
Post-Stroke Timing of ECM Hydrogel Implantation Affects Biodegradation and Tissue Restoration


 ,
,
Abstract
:1. Introduction
2. Results
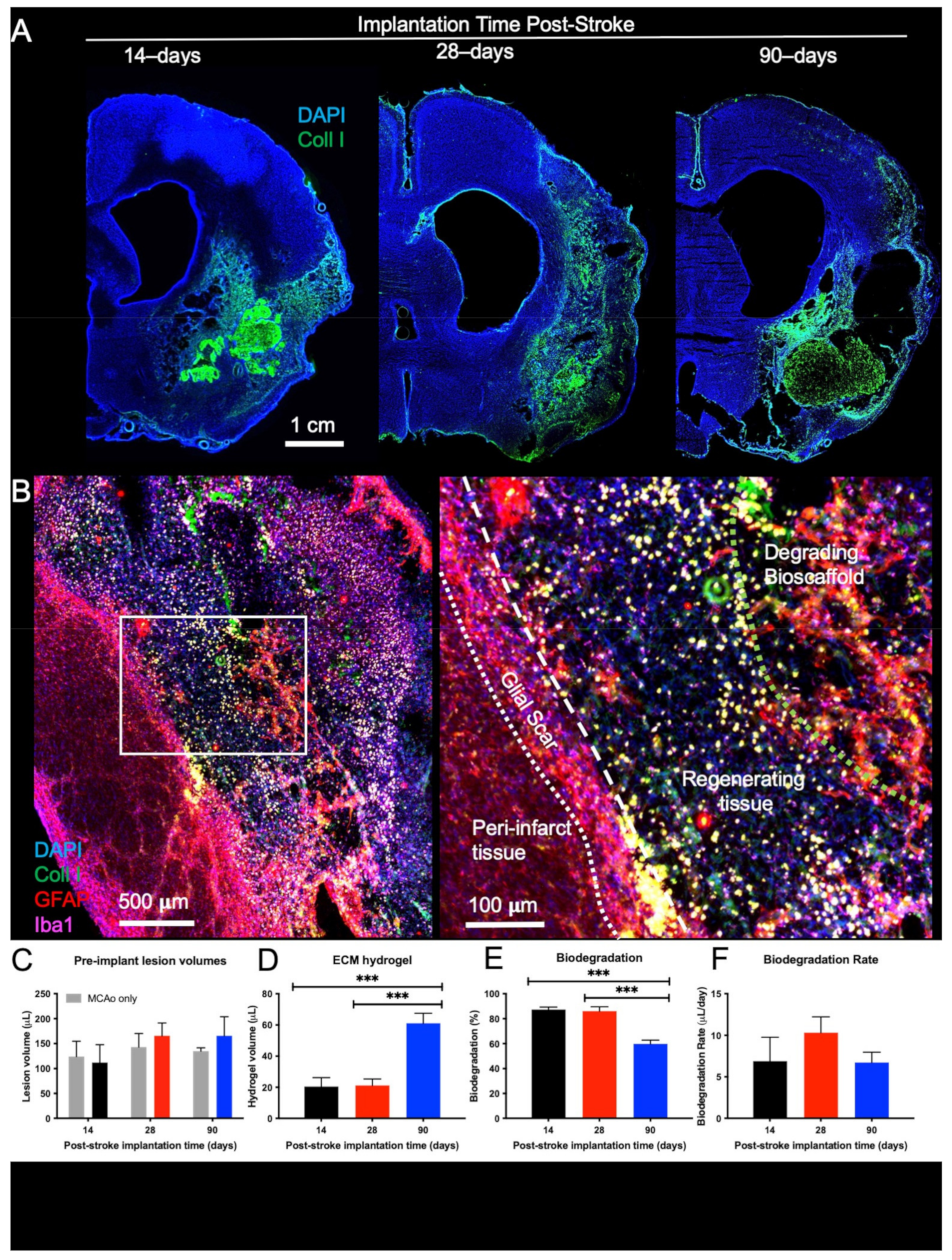
2.1. Biodegradation Is Influenced by Post-Stroke Implantation Timing
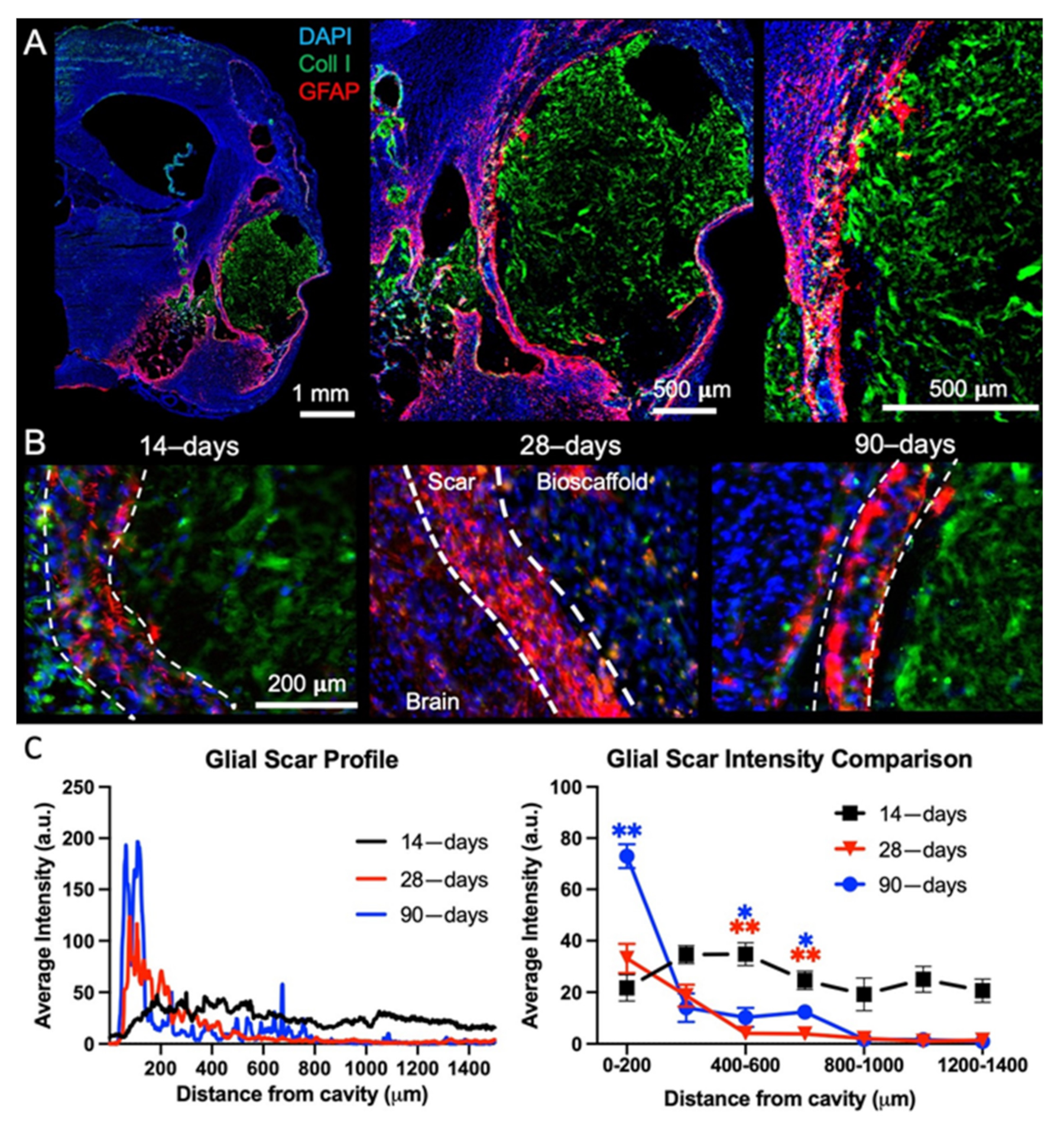
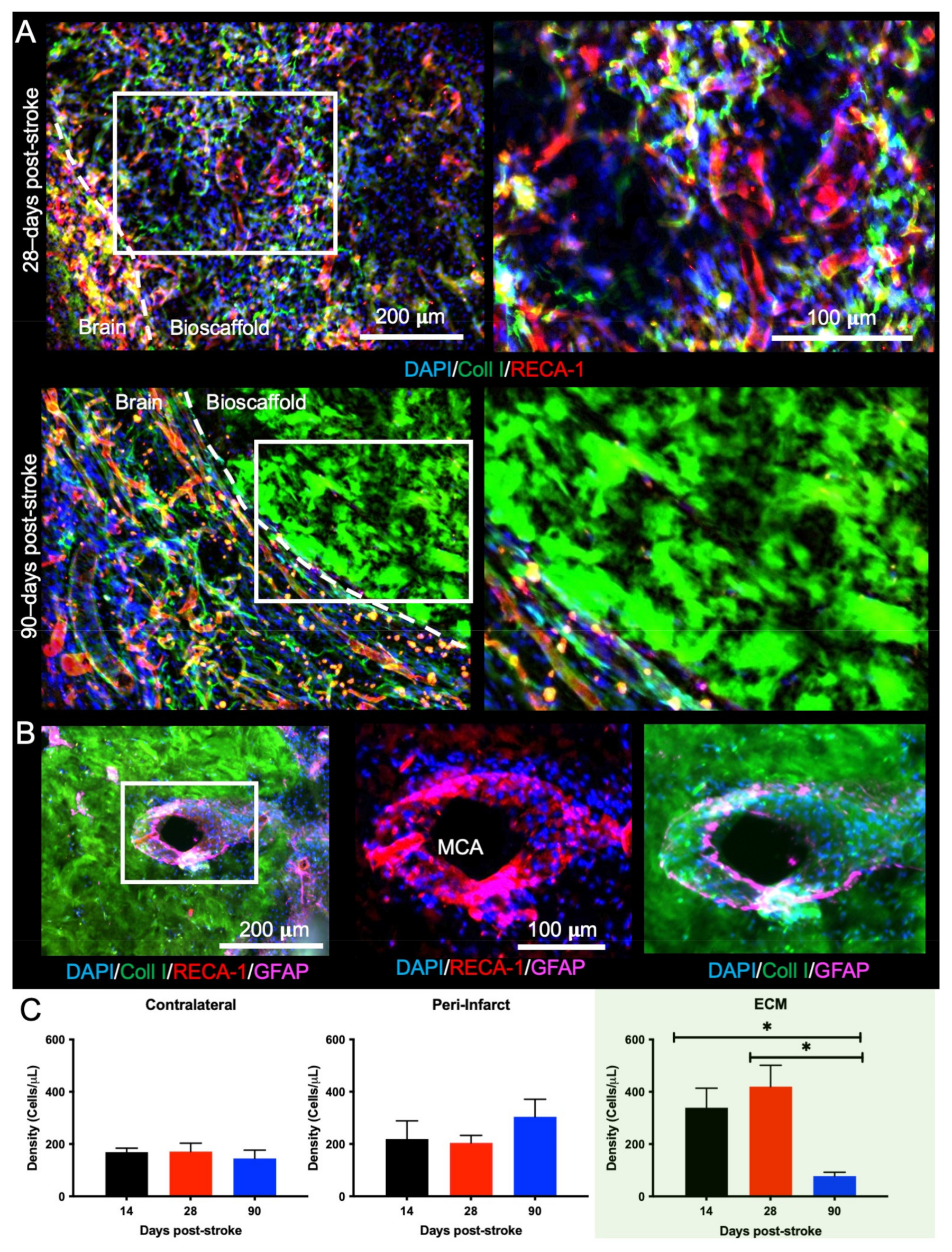
2.2. Post-Stroke Maturation of Glial Scar Affects Biodegradation
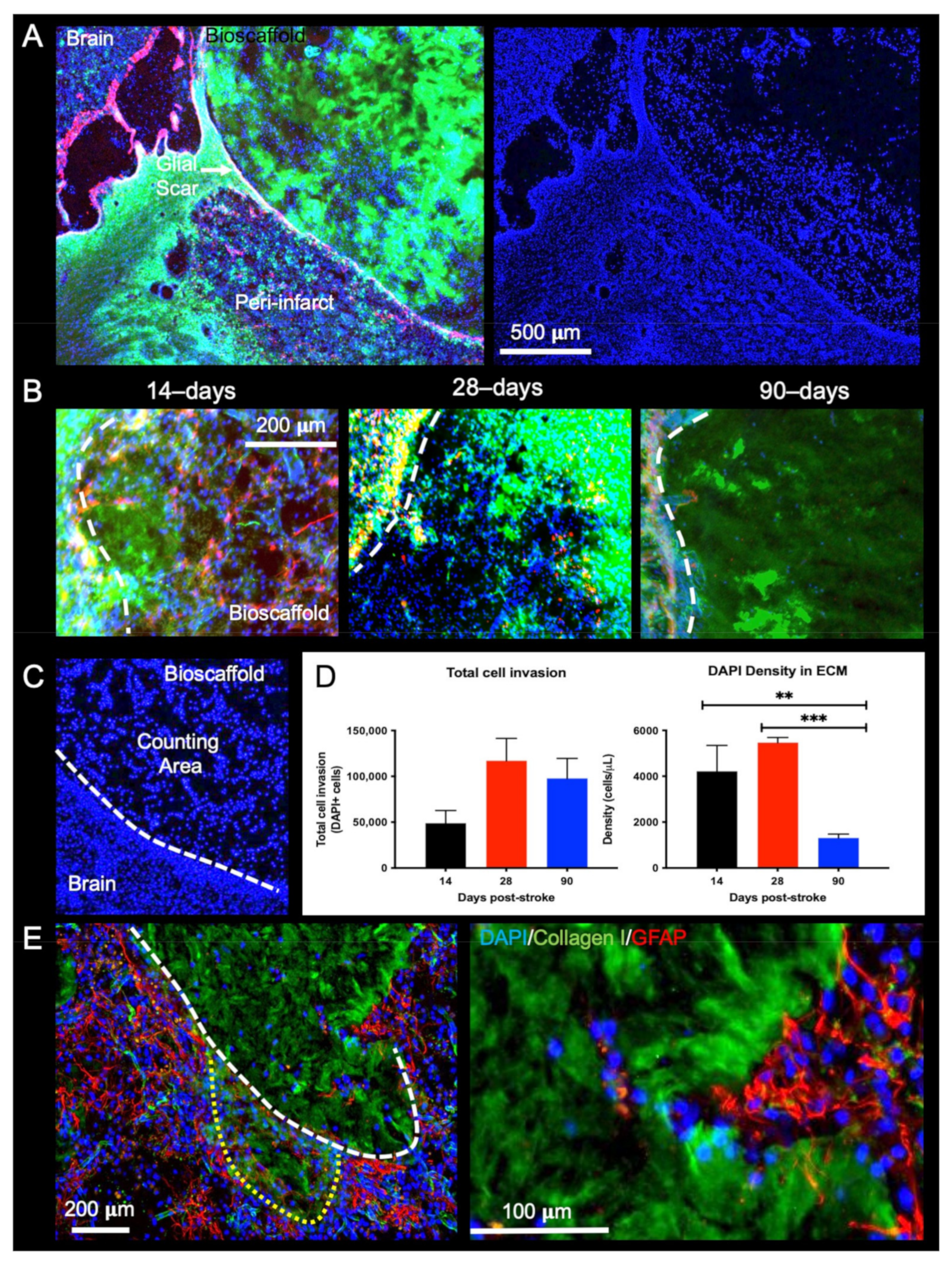
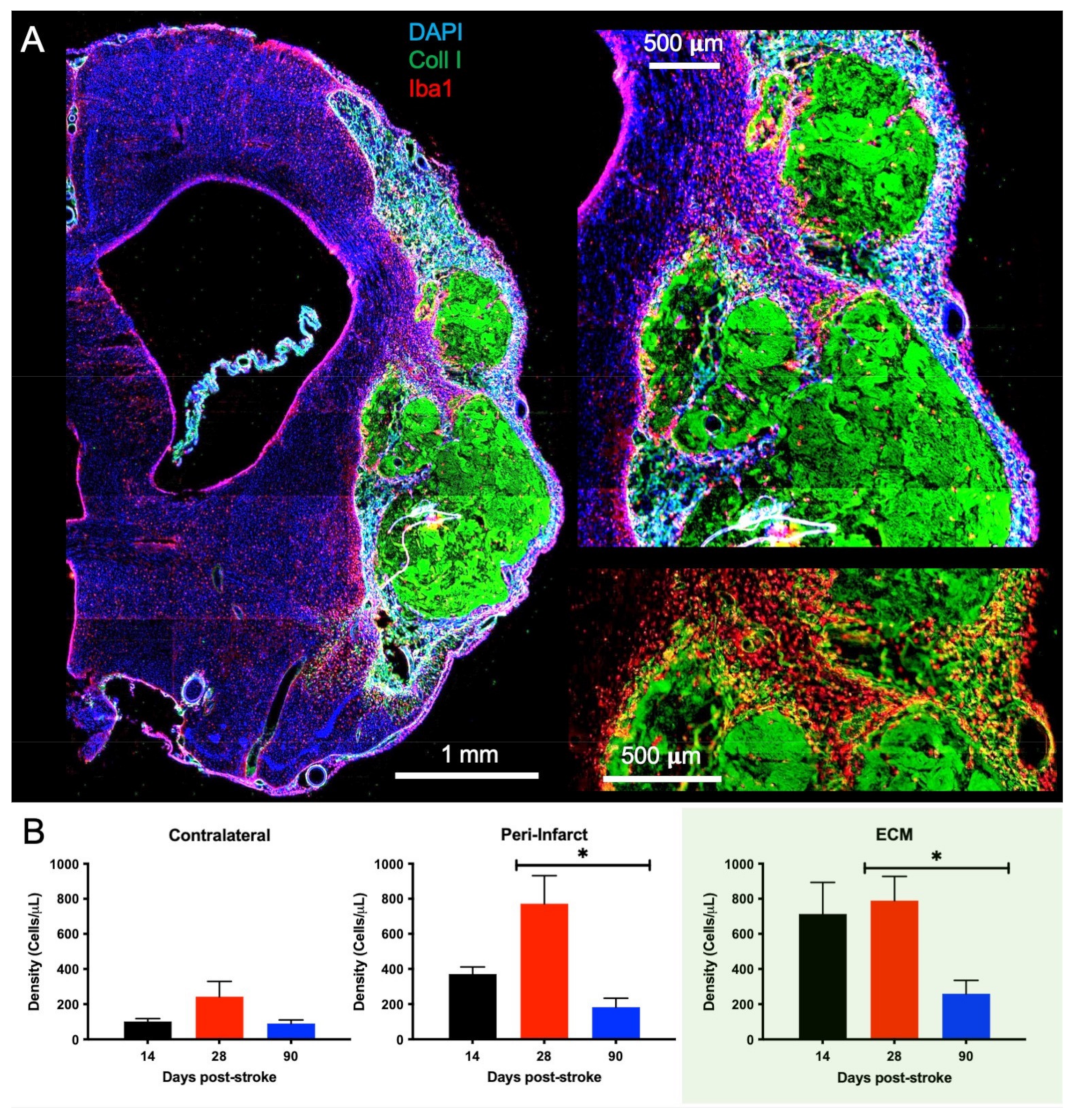
2.3. Post-Stroke Implantation Timing Affects Host Cell Infiltration
2.4. Macrophage Invasion and Pro-Repair Polarization Is Reduced with a Delay in Post-Stroke Implantation
2.5. Bioscaffold Vascularization Is Reduced with a 90-Day Post-Stroke Implantation
2.6. Delayed Implantation of ECM Hydrogel Reduces Oligodendrocytes in the Bioscaffold
3. Discussion
3.1. Impact of Post-Stroke Implantation Timing on Biodegradation
3.2. Brain Cell Invasion Is Reduced with Prolonged Post-Stroke Implantation
3.3. Translational Considerations for a Therapeutic Window of Bioscaffold Implantation
4. Materials and Methods
4.1. Extracellular Matrix (ECM) Hydrogel
4.2. Middle Cerebral Artery Occlusion (MCAo)
4.3. Magnetic Resonance Imaging (MRI)
4.3.1. Acquisition
4.3.2. Lesion Volume and Intensity Measurements
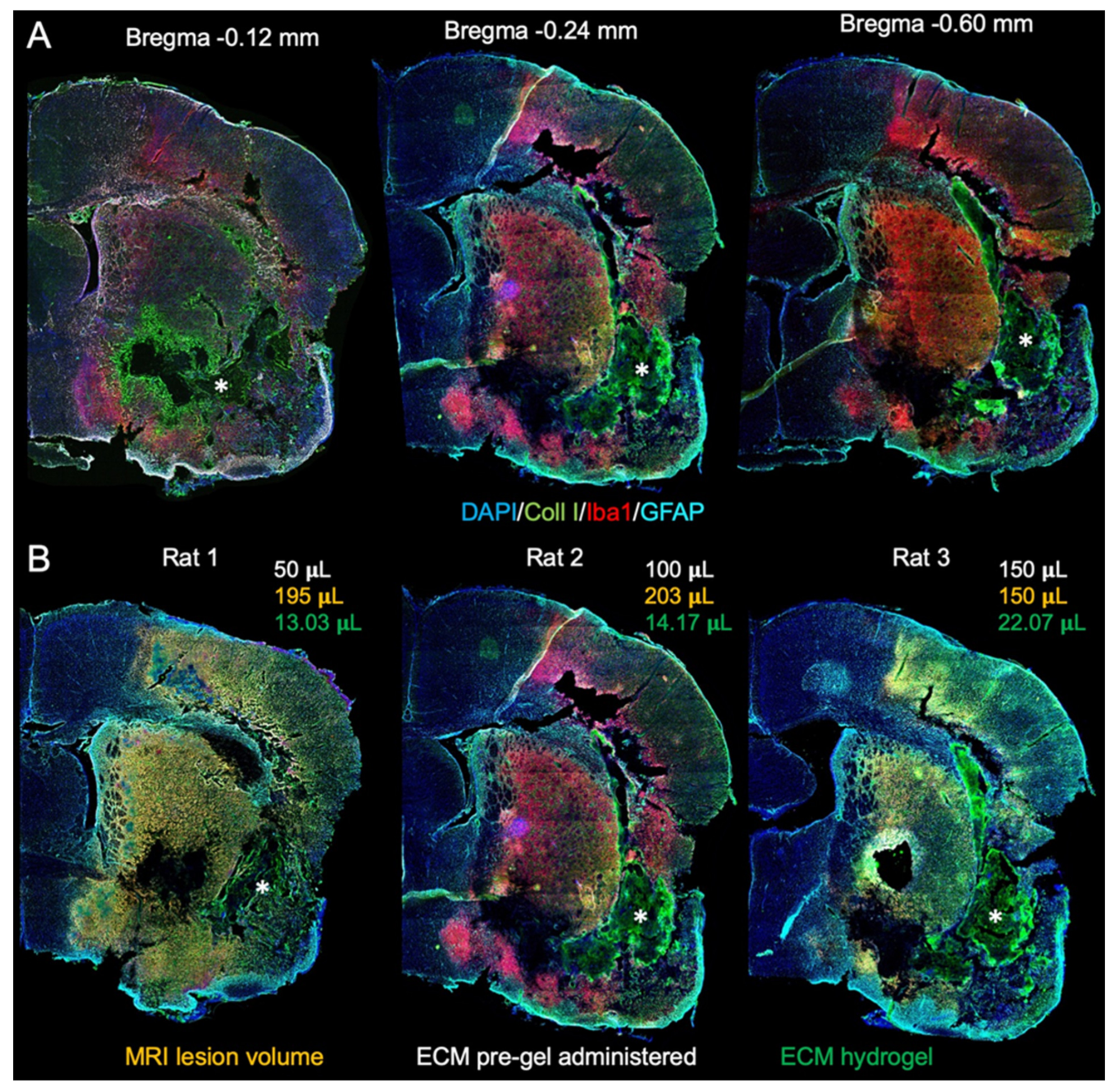
4.4. MRI-Guided Implantation of ECM Hydrogel
4.5. Histologic Analyses
4.5.1. Perfusion-Fixation of Tissue
4.5.2. Immunohistochemistry
4.5.3. ECM Hydrogel Volume
4.5.4. Glial Scarring
4.5.5. Cell Invasion
4.5.6. Cell Phenotypes
4.6. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baddour, J.A.; Sousounis, K.; Tsonis, P.A. Organ repair and regeneration: An overview. Birth Defects Res. C Embryo Today 2012, 96, 1–29. [Google Scholar] [CrossRef]
- Bechmann, I. Failed central nervous system regeneration: A downside of immune privilege? Neuromolecular Med. 2005, 7, 217–228. [Google Scholar] [CrossRef]
- Meairs, S.; Wahlgren, N.; Dirnagl, U.; Lindvall, O.; Rothwell, P.; Baron, J.C.; Hossmann, K.; Engelhardt, B.; Ferro, J.; McCulloch, J.; et al. Stroke research priorities for the next decade--A representative view of the European scientific community. Cerebrovasc. Dis. 2006, 22, 75–82. [Google Scholar] [CrossRef]
- Smith, E.J.; Stroemer, R.P.; Gorenkova, N.; Nakajima, M.; Crum, W.R.; Tang, E.; Stevanato, L.; Sinden, J.D.; Modo, M. Implantation site and lesion topology determine efficacy of a human neural stem cell line in a rat model of chronic stroke. Stem Cells 2012, 30, 785–796. [Google Scholar] [CrossRef]
- Moreau, F.; Patel, S.; Lauzon, M.L.; McCreary, C.R.; Goyal, M.; Frayne, R.; Demchuk, A.M.; Coutts, S.B.; Smith, E.E. Cavitation after acute symptomatic lacunar stroke depends on time, location, and MRI sequence. Stroke 2012, 43, 1837–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashioti, M.; Beech, J.S.; Lowe, A.S.; Hesselink, M.B.; Modo, M.; Williams, S.C. Multi-modal characterisation of the neocortical clip model of focal cerebral ischaemia by MRI, behaviour and immunohistochemistry. Brain Res. 2007, 1145, 177–189. [Google Scholar] [CrossRef]
- Modo, M.; Beech, J.S.; Meade, T.J.; Williams, S.C.; Price, J. A chronic 1 year assessment of MRI contrast agent-labelled neural stem cell transplants in stroke. Neuroimage 2009, 47 (Suppl. 2), T133–T142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Zbesko, J.C.; Nguyen, T.V.; Yang, T.; Frye, J.B.; Hussain, O.; Hayes, M.; Chung, A.; Day, W.A.; Stepanovic, K.; Krumberger, M.; et al. Glial scars are permeable to the neurotoxic environment of chronic stroke infarcts. Neurobiol. Dis. 2018, 112, 63–78. [Google Scholar] [CrossRef]
- Loos, C.M.; Staals, J.; Wardlaw, J.M.; van Oostenbrugge, R.J. Cavitation of deep lacunar infarcts in patients with first-ever lacunar stroke: A 2-year follow-up study with MR. Stroke 2012, 43, 2245–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitch, M.T.; Doller, C.; Combs, C.K.; Landreth, G.E.; Silver, J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: In vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J. Neurosci. 1999, 19, 8182–8198. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.W.; Asher, R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999, 49, 377–391. [Google Scholar] [CrossRef]
- Sherman, L.S.; Back, S.A. A ’GAG’ reflex prevents repair of the damaged CNS. Trends Neurosci. 2008, 31, 44–52. [Google Scholar] [CrossRef]
- Ridet, J.L.; Malhotra, S.K.; Privat, A.; Gage, F.H. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997, 20, 570–577. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kawabori, M.; Yenari, M.A. Innate inflammatory responses in stroke: Mechanisms and potential therapeutic targets. Curr. Med. Chem. 2014, 21, 2076–2097. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, H.; Mauney, C.; Donnelly, J.; Massensini, A.R.; Badylak, S.F.; Modo, M. Biodegradation of ECM hydrogel promotes endogenous brain tissue restoration in a rat model of stroke. Acta Biomater. 2018, 80, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Modo, M. Bioscaffold-Induced Brain Tissue Regeneration. Front. Neurosci. 2019, 13, 1156. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.N.; Valentin, J.E.; Stewart-Akers, A.M.; McCabe, G.P.; Badylak, S.F. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 2009, 30, 1482–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentin, J.E.; Stewart-Akers, A.M.; Gilbert, T.W.; Badylak, S.F. Macrophage participation in the degradation and remodeling of extracellular matrix scaffolds. Tissue Eng. Part A 2009, 15, 1687–1694. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, H.; Massensini, A.R.; Donnelly, J.; Kim, S.M.; Medberry, C.J.; Badylak, S.F.; Modo, M. ECM hydrogel for the treatment of stroke: Characterization of the host cell infiltrate. Biomaterials 2016, 91, 166–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahyouche, A.; Zhidao, X.; Czernuszka, J.T.; Clover, A.J. Macrophage-mediated degradation of crosslinked collagen scaffolds. Acta Biomater. 2011, 7, 278–286. [Google Scholar] [CrossRef]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jurgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, N.L.; Crowe, S.M. Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J. Leukoc. Biol. 2006, 80, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Liou, A.K.; Ren, Z.H.; Zhang, L.; Brown, B.N.; Cui, X.T.; Badylak, S.F.; Cai, Y.N.; Guan, Y.Q.; Leak, R.K.; et al. Neurorestorative effect of urinary bladder matrix-mediated neural stem cell transplantation following traumatic brain injury in rats. CNS Neurol. Disord. Drug Targets 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, J.; Shi, Y.; Pu, H.; Leak, R.K.; Liou, A.K.F.; Badylak, S.F.; Liu, Z.; Zhang, J.; Chen, J.; et al. Implantation of Brain-Derived Extracellular Matrix Enhances Neurological Recovery after Traumatic Brain Injury. Cell Transpl. 2017, 26, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, F.; Weng, Z.; Brown, B.N.; Yan, H.; Ma, X.M.; Vosler, P.S.; Badylak, S.F.; Dixon, C.E.; Cui, X.T.; et al. Effect of an inductive hydrogel composed of urinary bladder matrix upon functional recovery following traumatic brain injury. Tissue Eng. Part A 2013, 19, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Massensini, A.R.; Ghuman, H.; Saldin, L.T.; Medberry, C.J.; Keane, T.J.; Nicholls, F.J.; Velankar, S.S.; Badylak, S.F.; Modo, M. Concentration-dependent rheological properties of ECM hydrogel for intracerebral delivery to a stroke cavity. Acta Biomater. 2015, 27, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Bible, E.; Chau, D.Y.; Alexander, M.R.; Price, J.; Shakesheff, K.M.; Modo, M. Attachment of stem cells to scaffold particles for intra-cerebral transplantation. Nat. Protoc. 2009, 4, 1440–1453. [Google Scholar] [CrossRef]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 2006, 12, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A.; Estrada, E.Y.; Dencoff, J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 1998, 29, 2189–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciari, B.; Dziki, J.L.; Siu, B.F.; Medberry, C.J.; Dearth, C.L.; Badylak, S.F. The promotion of a constructive macrophage phenoytype by solubilized extracellular matrix. Biomaterials 2014, 35, 8605–8612. [Google Scholar]
- Slivka, P.; Badylak, S.F. Fractionation of an ECM hydrogel into structural and soluble components reveals distinctive role in regulating macrophage behavior. Biomater. Sci. 2014, 2, 1521–1534. [Google Scholar] [CrossRef]
- Dziki, J.L.; Wang, D.S.; Pineda, C.; Sicari, B.M.; Rausch, T.; Badylak, S.F. Solubilized extracellular matrix bioscaffolds derived from diverse source tissues differentially influence macrophage phenotype. J. Biomed. Mater. Res. A 2017, 105, 138–147. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.; Chen, G.; Cui, S. Corrigendum: Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front. Cell Neurosci. 2020, 14, 270. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, H.; Gerwig, M.; Nicholls, F.J.; Liu, J.R.; Donnelly, J.; Badylak, S.F.; Modo, M. Long-term retention of ECM hydrogel after implantation into a sub-acute stroke cavity reduces lesion volume. Acta Biomater. 2017, 63, 50–63. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, B.; ZhuGe, Q.; Jin, K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015, 1623, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, R.; Christensen, T.; Lehrmann, E.; Diemer, N.H.; Finsen, B. Focal cerebral ischemia induces increased myelin basic protein and growth-associated protein-43 gene transcription in peri-infarct areas in the rat brain. Exp. Brain Res. 2001, 138, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Jia, L.; Wei, M.; Lu, M.; Wang, X.; Pourabdollah, S.; Zhang, Z.G. Ascl1 lineage cells contribute to ischemia-induced neurogenesis and oligodendrogenesis. J. Cereb. Blood Flow Metab. 2011, 31, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinholm, J.E.; Hamilton, N.B.; Kessaris, N.; Richardson, W.D.; Bergersen, L.H.; Attwell, D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 2011, 31, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Chopp, M.; Zhang, Z.G. Oligodendrogenesis after cerebral ischemia. Front. Cell Neurosci. 2013, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- Stichel, C.C.; Muller, H.W. The CNS lesion scar: New vistas on an old regeneration barrier. Cell Tissue Res. 1998, 294, 1–9. [Google Scholar] [CrossRef]
- Galtrey, C.M.; Fawcett, J.W. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res. Rev. 2007, 54, 1–18. [Google Scholar] [CrossRef]
- Jin, T.; Nicholls, F.J.; Crum, W.R.; Ghuman, H.; Badylak, S.F.; Modo, M. Diamagnetic chemical exchange saturation transfer (diaCEST) affords magnetic resonance imaging of extracellular matrix hydrogel implantation in a rat model of stroke. Biomaterials 2017, 113, 176–190. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Bar-Shir, A.; Song, X.; Gilad, A.A.; Walczak, P.; Bulte, J.W. Label-free imaging of gelatin-containing hydrogel scaffolds. Biomaterials 2015, 42, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Chu, C.; Kuddannaya, S.; Yuan, Y.; Walczak, P.; Singh, A.; Song, X.; Bulte, J.W.M. In Vivo Imaging of Composite Hydrogel Scaffold Degradation Using CEST MRI and Two-Color NIR Imaging. Adv. Funct. Mater. 2019, 29. [Google Scholar] [CrossRef]
- Ghuman, H.; Hitchens, T.K.; Modo, M. A systematic optimization of (19)F MR image acquisition to detect macrophage invasion into an ECM hydrogel implanted in the stroke-damaged brain. Neuroimage 2019, 202, 116090. [Google Scholar] [CrossRef]
- Bible, E.; Dell’Acqua, F.; Solanky, B.; Balducci, A.; Crapo, P.M.; Badylak, S.F.; Ahrens, E.T.; Modo, M. Non-invasive imaging of transplanted human neural stem cells and ECM scaffold remodeling in the stroke-damaged rat brain by (19)F- and diffusion-MRI. Biomaterials 2012, 33, 2858–2871. [Google Scholar] [CrossRef] [Green Version]
- Ly, M.; Foley, L.; Manivannan, A.; Hitchens, T.K.; Richardson, R.M.; Modo, M. Mesoscale diffusion magnetic resonance imaging of the ex vivo human hippocampus. Hum. Brain Mapp. 2020, 41, 4200–4218. [Google Scholar] [CrossRef] [PubMed]
- Saldin, L.T.; Cramer, M.C.; Velankar, S.S.; White, L.J.; Badylak, S.F. Extracellular matrix hydrogels from decellularized tissues: Structure and function. Acta Biomater. 2017, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modo, M.; Lampe, K. Development and implementation of biomaterials to promote neural repair. Brain Res. Bull. 2019, 152, 297–298. [Google Scholar] [CrossRef]
- Freytes, D.O.; Martin, J.; Velankar, S.S.; Lee, A.S.; Badylak, S.F. Preparation and rheological characterization of a gel form of the porcine urinary bladder matrix. Biomaterials 2008, 29, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. [Google Scholar] [CrossRef] [Green Version]
- Medberry, C.J.; Crapo, P.M.; Siu, B.F.; Carruthers, C.A.; Wolf, M.T.; Nagarkar, S.P.; Agrawal, V.; Jones, K.E.; Kelly, J.; Johnson, S.A.; et al. Hydrogels derived from central nervous system extracellular matrix. Biomaterials 2013, 34, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Marcal, H.; Ahmed, T.; Badylak, S.F.; Tottey, S.; Foster, L.J. A comprehensive protein expression profile of extracellular matrix biomaterial derived from porcine urinary bladder. Regen Med. 2012, 7, 159–166. [Google Scholar] [CrossRef]
- Huleihel, L.; Hussey, G.S.; Naranjo, J.D.; Zhang, L.; Dziki, J.L.; Turner, N.J.; Stolz, D.B.; Badylak, S.F. Matrix-bound nanovesicles within ECM bioscaffolds. Sci. Adv. 2016, 2, e1600502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modo, M.; Stroemer, R.P.; Tang, E.; Veizovic, T.; Sowniski, P.; Hodges, H. Neurological sequelae and long-term behavioural assessment of rats with transient middle cerebral artery occlusion. J. Neurosci. Methods 2000, 104, 99–109. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [Green Version]
- Bogousslavsky, J.; Van Melle, G.; Regli, F. The Lausanne Stroke Registry: Analysis of 1,000 consecutive patients with first stroke. Stroke 1988, 19, 1083–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modo, M. Long-term survival and serial assessment of stroke damage and recovery—Practical and methodological considerations. J. Exp. Stroke Transl. Med. 2009, 2, 52–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stille, M.; Smith, E.J.; Crum, W.R.; Modo, M. 3D reconstruction of 2D fluorescence histology images and registration with in vivo MR images: Application in a rodent stroke model. J. Neurosci. Methods 2013, 219, 27–40. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Day 14 | Day 28 | Day 90 |
|---|---|---|---|
| MCAo only | 5 | 5 | 5 |
| MCAo + ECM | 5 | 5 | 4 |
| Antibody | Concentration | Company | Cat. Ref. | Clone |
|---|---|---|---|---|
| Collagen-I | 1:250 | Abcam | Ab34710 | Collagen I aa 1-1464 |
| Iba-1 | 1:300 | Abcam | Ab5076 | Iba1 aa 135-147 |
| GFAP | 1:3000 | Sigma | G3893 | G-A-5 |
| DCX | 1:150 | Abcam | Ab153668 | CAA0661′7.1 and AAT58219.1 |
| CNPase | 1:200 | Abcam | Ab6319 | 11-5B |
| Fox3 | 1:500 | Abcam | Ab104224 | 1B7 |
| RECA-1 | 1:100 | Abcam | Ab9774 | RECA-1 |
| CD86 | 1:200 | Abcam | Ab53004 | EP1159Y |
| CD206 | 1:200 | Santa Cruz | sc-34577 | C-20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damian, C.; Ghuman, H.; Mauney, C.; Azar, R.; Reinartz, J.; Badylak, S.F.; Modo, M. Post-Stroke Timing of ECM Hydrogel Implantation Affects Biodegradation and Tissue Restoration. Int. J. Mol. Sci. 2021, 22, 11372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111372
Damian C, Ghuman H, Mauney C, Azar R, Reinartz J, Badylak SF, Modo M. Post-Stroke Timing of ECM Hydrogel Implantation Affects Biodegradation and Tissue Restoration. International Journal of Molecular Sciences. 2021; 22(21):11372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111372
Chicago/Turabian StyleDamian, Corina, Harmanvir Ghuman, Carrinton Mauney, Reem Azar, Janina Reinartz, Stephen F. Badylak, and Michel Modo. 2021. "Post-Stroke Timing of ECM Hydrogel Implantation Affects Biodegradation and Tissue Restoration" International Journal of Molecular Sciences 22, no. 21: 11372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111372