
DisA Restrains the Processing and Cleavage of Reversed Replication Forks by the RuvAB-RecU Resolvasome


Abstract
:1. Introduction
2. Results
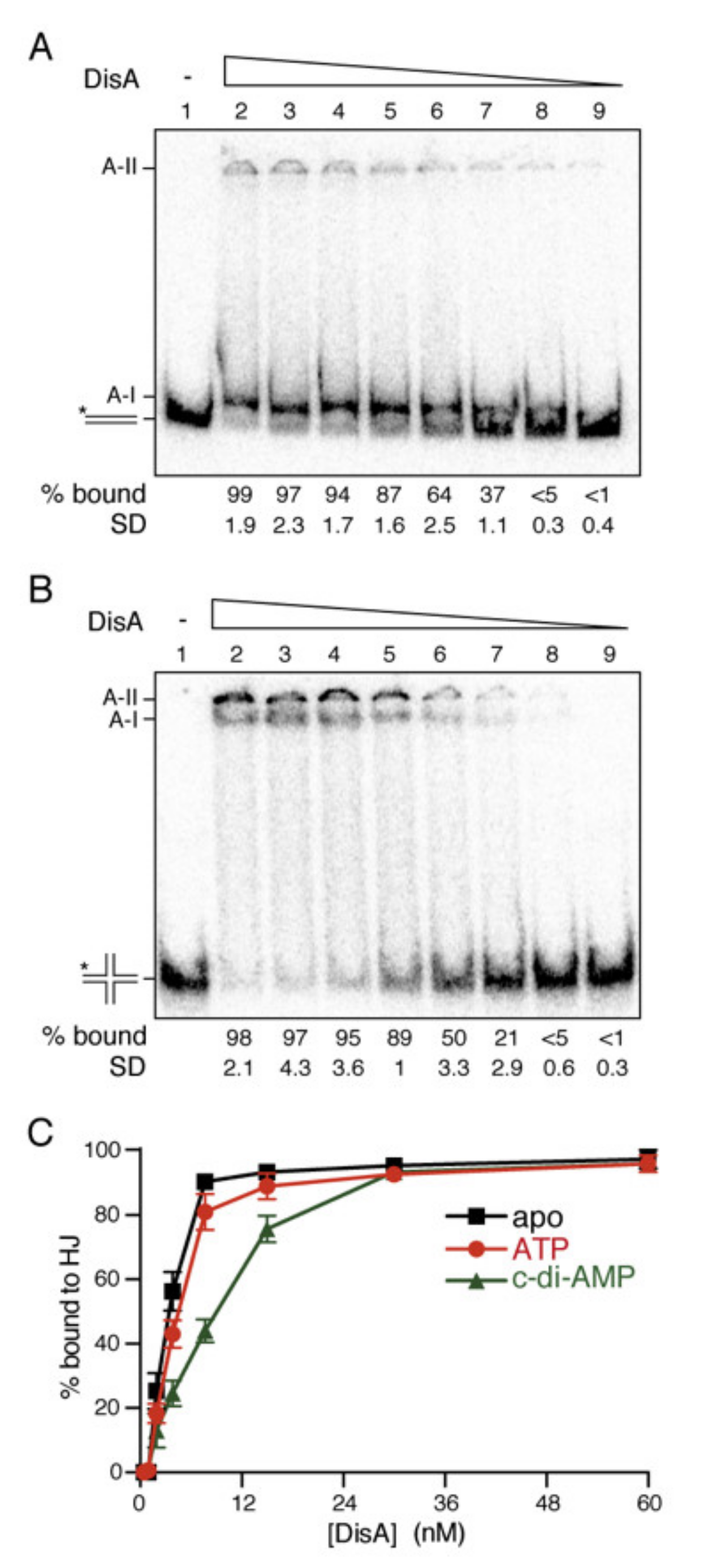
2.1. DisA Preferentially Binds DNA at High Mg2+ Concentrations
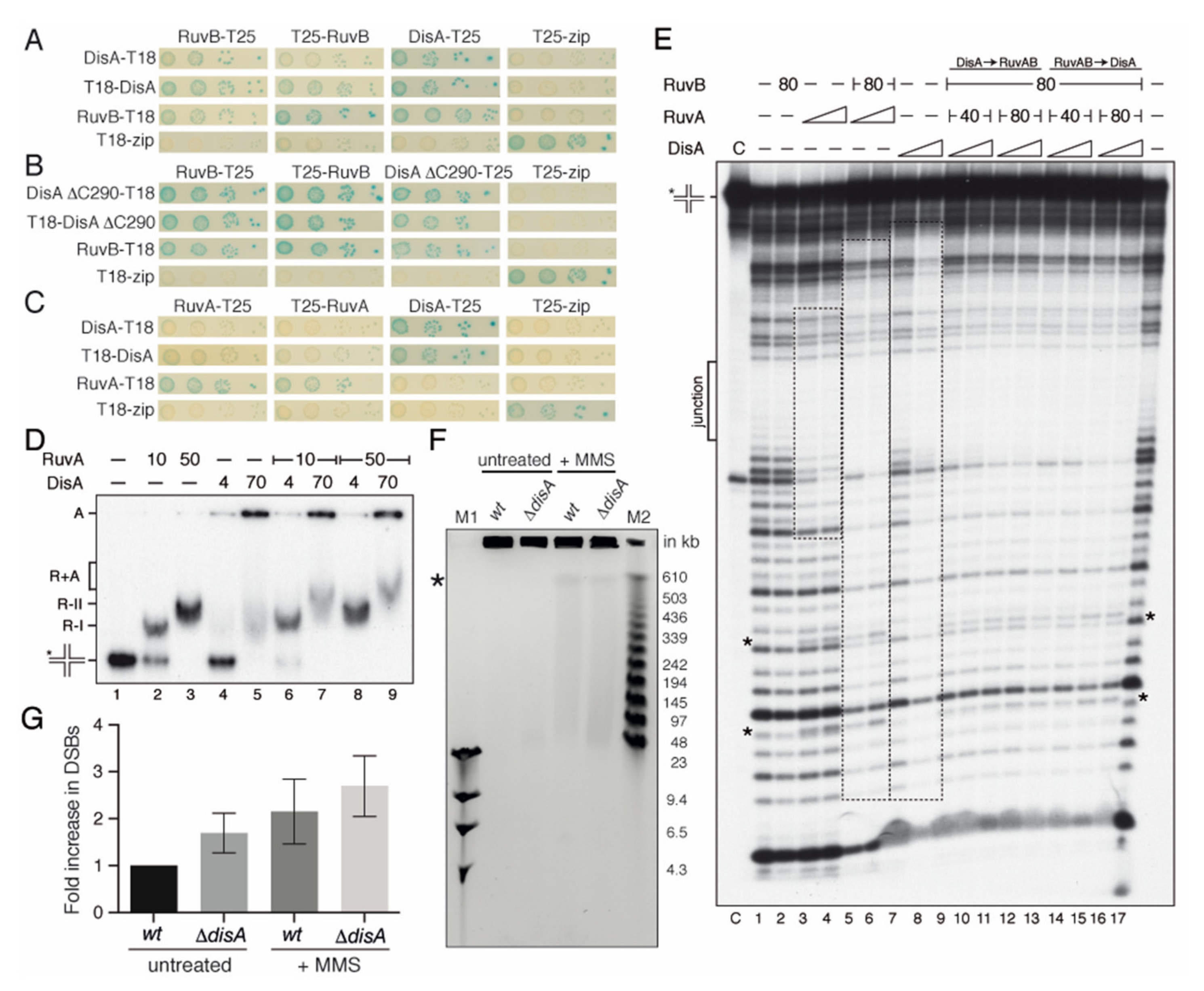
2.2. DisA Interacts with RuvB
2.3. DisA Coexists with RuvAB on HJ DNA
2.4. DisA May Protect Cells from Chromosome Breaks
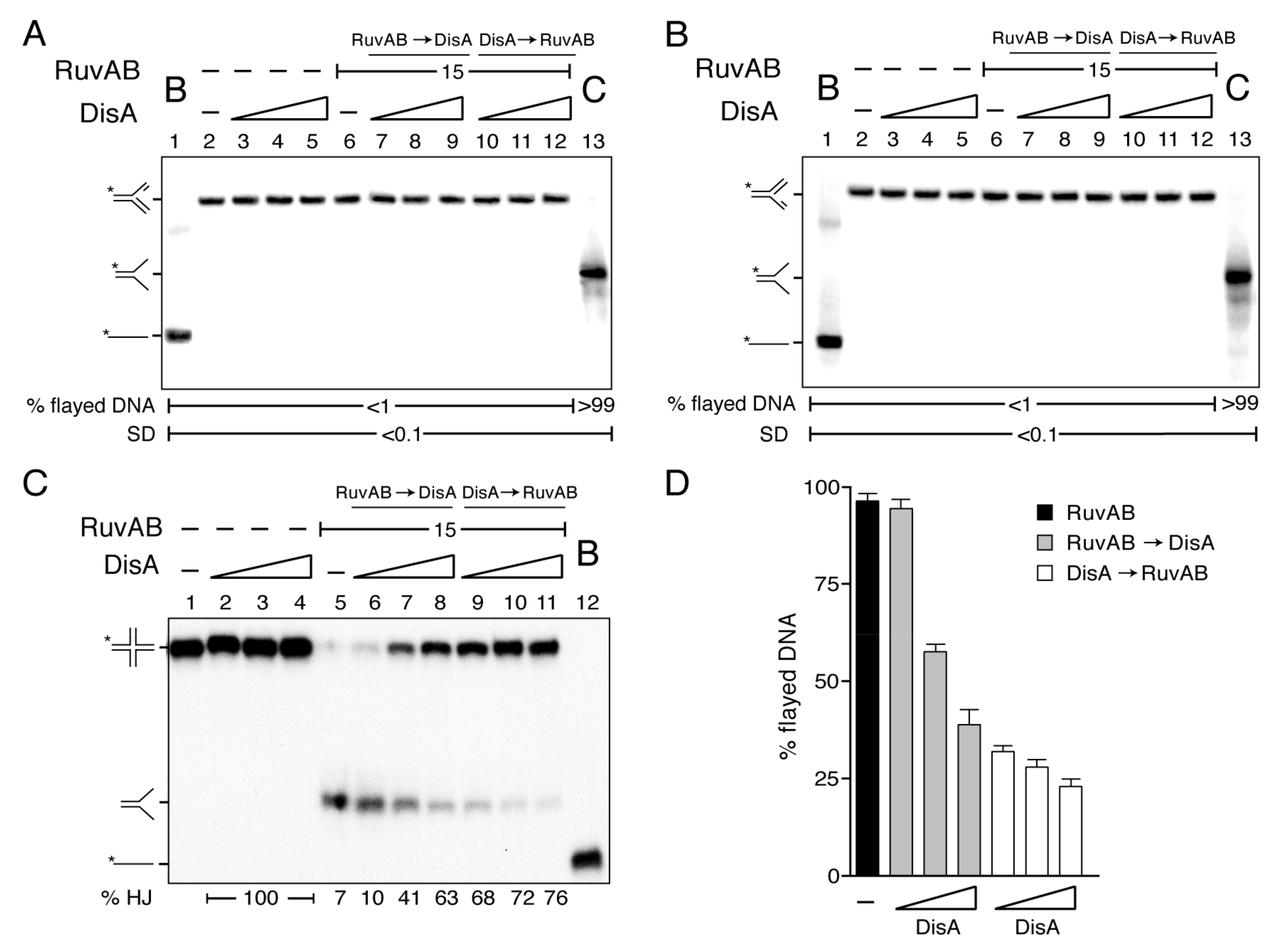
2.5. DisA Cannot Activate RuvAB to Catalyze Fork Reversal
2.6. DisA Bound to HJ DNA Inhibits RuvAB-Mediated Fork Restoration
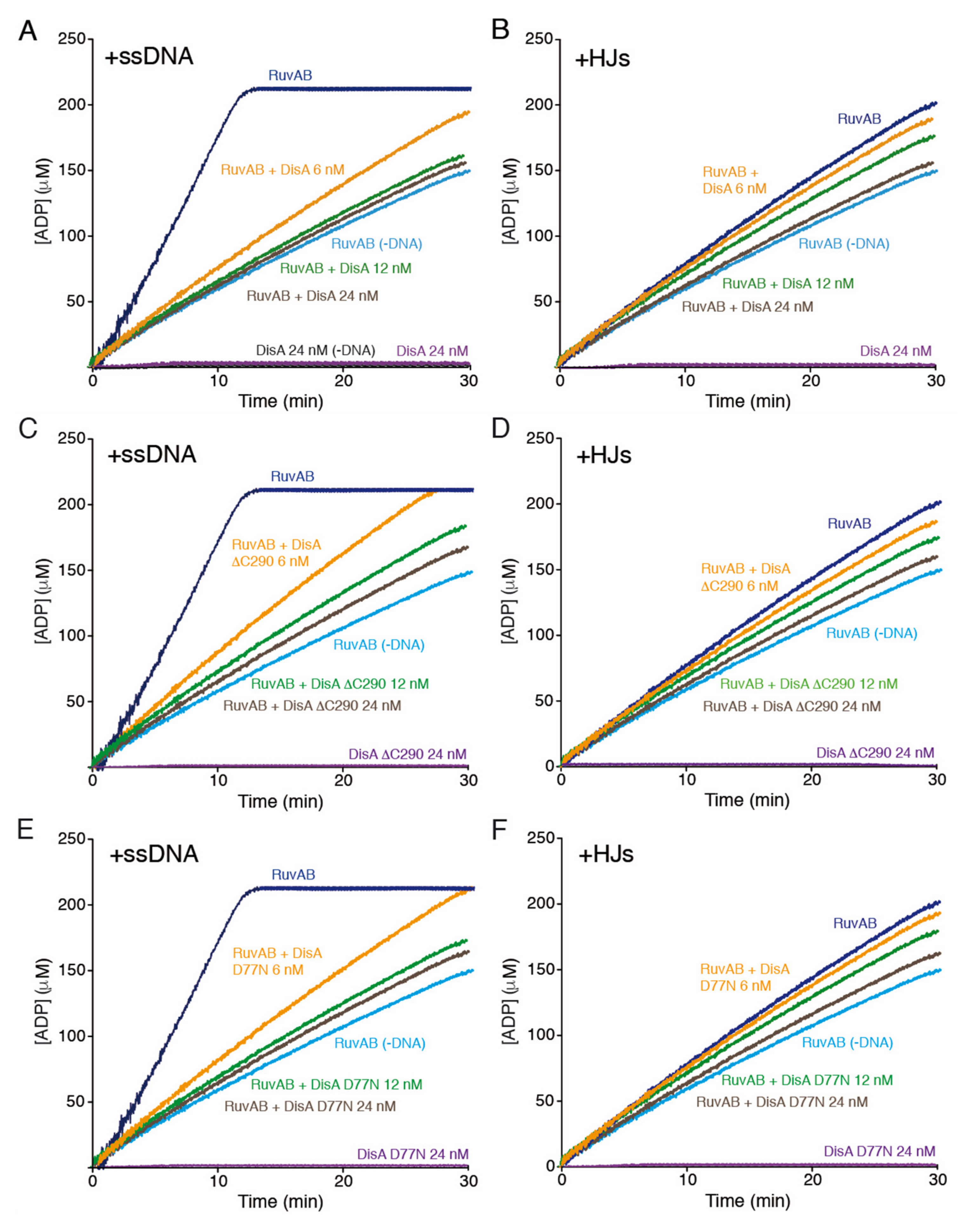
2.7. DisA Suppresses the DNA-Dependent ATPase Activity of RuvAB
2.8. DisA Does Not Compete with RuvAB for the Binding to DNA
2.9. The DAC Activity of DisA Does Not Compromise RuvAB-Mediated ATP Hydrolysis
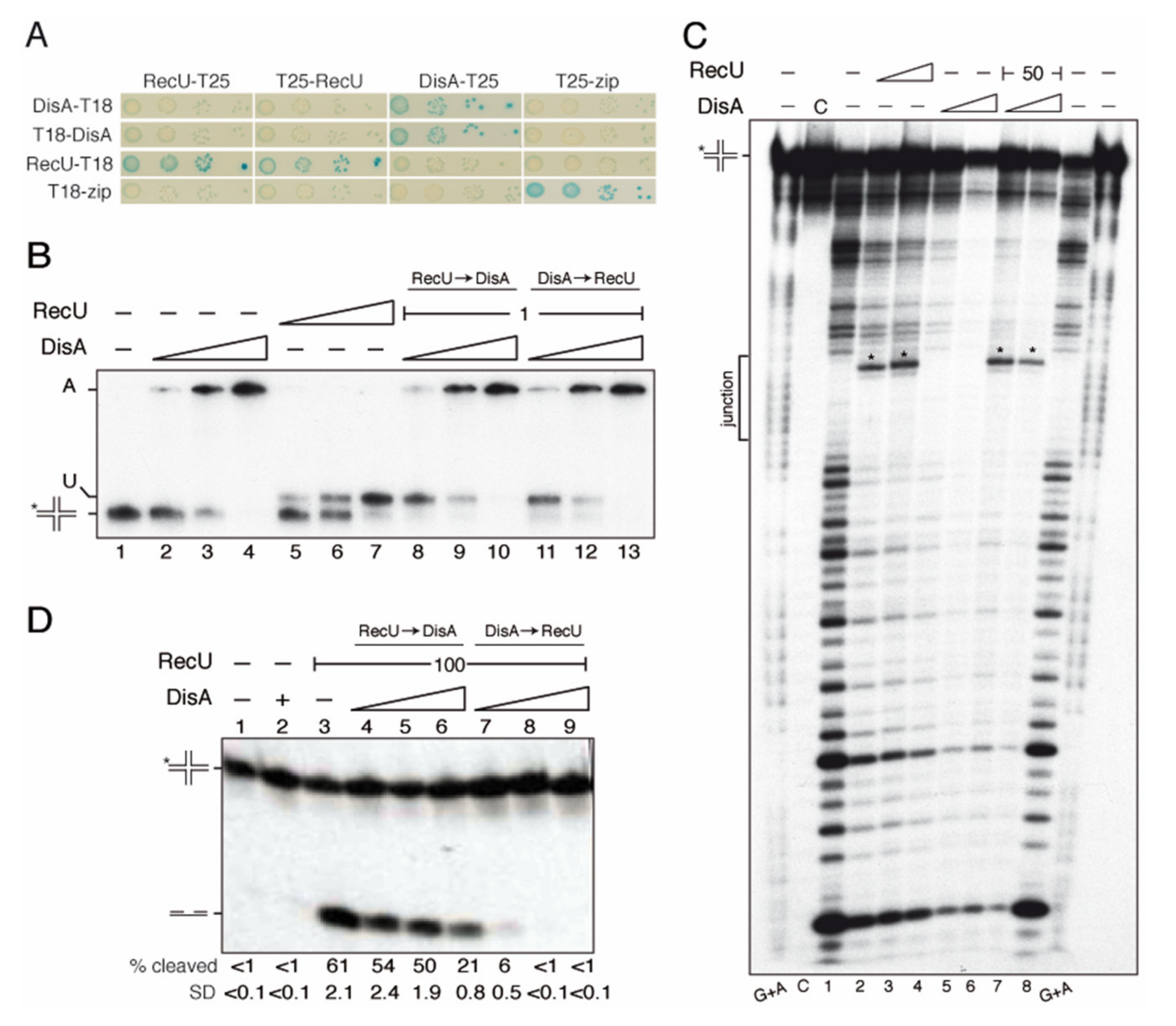
2.10. DisA Does Not Interact with RecU
2.11. DisA Limits RecU-Mediated HJ Cleavage
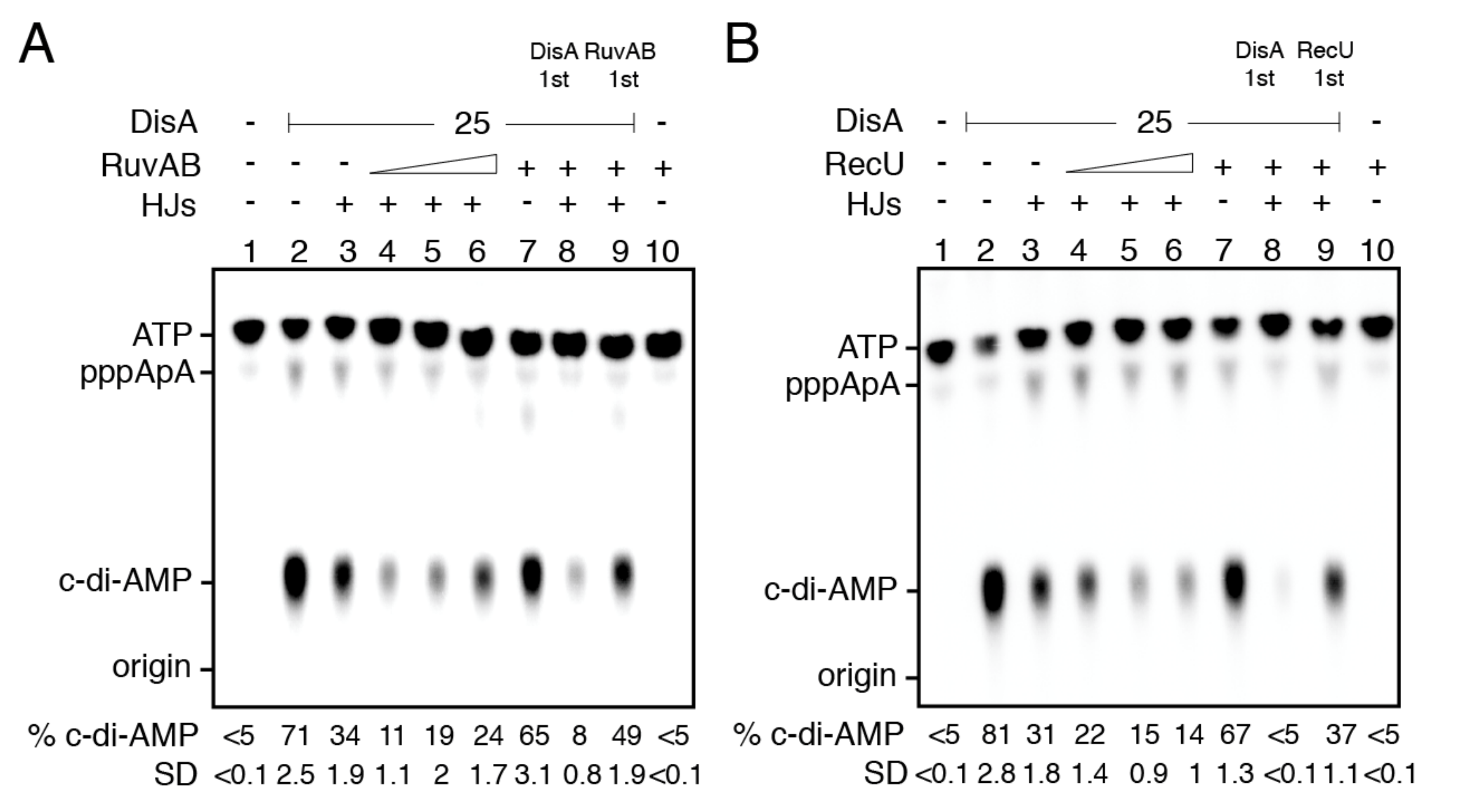
2.12. RuvAB- or RecU-Bound HJ-J3 DNA Blocks DisA-Mediated C-Di-AMP Synthesis
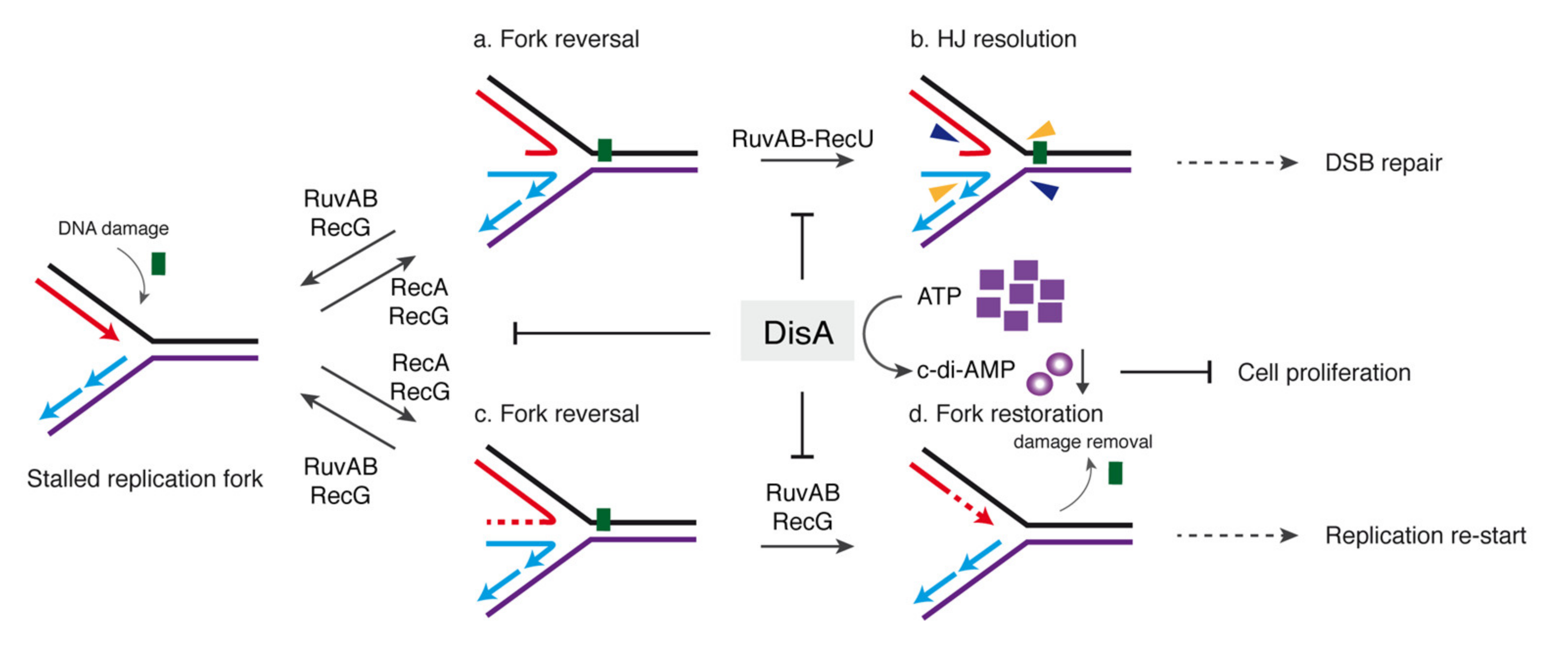
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Plasmids
4.2. Protein–Protein Interaction Assays
4.3. Pulse-Field Gel Electrophoresis
4.4. DNA Substrates
4.5. Protein Purification
4.6. ATPase Activity and c-di-AMP Synthesis
4.7. DNA Binding, HJ Branch Migration, and Cleavage Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cox, M.M.; Goodman, M.F.; Kreuzer, K.N.; Sherratt, D.J.; Sandler, S.J.; Marians, K.J. The importance of repairing stalled replication forks. Nature 2000, 404, 37–41. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczykowski, S.C. An Overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Marians, K.J. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Sinha, A.K.; Leach, D.R.F. Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 2018, 82, e00013-18. [Google Scholar] [CrossRef] [Green Version]
- Thakar, T.; Moldovan, G.L. The emerging determinants of replication fork stability. Nucleic. Acids Res. 2021, 49, 7224–7238. [Google Scholar] [CrossRef]
- Seigneur, M.; Bidnenko, V.; Ehrlich, S.D.; Michel, B. RuvAB acts at arrested replication forks. Cell 1998, 95, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Vlasic, I.; Mertens, R.; Seco, E.M.; Carrasco, B.; Ayora, S.; Reitz, G.; Commichau, F.M.; Alonso, J.C.; Moeller, R. Bacillus subtilis RecA and its accessory factors, RecF, RecO, RecR and RecX, are required for spore resistance to DNA double-strand break. Nucleic. Acids Res. 2014, 42, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Raguse, M.; Torres, R.; Seco, E.M.; Gándara, C.; Ayora, S.; Moeller, R.; Alonso, J.C. Bacillus subtilis DisA helps to circumvent replicative stress during spore revival. DNA Repair (Amst) 2017, 59, 57–68. [Google Scholar] [CrossRef]
- Nicolas, P.; Mader, U.; Dervyn, E.; Rochat, T.; Leduc, A.; Pigeonneau, N.; Bidnenko, E.; Marchadier, E.; Hoebeke, M.; Aymerich, S.; et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 2012, 335, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Sinai, L.; Rosenberg, A.; Smith, Y.; Segev, E.; Ben-Yehuda, S. The molecular timeline of a reviving bacterial spore. Mol. Cell 2015, 57, 695–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, R.M.; Grundling, A. Cyclic di-AMP: Another second messenger enters the fray. Nat. Rev. Microbiol. 2013, 11, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Bejerano-Sagie, M.; Oppenheimer-Shaanan, Y.; Berlatzky, I.; Rouvinski, A.; Meyerovich, M.; Ben-Yehuda, S. A checkpoint protein that scans the chromosome for damage at the start of sporulation in Bacillus subtilis. Cell 2006, 125, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Carrasco, B.; Gándara, C.; Baidya, A.K.; Ben-Yehuda, S.; Alonso, J.C. Bacillus subtilis DisA regulates RecA-mediated DNA strand exchange. Nucleic. Acids Res. 2019, 47, 5141–5154. [Google Scholar] [CrossRef] [Green Version]
- Oppenheimer-Shaanan, Y.; Wexselblatt, E.; Katzhendler, J.; Yavin, E.; Ben-Yehuda, S. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 2011, 12, 594–601. [Google Scholar] [CrossRef]
- Merrick, C.J.; Jackson, D.; Diffley, J.F. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004, 279, 20067–20075. [Google Scholar] [CrossRef] [Green Version]
- Gándara, C.; Alonso, J.C. DisA and c-di-AMP act at the intersection between DNA-damage response and stress homeostasis in exponentially growing Bacillus subtilis cells. DNA Repair (Amst) 2015, 27, 1–8. [Google Scholar] [CrossRef]
- Witte, G.; Hartung, S.; Buttner, K.; Hopfner, K.P. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 2008, 30, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Gándara, C.; de Lucena, D.K.C.; Torres, R.; Serrano, E.; Altenburger, S.; Graumann, P.L.; Alonso, J.C. Activity and in vivo dynamics of Bacillus subtilis DisA are affected by RadA/Sms and by Holliday junction-processing proteins. DNA Repair (Amst) 2017, 55, 17–30. [Google Scholar] [CrossRef]
- Torres, R.; Serrano, E.; Tramm, K.; Alonso, J.C. Bacillus subtilis RadA/Sms contributes to chromosomal transformation and DNA repair in concert with RecA and circumvents replicative stress in concert with DisA. DNA Repair (Amst) 2019, 77, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Kruger, L.; Herzberg, C.; Wicke, D.; Bahre, H.; Heidemann, J.L.; Dickmanns, A.; Schmitt, K.; Ficner, R.; Stulke, J. A meet-up of two second messengers: The c-di-AMP receptor DarB controls (p)ppGpp synthesis in Bacillus subtilis. Nat. Commun. 2021, 12, 1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Sanders, G.M.; Grossman, A.D. Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 2007, 128, 865–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, R.; Gándara, C.; Carrasco, B.; Baquedano, I.; Ayora, S.; Alonso, J.C. DisA limits RecG activities at stalled or reversed replication forks. Cells 2021, 10, 1357. [Google Scholar] [CrossRef]
- Robu, M.E.; Inman, R.B.; Cox, M.M. RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 8211–8218. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, P.; Mahdi, A.A.; Lloyd, R.G. Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 2000, 28, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, P.; Lloyd, R.G.; Marians, K.J. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. USA 2001, 98, 8235–8240. [Google Scholar] [CrossRef] [Green Version]
- Abd Wahab, S.; Choi, M.; Bianco, P.R. Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J. Biol. Chem. 2013, 288, 26397–26409. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, D.; Manosas, M.; Zhang, W.; Manthei, K.A.; Hodeib, S.; Ducos, B.; Keck, J.L.; Croquette, V. Single molecule kinetics uncover roles for E. coli RecQ DNA helicase domains and interaction with SSB. Nucleic. Acids Res. 2018, 46, 8500–8515. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Alonso, J.C. Bacillus subtilis RecA, DisA and RadA/Sms interplay prevents replication stress by regulating fork remodeling. Front. Microbiol. 2021. In press. [Google Scholar]
- Au, N.; Kuester-Schoeck, E.; Mandava, V.; Bothwell, L.E.; Canny, S.P.; Chachu, K.; Colavito, S.A.; Fuller, S.N.; Groban, E.S.; Hensley, L.A.; et al. Genetic composition of the Bacillus subtilis SOS system. J. Bacteriol. 2005, 187, 7655–7666. [Google Scholar] [CrossRef] [Green Version]
- Eiamphungporn, W.; Helmann, J.D. The Bacillus subtilis σM regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 2008, 67, 830–848. [Google Scholar] [CrossRef] [Green Version]
- Cañas, C.; Carrasco, B.; Garcia-Tirado, E.; Rafferty, J.B.; Alonso, J.C.; Ayora, S. The stalk region of the RecU resolvase is essential for Holliday junction recognition and distortion. J. Mol. Biol. 2011, 410, 39–49. [Google Scholar] [CrossRef]
- West, S.C. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 1997, 31, 213–244. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.; Rice, D.W.; Sedelnikova, S.E.; Artymiuk, P.J.; Lloyd, R.G.; Rafferty, J.B. Crystal structure of E. coli RuvA with bound DNA Holliday junction at 6 Å resolution. Nat. Struct. Biol. 1998, 5, 441–446. [Google Scholar] [CrossRef] [PubMed]
- van Gool, A.J.; Shah, R.; Mezard, C.; West, S.C. Functional interactions between the Holliday junction resolvase and the branch migration motor of Escherichia coli. EMBO J. 1998, 17, 1838–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Ariyoshi, M.; Iwasaki, H.; Shinagawa, H.; Morikawa, K. Functional analyses of the domain structure in the Holliday junction binding protein RuvA. Structure 1998, 6, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Cañas, C.; Suzuki, Y.; Marchisone, C.; Carrasco, B.; Freire-Beneitez, V.; Takeyasu, K.; Alonso, J.C.; Ayora, S. Interaction of branch migration translocases with the Holliday junction-resolving enzyme and their implications in Holliday junction resolution. J. Biol. Chem. 2014, 289, 17634–17646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, N.; Ayora, S.; Sedelnikova, S.; Carrasco, B.; Alonso, J.C.; Thaw, P.; Rafferty, J. The structure of Bacillus subtilis RecU Holliday junction resolvase and its role in substrate selection and sequence-specific cleavage. Structure 2005, 13, 1341–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khavnekar, S.; Dantu, S.C.; Sedelnikova, S.; Ayora, S.; Rafferty, J.; Kale, A. Structural insights into dynamics of RecU-HJ complex formation elucidates key role of NTR and stalk region toward formation of reactive state. Nucleic. Acids Res. 2017, 45, 975–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit, R.; Gileadi, O.; Stavans, J. Direct observation of RuvAB-catalyzed branch migration of single Holliday junctions. Proc. Natl. Acad. Sci. USA 2004, 101, 11605–11610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawid, A.; Croquette, V.; Grigoriev, M.; Heslot, F. Single-molecule study of RuvAB-mediated Holliday-junction migration. Proc. Natl. Acad. Sci. USA 2004, 101, 11611–11616. [Google Scholar] [CrossRef] [Green Version]
- Ayora, S.; Carrasco, B.; Doncel, E.; Lurz, R.; Alonso, J.C. Bacillus subtilis RecU protein cleaves Holliday junctions and anneals single-stranded DNA. Proc. Natl. Acad. Sci. USA 2004, 101, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, B.; Cañas, C.; Sharples, G.J.; Alonso, J.C.; Ayora, S. The N-terminal region of the RecU holliday junction resolvase is essential for homologous recombination. J. Mol. Biol. 2009, 390, 1–9. [Google Scholar] [CrossRef]
- Suzuki, Y.; Endo, M.; Cañas, C.; Ayora, S.; Alonso, J.C.; Sugiyama, H.; Takeyasu, K. Direct analysis of Holliday junction resolving enzyme in a DNA origami nanostructure. Nucleic. Acids Res. 2014, 42, 7421–7428. [Google Scholar] [CrossRef]
- Parsons, C.A.; Tsaneva, I.; Lloyd, R.G.; West, S.C. Interaction of Escherichia coli RuvA and RuvB proteins with synthetic Holliday junctions. Proc. Natl. Acad. Sci. USA 1992, 89, 5452–5456. [Google Scholar] [CrossRef] [Green Version]
- Ingleston, S.M.; Dickman, M.J.; Grasby, J.A.; Hornby, D.P.; Sharples, G.J.; Lloyd, R.G. Holliday junction binding and processing by the RuvA protein of Mycoplasma pneumoniae. Eur. J. Biochem. 2002, 269, 1525–1533. [Google Scholar] [CrossRef]
- Panyutin, I.G.; Biswas, I.; Hsieh, P. A pivotal role for the structure of the Holliday junction in DNA branch migration. EMBO J. 1995, 14, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K.; West, S.C. Branch migration during homologous recombination: Assembly of a RuvAB-Holliday junction complex in vitro. Cell 1995, 80, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.R.; Kuzminov, A. Trapping and breaking of in vivo nicked DNA during pulsed field gel electrophoresis. Anal. Biochem. 2013, 443, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawer, J.S.; Leach, D.R. Branch migration prevents DNA loss during double-strand break repair. PLoS Genet. 2014, 10, e1004485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrione, P.E.; Cox, M.M. RuvB protein-mediated ATP hydrolysis: Functional asymmetry in the RuvB hexamer. Biochemistry 1995, 34, 9809–9818. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Takahagi, M.; Nakata, A.; Shinagawa, H. Escherichia coli RuvA and RuvB proteins specifically interact with Holliday junctions and promote branch migration. Genes Dev. 1992, 6, 2214–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, B.; Ayora, S.; Lurz, R.; Alonso, J.C. Bacillus subtilis RecU Holliday-junction resolvase modulates RecA activities. Nucleic. Acids Res. 2005, 33, 3942–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiameli, S.M.; Merrikh, C.N.; Wiggins, P.A.; Merrikh, H. Transcription leads to pervasive replisome instability in bacteria. eLife 2017, 6. [Google Scholar] [CrossRef]
- Yadav, T.; Carrasco, B.; Myers, A.R.; George, N.P.; Keck, J.L.; Alonso, J.C. Genetic recombination in Bacillus subtilis: A division of labor between two single-strand DNA-binding proteins. Nucleic. Acids Res. 2012, 40, 5546–5559. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, B.; Yadav, T.; Serrano, E.; Alonso, J.C. Bacillus subtilis RecO and SsbA are crucial for RecA-mediated recombinational DNA repair. Nucleic. Acids Res. 2015, 43, 5984–5997. [Google Scholar] [CrossRef] [Green Version]
- Cañas, C.; Carrasco, B.; Ayora, S.; Alonso, J.C. The RecU Holliday junction resolvase acts at early stages of homologous recombination. Nucleic. Acids Res. 2008, 36, 5242–5249. [Google Scholar] [CrossRef] [PubMed]
- de la Hoz:, A.B.; Ayora, S.; Sitkiewicz, I.; Fernandez, S.; Pankiewicz, R.; Alonso, J.C.; Ceglowski, P. Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. USA 2000, 97, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ± DNA | Kcat (min−1) a |
|---|---|
| RuvA (15 nM), no DNA | <0.1 |
| RuvB (15 nM), no DNA | 283 ± 5 |
| RuvAB (15 nM), no DNA | 280 ± 7 |
| RuvAB + HJ DNA | 660 ± 13 |
| RuvAB + HJ DNA + 6 nM DisA b | 645 ± 11 |
| RuvAB + HJ DNA + 12 nM DisA c | 554 ± 9 |
| RuvAB + HJ DNA + 24 nM DisA d | 321 ± 11 |
| RuvAB + HJ DNA + 6 nM DisA D77N b | 651 ± 11 |
| RuvAB + HJ DNA + 12 nM DisA D77N c | 578 ± 7 |
| RuvAB + HJ DNA + 24 nM DisA D77N d | 354 ± 9 |
| RuvAB + HJ DNA + 6 nM DisA ΔC290 b | 640 ± 15 |
| RuvAB + HJ DNA + 12 nM DisA ΔC290 c | 547 ± 11 |
| RuvAB + HJ DNA + 24 nM DisA ΔC290 d | 351 ± 10 |
| RuvAB + ssDNA | 1253 ± 19 |
| RuvAB + ssDNA + 6 nM DisA b | 405 ± 8 |
| RuvAB + ssDNA + 12 nM DisA c | 329 ± 4 |
| RuvAB + ssDNA + 24 nM DisA d | 301 ± 5 |
| RuvAB + ssDNA + 6 nM DisA D77N b | 433 ± 13 |
| RuvAB + ssDNA + 12 nM DisA D77N c | 350 ± 9 |
| RuvAB + ssDNA + 24 nM DisA D77N d | 330 ± 8 |
| RuvAB + ssDNA + 6 nM DisA ΔC290 b | 461 ± 14 |
| RuvAB + ssDNA + 12 nM DisA ΔC290 c | 375 ± 11 |
| RuvAB + ssDNA + 24 nM DisA ΔC290 d | 341 ± 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gándara, C.; Torres, R.; Carrasco, B.; Ayora, S.; Alonso, J.C. DisA Restrains the Processing and Cleavage of Reversed Replication Forks by the RuvAB-RecU Resolvasome. Int. J. Mol. Sci. 2021, 22, 11323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111323
Gándara C, Torres R, Carrasco B, Ayora S, Alonso JC. DisA Restrains the Processing and Cleavage of Reversed Replication Forks by the RuvAB-RecU Resolvasome. International Journal of Molecular Sciences. 2021; 22(21):11323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111323
Chicago/Turabian StyleGándara, Carolina, Rubén Torres, Begoña Carrasco, Silvia Ayora, and Juan C. Alonso. 2021. "DisA Restrains the Processing and Cleavage of Reversed Replication Forks by the RuvAB-RecU Resolvasome" International Journal of Molecular Sciences 22, no. 21: 11323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111323