |
|---|---|---|---|
| HQCl-L-Pro a | 20.92 ± 0.80 | 12.87 ± 0.74 | 4.57 ± 0.21 |
| HQCl-D-Pro | 17.0 ± 1.0 | 13.41 ± 0.79 | 5.33 ± 0.44 |
| HQCl-D-hPro | 18.9 ± 1.1 | 11.9 ± 1.5 | 3.63 ± 0.10 |
| RhCp*–HQCl-D-Pro | 31.5 ± 1.4 | 34.3 ± 1.0 | 82.1 ± 5.1 |
| RhCp*–HQCl-D-hPro | 21.54 ± 0.60 | 18.90 ± 0.54 | 37.6 ± 2.3 |
| RuCym–HQCl-D-Pro | >100 | >100 | >100 |
| RuCym–HQCl-D-hPro | >100 | >100 | >100 |
| doxorubicin | 0.05 ± 0.01 | 0.19 ± 0.02 | 0.82 ± 0.16 |
| Method | Ligand | pKa (NqH+) | pKa (OH) | pKa (NhPro/ProH+) |
|---|---|---|---|---|
| pH-potentiometry | HQCl-D-Pro a | < 2 | 7.71 ± 0.06 | >11 |
| HQCl-D-hPro | 2.42 ± 0.04 | 7.52 ± 0.06 | >11 | |
| UV-vis | HQCl-D-Pro | 2.13 ± 0.03 | 7.77 ± 0.04 | >11 |
| HQCl-D-hPro | 2.43 ± 0.01 | 7.49 ± 0.04 | 11.43 ± 0.01 | |
| 1H NMR | HQCl-D-Pro | 2.32 ± 0.03 | 7.75 ± 0.02 | >11 |
| HQCl-D-hPro | 2.52 ± 0.03 | 7.54 ± 0.02 | >11 |
| Complex | Method | pKa1 | pKa2 | logK′ (H2O/Cl−) |
|---|---|---|---|---|
| RhCp*–HQCl-D-Pro | UV-vis | 10.08 ± 0.02 | – | 1.72 ± 0.01 |
| RhCp*–HQCl-D-hPro | UV-vis | 10.11 ± 0.02 | – | 1.79 ± 0.01 |
| 1H NMR | 9.82 ± 0.13 a | 10.06 ± 0.06 b | – | |
| RuCym–HQCl-D-Pro | UV-vis | 8.96 ± 0.01 | 10.67 ± 0.01 | 1.18 ± 0.01 |
| 1H NMR | 9.02 ± 0.07 | 10.69 ± 0.06 | – | |
| RuCym–HQCl-D-hPro | UV-vis | 8.78 ± 0.01 | 10.03 ± 0.02 | 1.28 ± 0.01 |
| 1H NMR | 8.77 ± 0.05 | 10.13 ± 0.04 | – |
| FHA logKFHA′ | Trp-214 Quenching logKQ′, Site I | DG Displacement logKDG′, Site II | |
|---|---|---|---|
| RhCp*–HQCl-L-Pro a | 4.7 | 5.5 | 5.6 |
| RuCym–HQCl-L-Pro a | 5.1 | 5.7 | 6.1 |
| RhCp*–HQCl-D-Pro | 4.4 ± 0.2 | 5.6 ± 0.1 | 5.5 ± 0.1 |
| RuCym–HQCl-D-Pro | 4.4 ± 0.3 | 5.5 ± 0.1 | 5.6 ± 0.1 |
| RhCp*–HQCl-D-hPro | 4.5 ± 0.2 | 5.6 ± 0.1 | 5.4 ± 0.1 |
| RuCym–HQCl-D-hPro | 4.6 ± 0.2 | 5.3 ± 0.1 | 5.7 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pivarcsik, T.; Dömötör, O.; Mészáros, J.P.; May, N.V.; Spengler, G.; Csuvik, O.; Szatmári, I.; Enyedy, É.A. 8-Hydroxyquinoline-Amino Acid Hybrids and Their Half-Sandwich Rh and Ru Complexes: Synthesis, Anticancer Activities, Solution Chemistry and Interaction with Biomolecules. Int. J. Mol. Sci. 2021, 22, 11281. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011281
Pivarcsik T, Dömötör O, Mészáros JP, May NV, Spengler G, Csuvik O, Szatmári I, Enyedy ÉA. 8-Hydroxyquinoline-Amino Acid Hybrids and Their Half-Sandwich Rh and Ru Complexes: Synthesis, Anticancer Activities, Solution Chemistry and Interaction with Biomolecules. International Journal of Molecular Sciences. 2021; 22(20):11281. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011281
Chicago/Turabian StylePivarcsik, Tamás, Orsolya Dömötör, János P. Mészáros, Nóra V. May, Gabriella Spengler, Oszkár Csuvik, István Szatmári, and Éva A. Enyedy. 2021. "8-Hydroxyquinoline-Amino Acid Hybrids and Their Half-Sandwich Rh and Ru Complexes: Synthesis, Anticancer Activities, Solution Chemistry and Interaction with Biomolecules" International Journal of Molecular Sciences 22, no. 20: 11281. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011281