
The Effect of tRNA[Ser]Sec Isopentenylation on Selenoprotein Expression

 , ,
, ,  , , , ,
, , , ,
Abstract
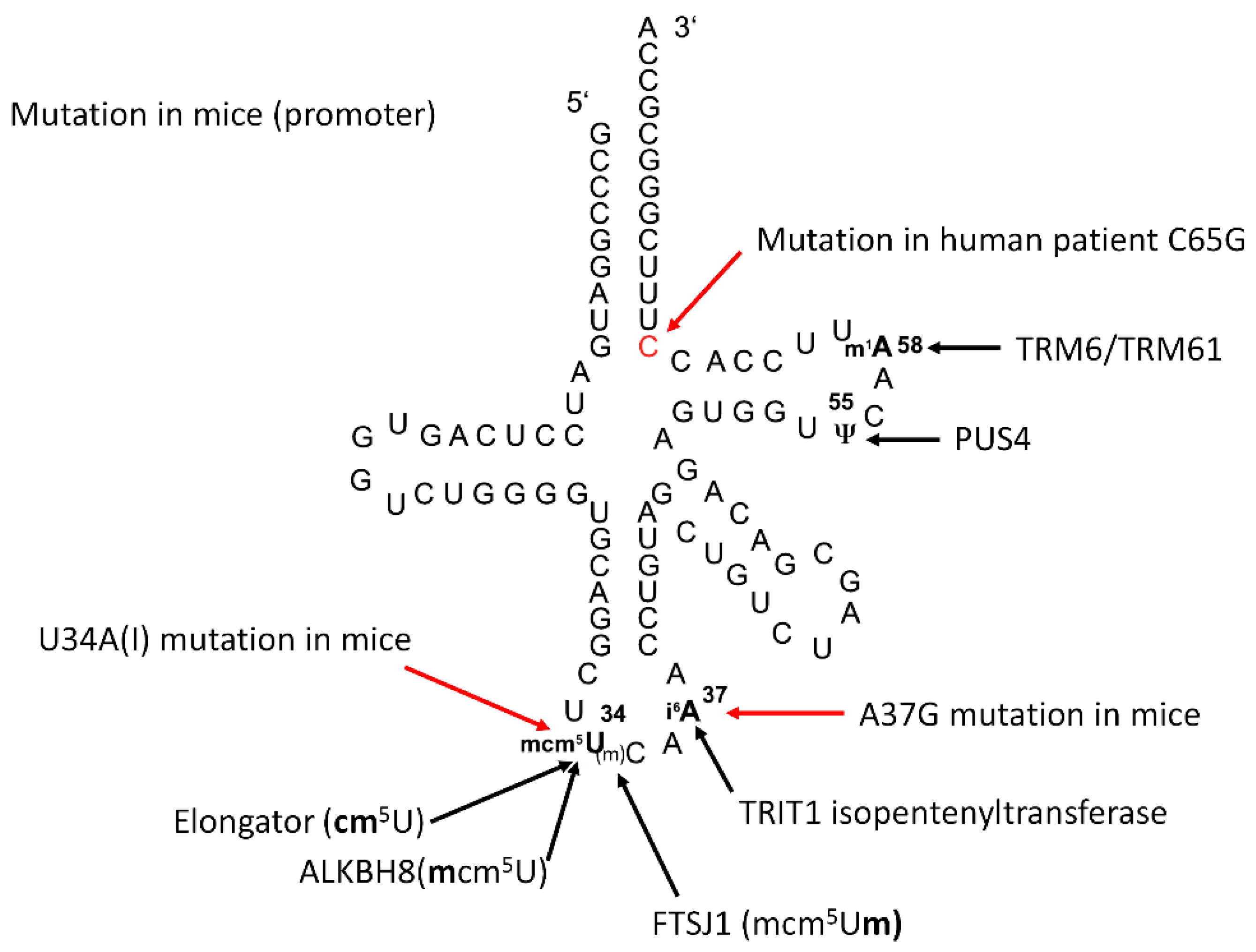
:1. Introduction
2. Results
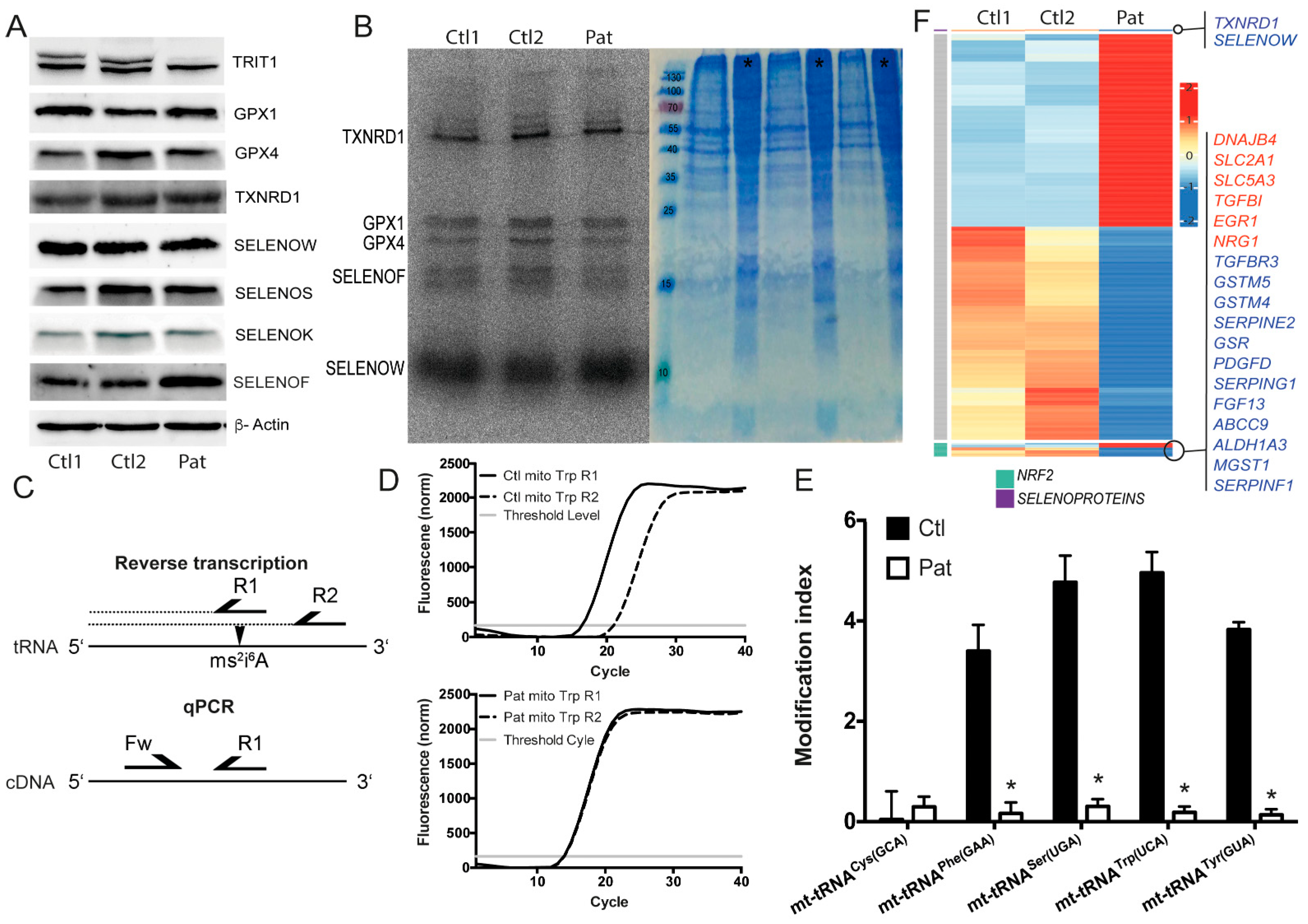
2.1. TRIT1-Mutant Human Fibroblasts
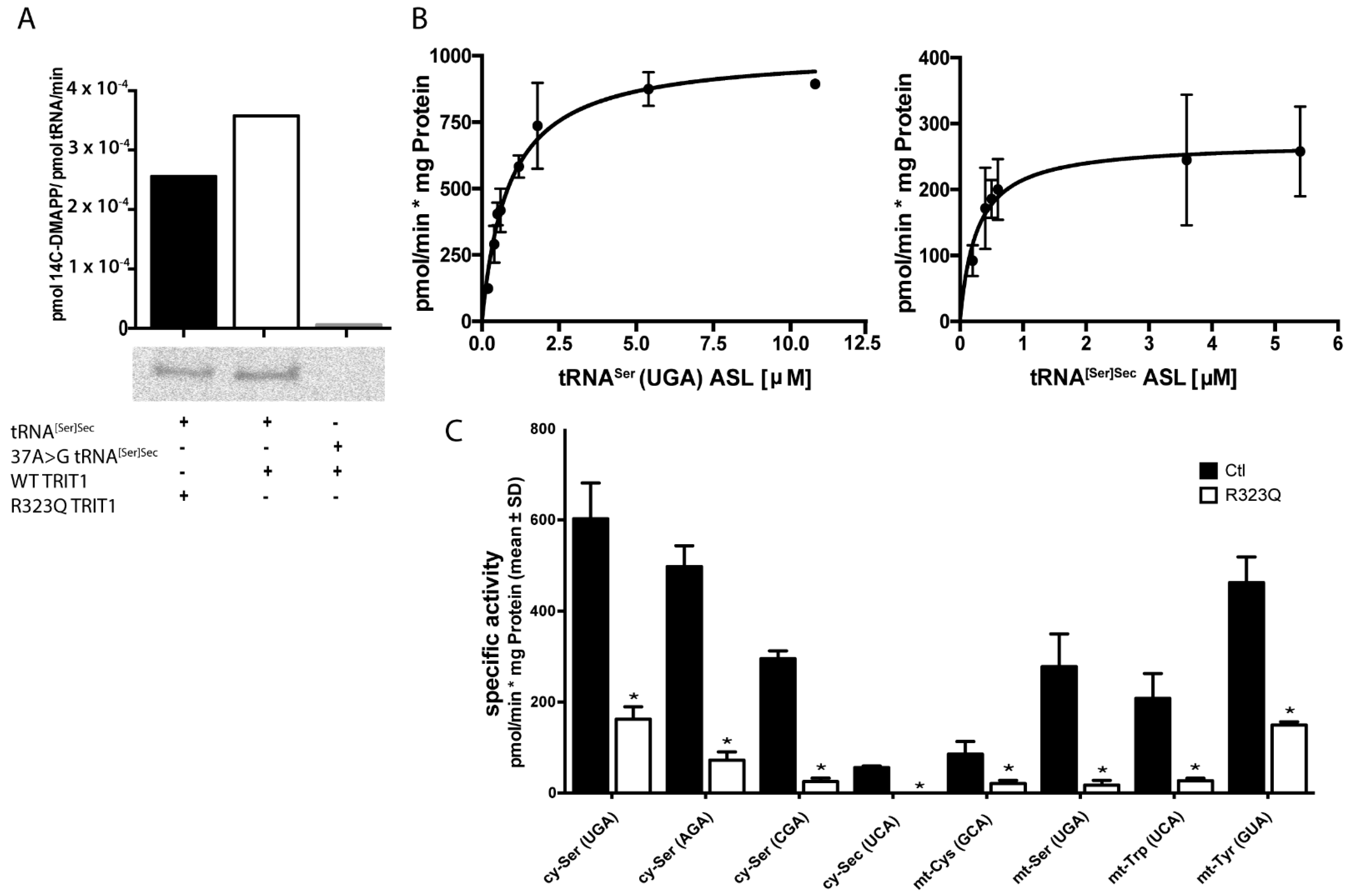
2.2. In Vitro Activity of TRIT1 and TRIT1R323Q
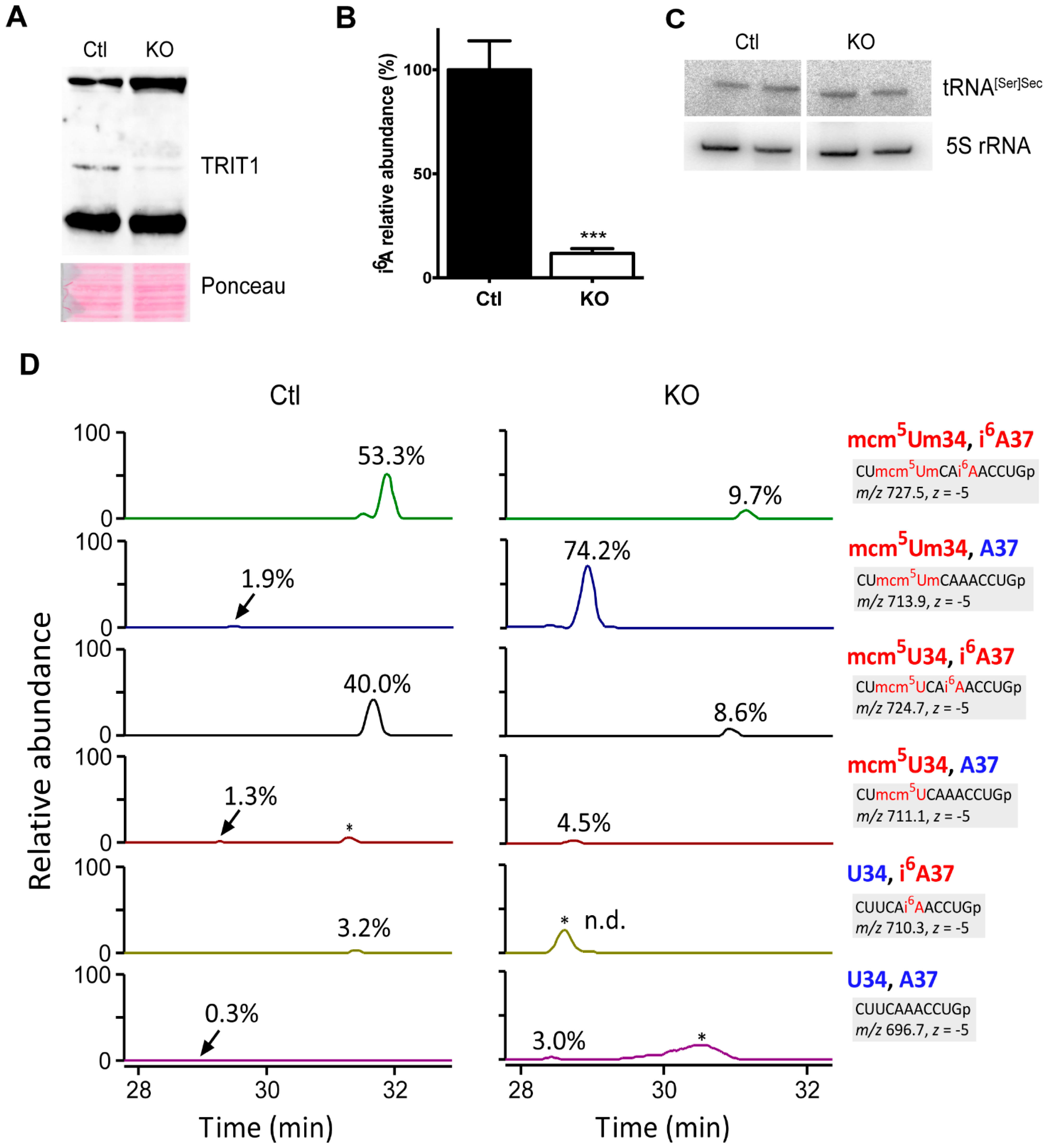
2.3. Inactivation of Trit1 in the Mouse
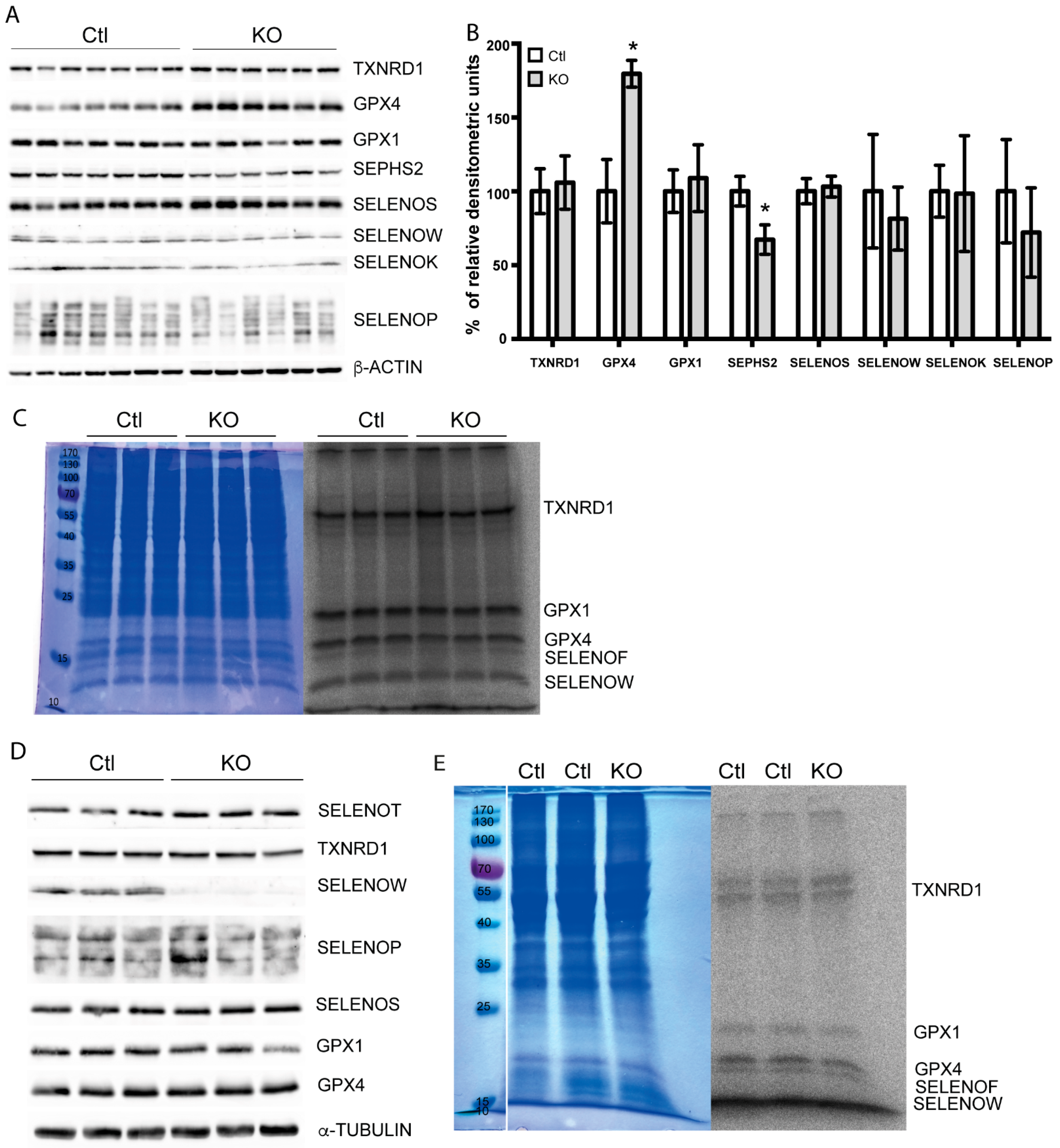
2.4. Selenoprotein Expression in Trit1-KO Mice
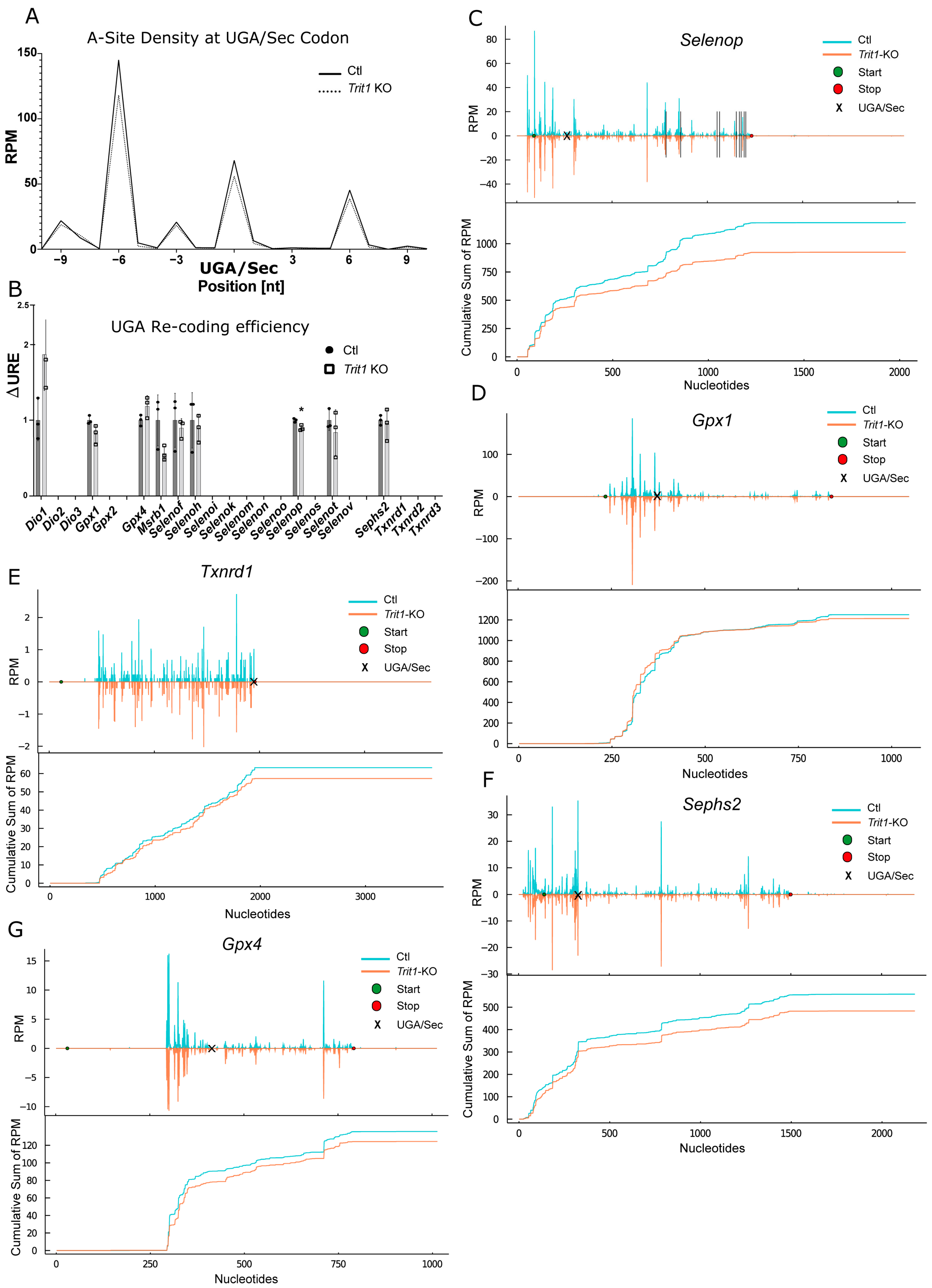
2.5. Ribosomal Profiling for Selenoproteins in Trit1-KO
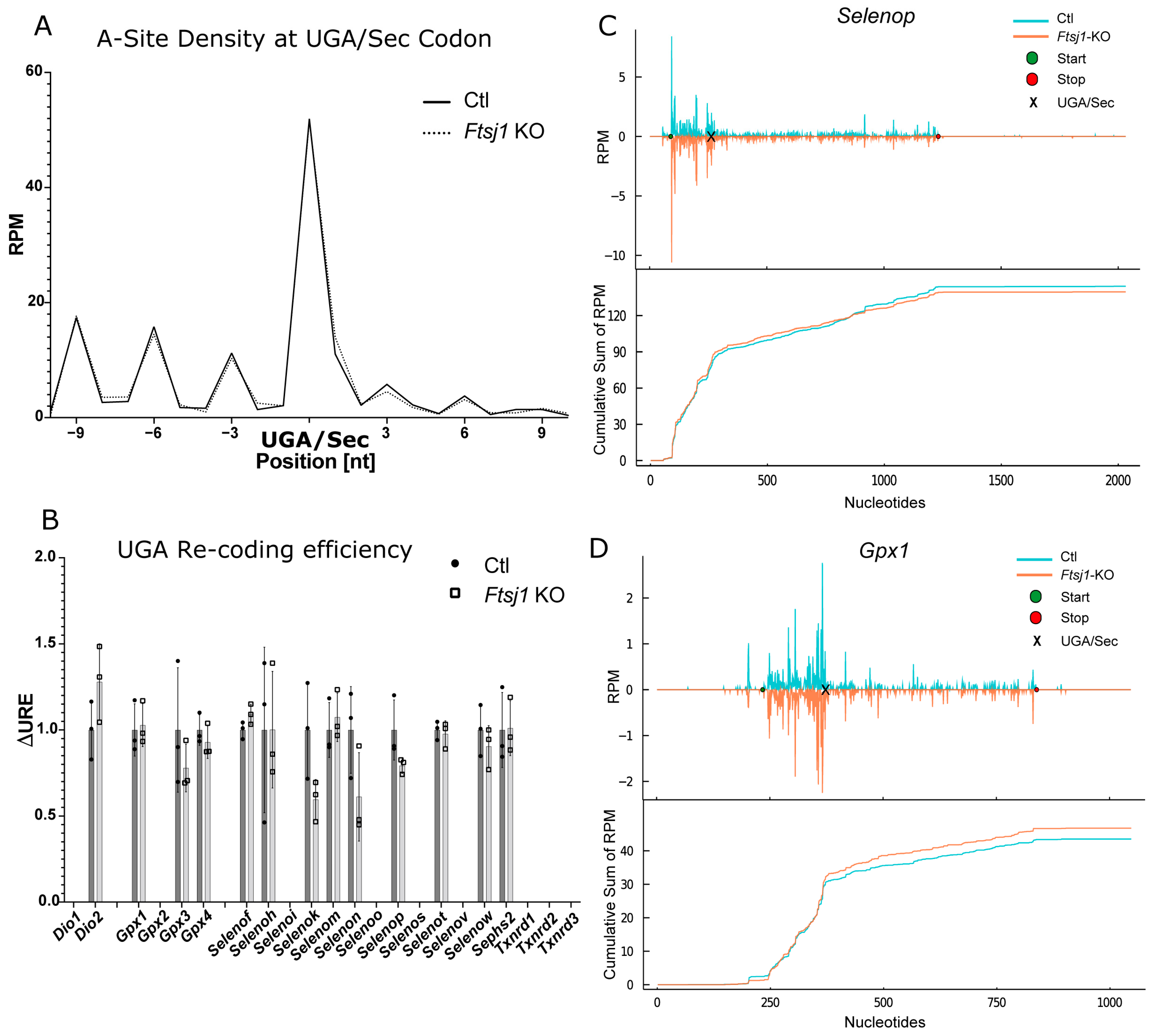
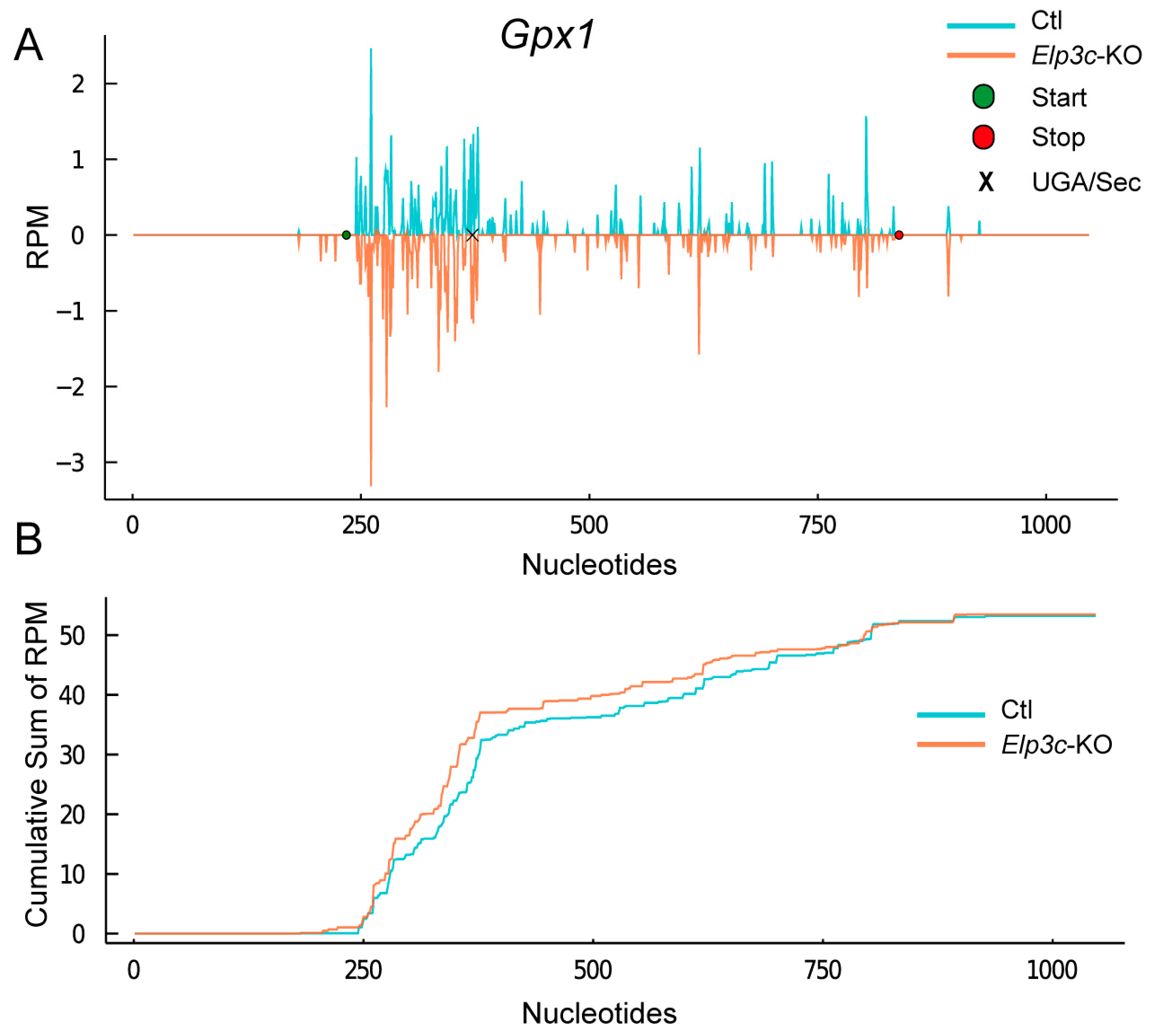
2.6. Effect of 2′O-Methylation of U34 in tRNA[Ser]Sec
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Human Fibroblast Culture
4.3. Hepatocyte Culture
4.4. Neuron Culture
4.5. Western Blot
4.6. 75Se Labeling
4.7. Transfer RNA Modification Index by RT-PCR
4.8. TRIT1 In Vitro Assay
4.9. tRNA[Ser]Sec Northern Blot
4.10. Quantification of i6A by LC-MS
4.11. Isolation and LC/MS Analysis of tRNA[Ser]Sec
4.12. 3′-. RNA Sequencing
4.13. RiboSeq
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Behne, D.; Hilmert, H.; Scheid, S.; Gessner, H.; Elger, W. Evidence for specific selenium target tissues and new biologically important selenoproteins. Biochim. Biophys. Acta 1988, 966, 12–21. [Google Scholar] [CrossRef]
- Schomburg, L.; Schweizer, U. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim. Biophys. Acta 2009, 1790, 1453–1462. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The neurobiology of selenium: Looking back and to the future. Front. Neurosci 2021, 15, 652099. [Google Scholar] [CrossRef]
- Wingler, K.; Bocher, M.; Flohe, L.; Kollmus, H.; Brigelius-Flohe, R. Mrna stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 1999, 259, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Wingler, K.; Brigelius-Flohé, R. 3′utrs of glutathione peroxidases differentially affect selenium-dependent mrna stability and selenocysteine incorporation efficiency. Biol. Chem. 2003, 384, 11–18. [Google Scholar] [CrossRef]
- Squires, J.E.; Stoytchev, I.; Forry, E.P.; Berry, M.J. Sbp2 binding affinity is a major determinant in differential selenoprotein mrna translation and sensitivity to nonsense-mediated decay. Mol. Cell. Biol. 2007, 27, 7848–7855. [Google Scholar] [CrossRef] [Green Version]
- Chavatte, L.; Brown, B.A.; Driscoll, D.M. Ribosomal protein l30 is a component of the uga-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 2005, 12, 408–416. [Google Scholar] [CrossRef]
- Budiman, M.E.; Bubenik, J.L.; Miniard, A.C.; Middleton, L.M.; Gerber, C.A.; Cash, A.; Driscoll, D.M. Eukaryotic initiation factor 4a3 is a selenium-regulated rna-binding protein that selectively inhibits selenocysteine incorporation. Mol. Cell. 2009, 35, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Shen, Q.; Newburger, P.E. Recognition and binding of the human selenocysteine insertion sequence by nucleolin. J. Cell. Biochem. 2000, 77, 507–516. [Google Scholar] [CrossRef]
- Hatfield, D.; Portugal, F.H. Seryl-trna in mammalian tissues: Chromatographic differences in brain and liver and a specific response to the codon, uga. Proc. Natl. Acad. Sci. USA 1970, 67, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Worland, P.J.; Davis, J.N.; Stadtman, T.C.; Hatfield, D.L. Identification of a selenocysteyl-trna(ser) in mammalian cells that recognizes the nonsense codon, uga. J. Biol. Chem. 1989, 264, 9724–9727. [Google Scholar] [CrossRef]
- Carlson, B.A.; Xu, X.M.; Kryukov, G.V.; Rao, M.; Berry, M.J.; Gladyshev, V.N.; Hatfield, D.L. Identification and characterization of phosphoseryl-trna[Ser]sec kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 12848–12853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.M.; Mix, H.; Carlson, B.A.; Grabowski, P.J.; Gladyshev, V.N.; Berry, M.J.; Hatfield, D.L. Evidence for direct roles of two additional factors, secp43 and soluble liver antigen, in the selenoprotein synthesis machinery. J. Biol. Chem. 2005, 280, 41568–41575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.M.; Carlson, B.A.; Mix, H.; Zhang, Y.; Saira, K.; Glass, R.S.; Berry, M.J.; Gladyshev, V.N.; Hatfield, D.L. Biosynthesis of selenocysteine on its trna in eukaryotes. PLoS Biol. 2007, 5, e4. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Palioura, S.; Salazar, J.C.; Su, D.; O’Donoghue, P.; Hohn, M.J.; Cardoso, A.M.; Whitman, W.B.; Söll, D. Rna-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc. Natl. Acad. Sci. USA 2006, 103, 18923–18927. [Google Scholar] [CrossRef] [Green Version]
- Sturchler, C.; Lescure, A.; Keith, G.; Carbon, P.; Krol, A. Base modification pattern at the wobble position of xenopus selenocysteine trna(sec). Nucleic Acids Res. 1994, 22, 1354–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schön, A.; Böck, A.; Ott, G.; Sprinzl, M.; Söll, D. The selenocysteine-inserting opal suppressor serine trna from e. Coli is highly unusual in structure and modification. Nucleic Acids Res. 1989, 17, 7159–7165. [Google Scholar] [CrossRef]
- Diamond, A.M.; Choi, I.S.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; Cruz, R.; Steer, C.J.; Hill, K.E.; Burk, R.F.; McCloskey, J.A. Dietary selenium affects methylation of the wobble nucleoside in the anticodon of selenocysteine trna([Ser]sec). J. Biol. Chem. 1993, 268, 14215–14223. [Google Scholar] [CrossRef]
- Kim, L.K.; Matsufuji, T.; Matsufuji, S.; Carlson, B.A.; Kim, S.S.; Hatfield, D.L.; Lee, B.J. Methylation of the ribosyl moiety at position 34 of selenocysteine trna[Ser]sec is governed by both primary and tertiary structure. RNA 2000, 6, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T. The expanding world of trna modifications and their disease relevance. Nat. Rev. Mol. Cell. Biol. 2021, 22, 375–392. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Lee, B.J.; Tsuji, P.A.; Copeland, P.R.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine trna([Ser]sec), the central component of selenoprotein biosynthesis: Isolation, identification, modification, and sequencing. Methods Mol. Biol. 2018, 1661, 43–60. [Google Scholar]
- Carlson, B.A.; Schweizer, U.; Perella, C.; Shrimali, R.K.; Feigenbaum, L.; Shen, L.; Speransky, S.; Floss, T.; Jeong, S.J.; Watts, J.; et al. The selenocysteine trna staf-binding region is essential for adequate selenocysteine trna status, selenoprotein expression and early age survival of mice. Biochem. J. 2009, 418, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bösl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine trna gene (trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, E.; Carlson, B.A.; Morgan, F.; Miyoshi, K.; Robinson, G.W.; Su, D.; Wang, S.; Southon, E.; Tessarollo, L.; Lee, B.J.; et al. Selective removal of the selenocysteine trna [Ser]sec gene (trsp) in mouse mammary epithelium. Mol. Cell. Biol. 2003, 23, 1477–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Streckfuss, F.; Pelt, P.; Carlson, B.A.; Hatfield, D.L.; Köhrle, J.; Schomburg, L. Hepatically derived selenoprotein p is a key factor for kidney but not for brain selenium supply. Biochem. J. 2005, 386, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, A.M.; Liao, X.H.; Abdullah, M.S.; Lado-Abeal, J.; Majed, F.A.; Moeller, L.C.; Boran, G.; Schomburg, L.; Weiss, R.E.; Refetoff, S. Mutations in secisbp2 result in abnormal thyroid hormone metabolism. Nat. Genet. 2005, 37, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Schoenmakers, E.; Agostini, M.; Mitchell, C.; Schoenmakers, N.; Papp, L.; Rajanayagam, O.; Padidela, R.; Ceron-Gutierrez, L.; Doffinger, R.; Prevosto, C.; et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Investig. 2010, 120, 4220–4235. [Google Scholar] [CrossRef] [Green Version]
- Schoenmakers, E.; Carlson, B.; Agostini, M.; Moran, C.; Rajanayagam, O.; Bochukova, E.; Tobe, R.; Peat, R.; Gevers, E.; Muntoni, F.; et al. Mutation in human selenocysteine transfer rna selectively disrupts selenoprotein synthesis. J. Clin. Investig. 2016, 126, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.A.; Xu, X.M.; Gladyshev, V.N.; Hatfield, D.L. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine trna. J. Biol. Chem. 2005, 280, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Carlson, B.A.; Moustafa, M.E.; Sengupta, A.; Schweizer, U.; Shrimali, R.; Rao, M.; Zhong, N.; Wang, S.; Feigenbaum, L.; Lee, B.J.; et al. Selective restoration of the selenoprotein population in a mouse hepatocyte selenoproteinless background with different mutant selenocysteine trnas lacking um34. J. Biol. Chem. 2007, 282, 32591–32602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasaikina, M.V.; Turanov, A.A.; Avanesov, A.; Schweizer, U.; Seeher, S.; Bronson, R.T.; Novoselov, S.N.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Contrasting roles of dietary selenium and selenoproteins in chemically induced hepatocarcinogenesis. Carcinogenesis 2013, 34, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, M.T.; Carlson, B.A.; Anderson, C.B.; Hatfield, D.L. Translational redefinition of uga codons is regulated by selenium availability. J. Biol. Chem. 2013, 288, 19401–19413. [Google Scholar] [CrossRef] [Green Version]
- Ganichkin, O.M.; Anedchenko, E.A.; Wahl, M.C. Crystal structure analysis reveals functional flexibility in the selenocysteine-specific trna from mouse. PLoS ONE 2011, 6, e20032. [Google Scholar] [CrossRef] [Green Version]
- Fischer, N.; Neumann, P.; Bock, L.V.; Maracci, C.; Wang, Z.; Paleskava, A.; Konevega, A.L.; Schroder, G.F.; Grubmuller, H.; Ficner, R.; et al. The pathway to gtpase activation of elongation factor selb on the ribosome. Nature 2016, 540, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Bohleber, S.; Fradejas-Villar, N. The modified base isopentenyladenosine and its derivatives in trna. RNA Biol. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, D.; Lee, B.J.; Hampton, L.; Diamond, A.M. Selenium induces changes in the selenocysteine trna[Ser]sec population in mammalian cells. Nucleic Acids Res. 1991, 19, 939–943. [Google Scholar] [CrossRef] [Green Version]
- Songe-Moller, L.; van den Born, E.; Leihne, V.; Vagbo, C.B.; Kristoffersen, T.; Krokan, H.E.; Kirpekar, F.; Falnes, P.O.; Klungland, A. Mammalian alkbh8 possesses trna methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 2010, 30, 1814–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, L.; Begley, U.; Clark, R.; Gu, C.; Dziergowska, A.; Malkiewicz, A.; Melendez, J.A.; Dedon, P.C.; Begley, T.J. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS ONE 2015, 10, e0131335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, A.M.; Jaffe, D.; Murray, J.L.; Safa, A.R.; Samuels, B.L.; Hatfield, D.L. Lovastatin effects on human breast carcinoma cells. Differential toxicity of an adriamycin-resistant derivative and influence on selenocysteine trnas. Biochem. Mol. Biol. Int. 1996, 38, 345–355. [Google Scholar]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine trna[Ser]sec lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [Green Version]
- Fradejas, N.; Carlson, B.A.; Rijntjes, E.; Becker, N.P.; Tobe, R.; Schweizer, U. Mammalian trit1 is a trna([Ser]sec)-isopentenyl transferase required for full selenoprotein expression. Biochem. J. 2013, 450, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective i6a37 modification of mitochondrial and cytosolic trnas results from pathogenic mutations in trit1 and its substrate trna. PLoS Genet 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, E.K.; Conrad, M.; Winterer, J.; Wozny, C.; Carlson, B.A.; Roth, S.; Schmitz, D.; Bornkamm, G.W.; Coppola, V.; Tessarollo, L.; et al. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 2010, 24, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Xie, P.; Wei, F.Y.; Hirata, S.; Kaitsuka, T.; Suzuki, T.; Suzuki, T.; Tomizawa, K. Quantitative pcr measurement of trna 2-methylthio modification for assessing type 2 diabetes risk. Clinical. Chem. 2013, 59, 1604–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Huang, R.H. Crystallographic snapshots of eukaryotic dimethylallyltransferase acting on trna: Insight into trna recognition and reaction mechanism. Proc. Natl. Acad. Sci. USA 2008, 105, 16142–16147. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Bohleber, S.; Schmidt, H.; Seeher, S.; Howard, M.T.; Braun, D.; Arndt, S.; Reuter, U.; Wende, H.; Birchmeier, C.; et al. Ribosome profiling of selenoproteins in vivo reveals consequences of pathogenic secisbp2 missense mutations. J. Biol. Chem. 2019, 294, 14185–14200. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Ohara, T.; Suzuki, T. Automated parallel isolation of multiple species of non-coding rnas by the reciprocal circulating chromatography method. Nucleic Acids Res. 2007, 35, e24. [Google Scholar] [CrossRef]
- Suzuki, T.; Ikeuchi, Y.; Noma, A.; Suzuki, T.; Sakaguchi, Y. Mass spectrometric identification and characterization of rna-modifying enzymes. Methods Enzym. 2007, 425, 211–229. [Google Scholar]
- Suzuki, T.; Yashiro, Y.; Kikuchi, I.; Ishigami, Y.; Saito, H.; Matsuzawa, I.; Okada, S.; Mito, M.; Iwasaki, S.; Ma, D.; et al. Complete chemical structures of human mitochondrial trnas. Nat. Commun. 2020, 11, 4269. [Google Scholar] [CrossRef] [PubMed]
- Fradejas-Villar, N.; Seeher, S.; Anderson, C.B.; Doengi, M.; Carlson, B.A.; Hatfield, D.L.; Schweizer, U.; Howard, M.T. The rna-binding protein secisbp2 differentially modulates uga codon reassignment and rna decay. Nucleic Acids Res. 2017, 45, 4094–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freude, K.; Hoffmann, K.; Jensen, L.R.; Delatycki, M.B.; des Portes, V.; Moser, B.; Hamel, B.; van Bokhoven, H.; Moraine, C.; Fryns, J.P.; et al. Mutations in the ftsj1 gene coding for a novel s-adenosylmethionine-binding protein cause nonsyndromic x-linked mental retardation. Am. J. Hum. Genet. 2004, 75, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, Y.; Chujo, T.; Hirata, S.; Nakatsuka, H.; Chen, C.W.; Takakura, M.; Miyauchi, K.; Ikeuchi, Y.; Carlyle, B.C.; Kitchen, R.R.; et al. Loss of ftsj1 perturbs codon-specific translation efficiency in the brain and is associated with x-linked intellectual disability. Sci. Adv. 2021, 7, eabf3072. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial trnas. Nucleic Acids Res. 2014, 42, 7346–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Dyment, D.A.; Pupavac, M.; Cramer, Z.; McBride, A.; Bernard, G.; Straub, I.; Tetreault, M.; Hartley, T.; Huang, L.; et al. Matchmaking facilitates the diagnosis of an autosomal-recessive mitochondrial disease caused by biallelic mutation of the trna isopentenyltransferase (trit1) gene. Hum. Mutat. 2017, 38, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Carlson, B.A.; Weaver, J.A.; Novoselov, S.V.; Fomenko, D.E.; Gladyshev, V.N.; Hatfield, D.L. A functional link between housekeeping selenoproteins and phase ii enzymes. Biochem. J. 2008, 413, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Seeher, S.; Atassi, T.; Mahdi, Y.; Carlson, B.A.; Braun, D.; Wirth, E.K.; Klein, M.O.; Reix, N.; Miniard, A.C.; Schomburg, L.; et al. Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid. Redox. Signal 2014, 21, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Barroso, M.; Florindo, C.; Kalwa, H.; Silva, Z.; Turanov, A.A.; Carlson, B.A.; Tavares de Almeida, I.; Blom, H.J.; Gladyshev, V.N.; Hatfield, D.L.; et al. Inhibition of cellular methyltransferases promotes endothelial cell activation by suppressing glutathione peroxidase-1 expression. J. Biol. Chem. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, A.; Kovalchuk, N.; Yin, L.; Endres, L.; Evke, S.; Nevins, S.; Martin, S.; Dedon, P.C.; Melendez, J.A.; Van Winkle, L.; et al. The epitranscriptomic writer alkbh8 drives tolerance and protects mouse lungs from the environmental pollutant naphthalene. Epigenetics 2020, 15, 1121–1138. [Google Scholar] [CrossRef] [Green Version]
- Endres, L.; Dedon, P.C.; Begley, T.J. Codon-biased translation can be regulated by wobble-base trna modification systems during cellular stress responses. RNA Biol. 2015, 12, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Leonardi, A.; Begley, T.J.; Melendez, J.A. Loss of epitranscriptomic control of selenocysteine utilization engages senescence and mitochondrial reprogramming. Redox Biol. 2020, 28, 101375. [Google Scholar] [CrossRef]
- Laguesse, S.; Creppe, C.; Nedialkova, D.D.; Prevot, P.P.; Borgs, L.; Huysseune, S.; Franco, B.; Duysens, G.; Krusy, N.; Lee, G.; et al. A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell. 2015, 35, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Beaudoin, G.M., 3rd; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Tomizawa, K. Measurement of 2-methylthio modifications in mitochondrial transfer rnas by reverse-transcription quantitative pcr. Bio-Protoc. 2016, 6, e1695. [Google Scholar] [CrossRef]
- Igloi, G.L.; von der Haar, F.; Cramer, F. Experimental proof for the misactivation of amino acids by aminoacyl-trna synthetases. Methods Enzym. 1979, 59, 282–291. [Google Scholar]
- Yoshida, M.; Kataoka, N.; Miyauchi, K.; Ohe, K.; Iida, K.; Yoshida, S.; Nojima, T.; Okuno, Y.; Onogi, H.; Usui, T.; et al. Rectifier of aberrant mrna splicing recovers trna modification in familial dysautonomia. Proc. Natl. Acad. Sci. USA 2015, 112, 2764–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tRNA | KM [µM] | Vmax [pmol/min * mg Protein] | Sequence | |
|---|---|---|---|---|
| cytosolic | Ser AGA | 0.7980 ± 0.1091 | 989.0 ± 45.27 | GA-UGG-ACU-AGA-AAU-CCA-UU |
| Ser CGA | 0.4384 ± 0.0849 | 465.5 ± 27.17 | GU-UGG-ACU-CGA-AAU-CCA-AU | |
| Ser UGA | 0.8690 ± 0.1210 | 1016 ± 54.85 | GA-UGG-ACU-UGA-AAU-CCA-UU | |
| Sec UCA | 0.3848 ± 0.0934 | 321.5 ± 25.72 | UG-CAG-GCU-UCA-AAC-CUG-UA | |
| mitochondrial | Cys GCA | 5.293 ± 4.294 | 73.94 ± 28.37 | AU-UGA-AUU-GCA-AAU-UCG-AA |
| Ser UGA | 1.673 ± 0.2683 | 523.6 ± 32.23 | GG-UUG-GCU-UGA-AAC-CAG-CU | |
| Trp UCA | 3.710 ± 0.5627 | 442.2 ± 31.37 | AA-GAG-CCU-UCA-AAG-CCC-UC | |
| Tyr GUA | 0.7735 ± 0.1127 | 549.7 ± 26.37 | AU-UGG-ACU-GUA-AAU-CUA-AA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fradejas-Villar, N.; Bohleber, S.; Zhao, W.; Reuter, U.; Kotter, A.; Helm, M.; Knoll, R.; McFarland, R.; Taylor, R.W.; Mo, Y.; et al. The Effect of tRNA[Ser]Sec Isopentenylation on Selenoprotein Expression. Int. J. Mol. Sci. 2021, 22, 11454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111454
Fradejas-Villar N, Bohleber S, Zhao W, Reuter U, Kotter A, Helm M, Knoll R, McFarland R, Taylor RW, Mo Y, et al. The Effect of tRNA[Ser]Sec Isopentenylation on Selenoprotein Expression. International Journal of Molecular Sciences. 2021; 22(21):11454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111454
Chicago/Turabian StyleFradejas-Villar, Noelia, Simon Bohleber, Wenchao Zhao, Uschi Reuter, Annika Kotter, Mark Helm, Rainer Knoll, Robert McFarland, Robert W. Taylor, Yufeng Mo, and et al. 2021. "The Effect of tRNA[Ser]Sec Isopentenylation on Selenoprotein Expression" International Journal of Molecular Sciences 22, no. 21: 11454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111454