
Interferon Lambda Regulates Cellular and Humoral Immunity in Pristane-Induced Lupus

 , and
, and 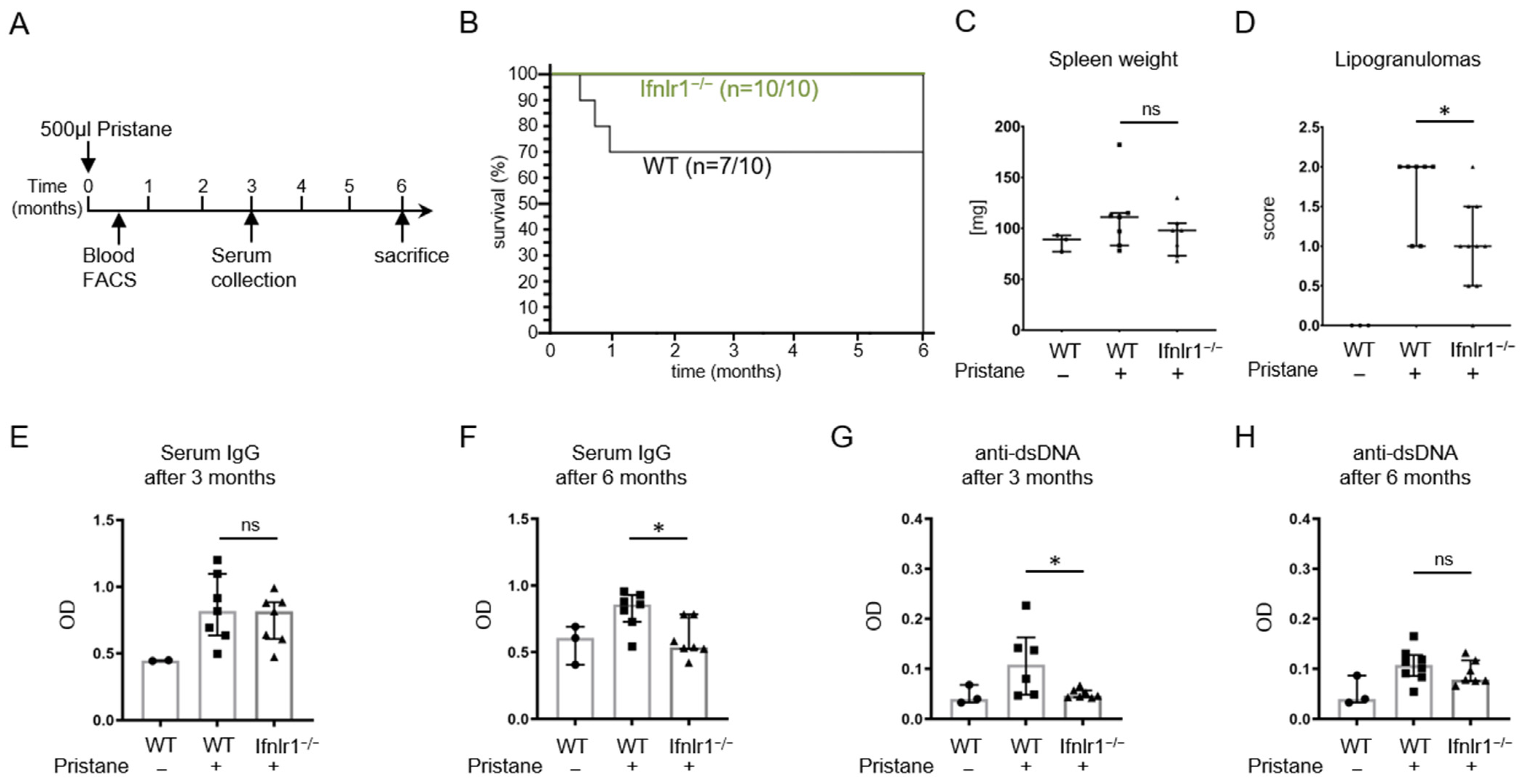{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pristane-Treated Ifnlr1−/− Mice Show Reduced Numbers of Lipogranulomas and a Better Survival Rate
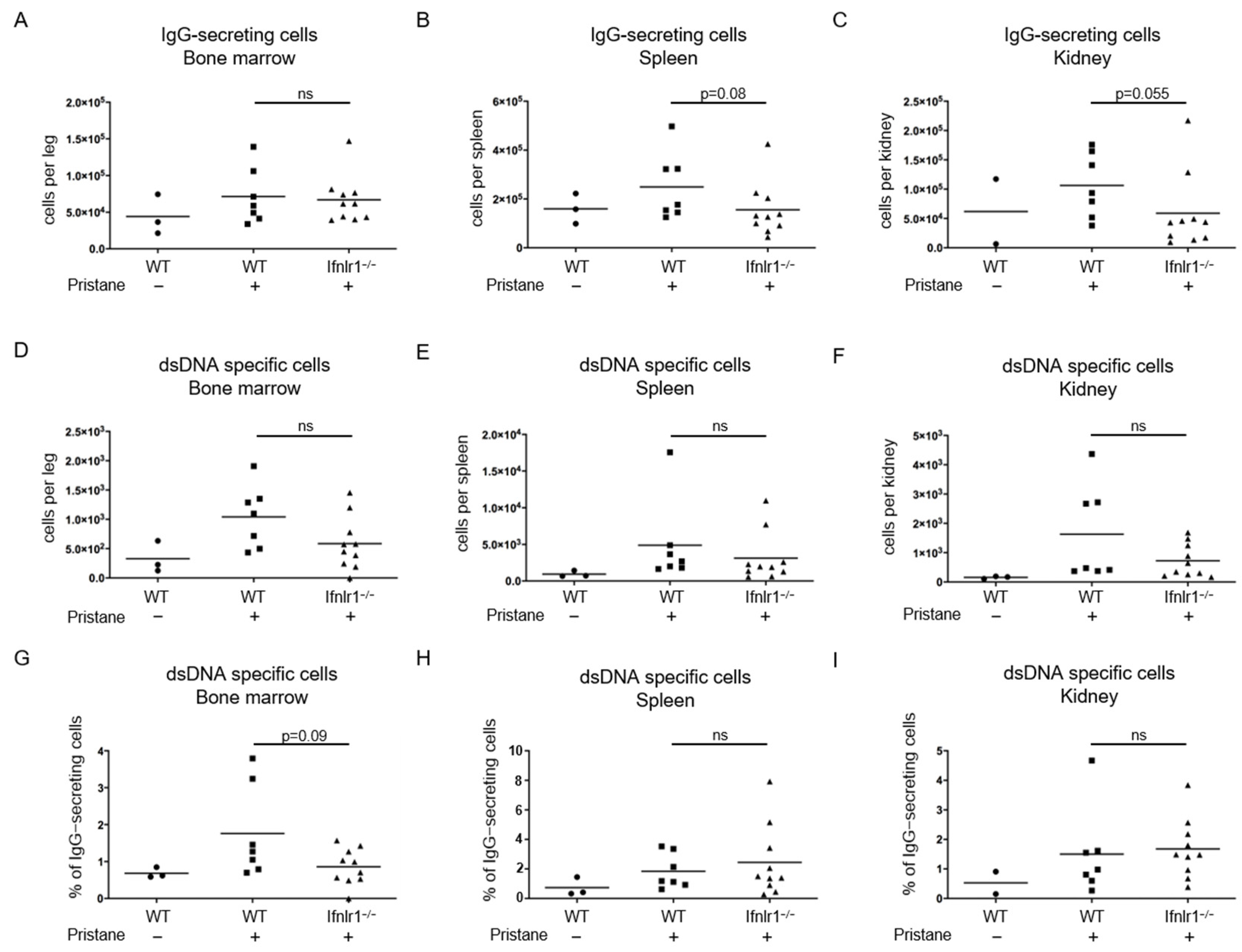
2.2. Trend toward Reduced Numbers of Antibody-Secreting Cells in the Spleens and Kidneys of Pristane-Treated Ifnlr1−/− Mice
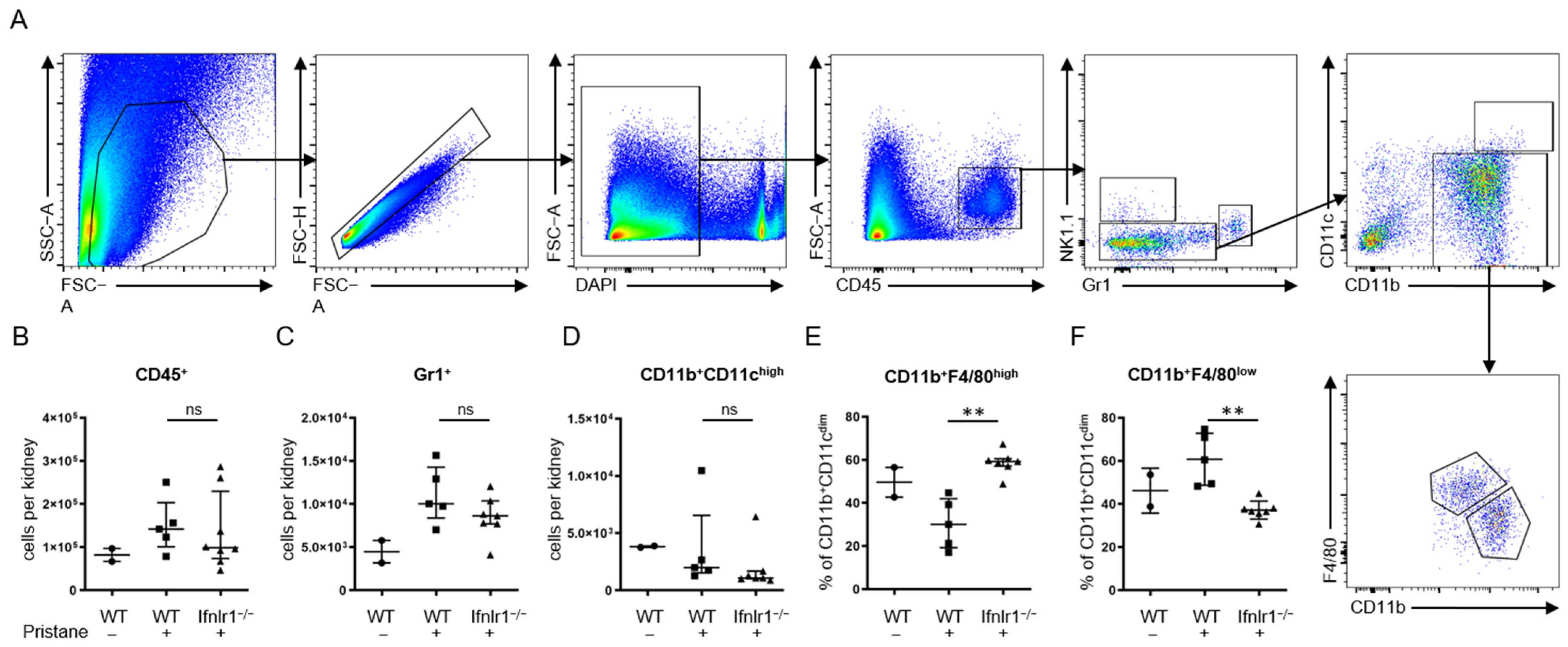
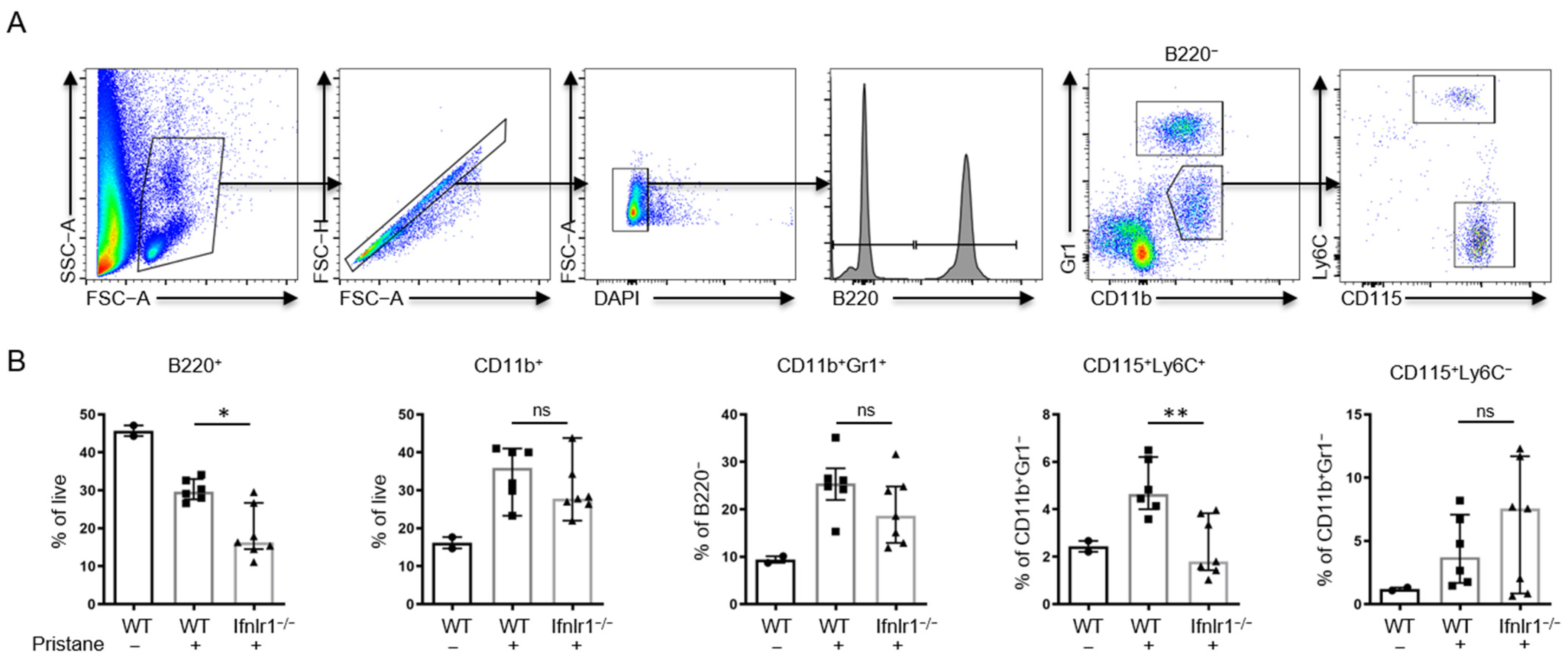
2.3. Interferon Lambda Promotes Replacement of Tissue Resident Macrophages with Inflammatory Monocytes
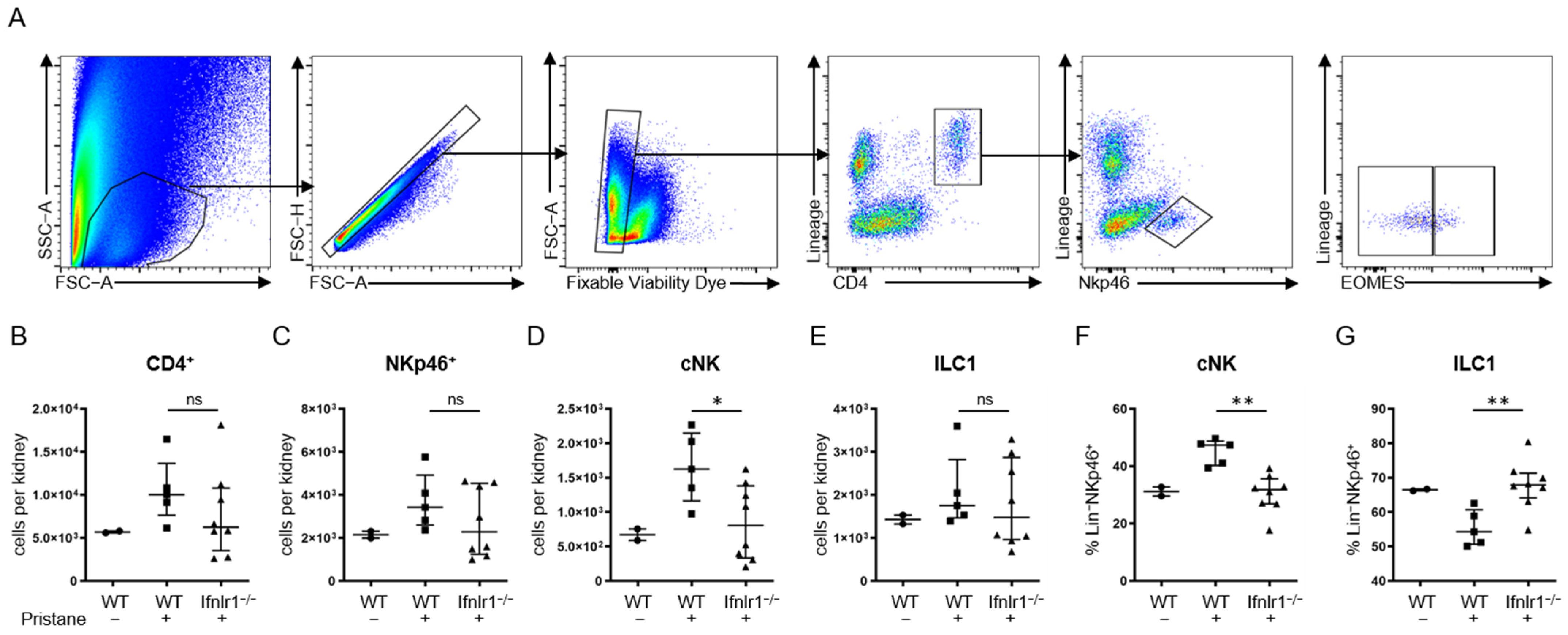
2.4. After Pristane Treatment, Ifnlr1−/− Mice Show Reduced Numbers of Conventional NK Cells in the Kidney Compared to Wild-Type Mice
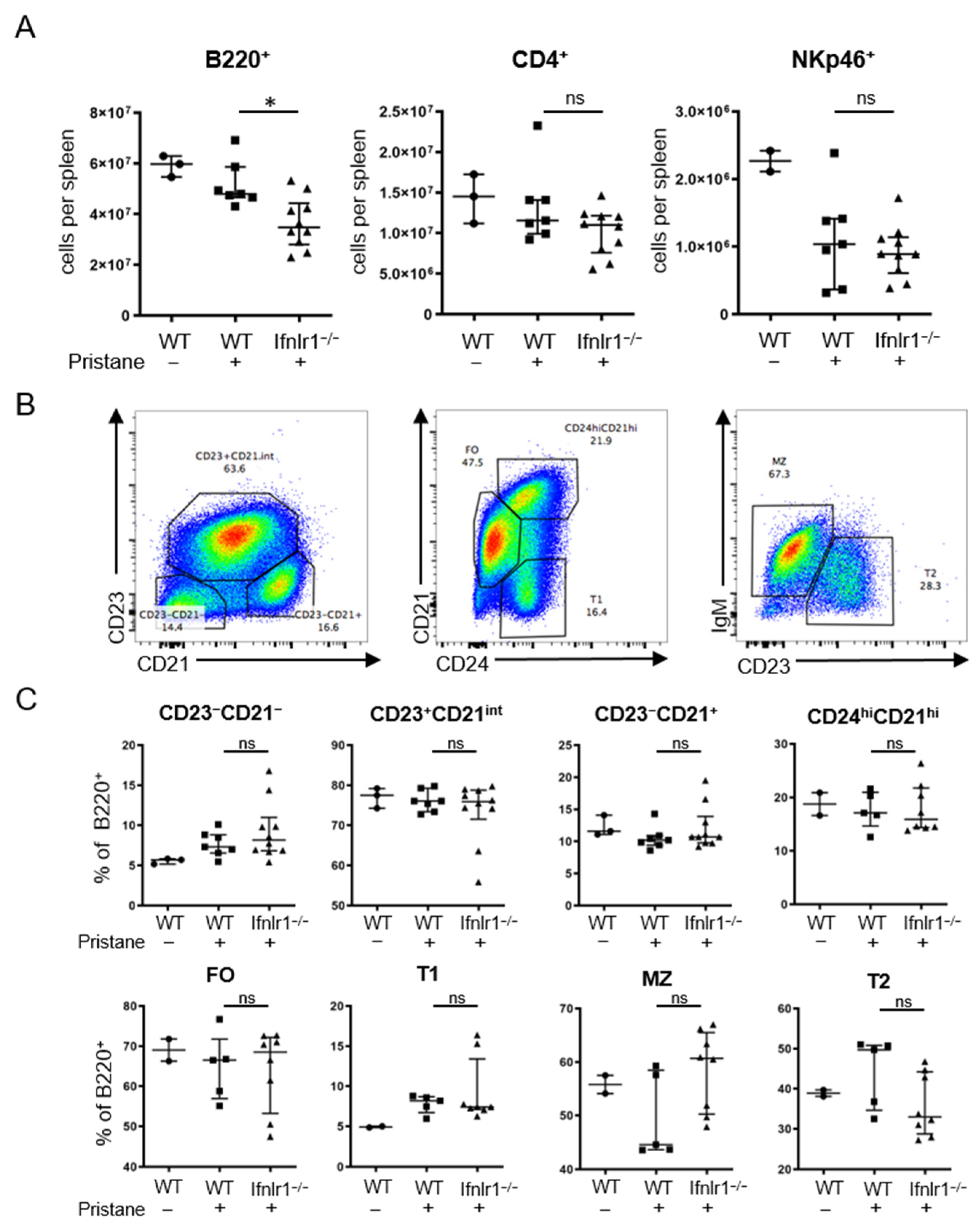
2.5. Ifnlr1−/− Mice Display Reduced Numbers of B Cells in the Spleen after Pristane Treatment
2.6. Ifnlr1−/− Mice Show an Increased Proportion of Peripheral CD115+Ly6C+ Monocytes at 2 Weeks after Pristane Treatment
3. Discussion
4. Materials and Methods
4.1. Mice and Animal Housing
4.2. In Vivo Intraperitoneal Injections
4.3. Collection Peripheral Blood and Serum
4.4. Euthanasia of Mice and Harvesting of Organs
4.5. Semi-Quantitative Scoring of Peritoneal Lipogranulomas
4.6. Quantification of Total Serum IgG Levels
4.7. Quantification of Serum Anti-dsDNA Levels
4.8. Quantification of Antibody Secreting Cells
4.9. Preparation of Kidney Tissue for Flow Cytometry
4.10. Preparation of Spleen Tissue for Flow Cytometry
4.11. Preparation of Bone Marrow Single Cell Suspensions
4.12. Preparation of Peripheral Blood Cells
4.13. Flow Cytometry of Single Cell Suspensions
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global epidemiology of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Yen, E.Y.; Singh, R.R. Brief Report: Lupus-An Unrecognized Leading Cause of Death in Young Females: A Population-Based Study Using Nationwide Death Certificates, 2000-2015. Arthritis Rheumatol. 2018, 70, 1251–1255. [Google Scholar] [CrossRef] [Green Version]
- Izmirly, P.M.; Parton, H.; Wang, L.; McCune, W.J.; Lim, S.S.; Drenkard, C.; Ferucci, E.D.; Dall’Era, M.; Gordon, C.; Helmick, C.G.; et al. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates From a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol. 2021, 73, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Goodnow, C.C. Multistep pathogenesis of autoimmune disease. Cell 2007, 130, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Mumtaz, I.M.; Khodadadi, L.; Radbruch, A.; Hoyer, B.F.; Hiepe, F. Autoantibodies from long-lived ‘memory’ plasma cells of NZB/W mice drive immune complex nephritis. Ann. Rheum. Dis. 2013, 72, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Seredkina, N.; Van Der Vlag, J.; Berden, J.; Mortensen, E.; Rekvig, O.P. Lupus nephritis: Enigmas, conflicting models and an emerging concept. Mol. Med. 2013, 19, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ytterberg, S.R.; Schnitzer, T.J. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 401–406. [Google Scholar] [CrossRef]
- Santiago-Raber, M.L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, H.; Jacob, N.; Carreras, E.; Bajana, S.; Putterman, C.; Turner, S.; Neas, B.; Mathian, A.; Koss, M.N.; Stohl, W.; et al. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J. Immunol. 2009, 183, 6021–6029. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, A.D.; Baron, S.; Talal, N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc. Natl. Acad. Sci. USA 1969, 63, 1102–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, D.F.; Steinberg, A.D.; Schur, P.H.; Talal, N. The pathogenesis of autoimmunity in New Zealand mice. II. Acceleration of glomerulonephritis by polyinosinic-polycytidylic acid. Lab. Investig. 1970, 23, 628–634. [Google Scholar] [PubMed]
- Mathian, A.; Weinberg, A.; Gallegos, M.; Banchereau, J.; Koutouzov, S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J. Immunol. 2005, 174, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Adam, C.; Thoua, Y.; Ronco, P.; Verroust, P.; Tovey, M.; Morel-Maroger, L. The effect of exogenous interferon: Acceleration of autoimmune and renal diseases in (NZB/W) F1 mice. Clin. Exp. Immunol. 1980, 40, 373–382. [Google Scholar] [PubMed]
- Triantafyllopoulou, A.; Franzke, C.W.; Seshan, S.V.; Perino, G.; Kalliolias, G.D.; Ramanujam, M.; van Rooijen, N.; Davidson, A.; Ivashkiv, L.B. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl. Acad. Sci. USA 2010, 107, 3012–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Uze, G.; Monneron, D. IL-28 and IL-29: Newcomers to the interferon family. Biochimie 2007, 89, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Kempuraj, D.; Donelan, J.; Frydas, S.; Iezzi, T.; Conti, F.; Boucher, W.; Papadopoulou, N.G.; Madhappan, B.; Letourneau, L.; Cao, J.; et al. Interleukin-28 and 29 (IL-28 and IL-29): New cytokines with anti-viral activities. Int. J. Immunopathol. Pharmacol. 2004, 17, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Q.; Lourenco, E.; Sun, H.; Zhang, Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: Its correlation with disease activity. Arthritis Res. Ther. 2011, 13, R88. [Google Scholar] [CrossRef] [Green Version]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjall, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [Green Version]
- Oke, V.; Brauner, S.; Larsson, A.; Gustafsson, J.; Zickert, A.; Gunnarsson, I.; Svenungsson, E. IFN-lambda1 with Th17 axis cytokines and IFN-alpha define different subsets in systemic lupus erythematosus (SLE). Arthritis Res. Ther. 2017, 19, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amezcua-Guerra, L.M.; Marquez-Velasco, R.; Chavez-Rueda, A.K.; Castillo-Martinez, D.; Masso, F.; Paez, A.; Colin-Fuentes, J.; Bojalil, R. Type III Interferons in Systemic Lupus Erythematosus: Association Between Interferon lambda3, Disease Activity, and Anti-Ro/SSA Antibodies. J. Clin. Rheumatol. 2017, 23, 368–375. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, C.M.; Chen, T.D.; Jan Wu, Y.J.; Lin, J.C.; Lu, L.Y.; Wu, J. Interferon-lambda3/4 genetic variants and interferon-lambda3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese. Arthritis Res. Ther. 2018, 20, 193. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.R.; Wang, X.; O’Neil, L.J.; Nakabo, S.; Hasneen, K.; Gupta, S.; Wigerblad, G.; Blanco, L.P.; Kopp, J.B.; Morasso, M.I.; et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc. Natl. Acad. Sci. USA 2020, 117, 5409–5419. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, M.; Boisgard, A.S.; Danoy, A.; El Kholti, N.; Salvi, J.P.; Boulieu, R.; Fromy, B.; Verrier, B.; Lamrayah, M. Advanced Characterization of Imiquimod-Induced Psoriasis-Like Mouse Model. Pharmaceutics 2020, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: A new model of systemic Lupus erythematosus. Arthritis Rheumatol. 2014, 66, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Richards, H.B.; Shaheen, V.M.; Yoshida, H.; Shaw, M.; Naim, J.O.; Wooley, P.H.; Reeves, W.H. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin. Exp. Immunol. 2000, 121, 399–405. [Google Scholar] [CrossRef]
- Satoh, M.; Reeves, W.H. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J. Exp. Med. 1994, 180, 2341–2346. [Google Scholar] [CrossRef] [Green Version]
- Urbonaviciute, V.; Starke, C.; Pirschel, W.; Pohle, S.; Frey, S.; Daniel, C.; Amann, K.; Schett, G.; Herrmann, M.; Voll, R.E. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis Rheum. 2013, 65, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Kumar, A.; Kanwar, Y.S.; Reeves, W.H. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc. Natl. Acad. Sci. USA 1995, 92, 10934–10938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, W.H.; Lee, P.Y.; Weinstein, J.S.; Satoh, M.; Lu, L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009, 30, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Chowdhary, V.R.; Grande, J.P.; Luthra, H.S.; David, C.S. Characterization of haemorrhagic pulmonary capillaritis: Another manifestation of Pristane-induced lupus. Rheumatology 2007, 46, 1405–1410. [Google Scholar] [CrossRef] [Green Version]
- Barker, T.T.; Lee, P.Y.; Kelly-Scumpia, K.M.; Weinstein, J.S.; Nacionales, D.C.; Kumagai, Y.; Akira, S.; Croker, B.P.; Sobel, E.S.; Reeves, W.H.; et al. Pathogenic role of B cells in the development of diffuse alveolar hemorrhage induced by pristane. Lab. Investig. 2011, 91, 1540–1550. [Google Scholar] [CrossRef]
- Nacionales, D.C.; Kelly-Scumpia, K.M.; Lee, P.Y.; Weinstein, J.S.; Lyons, R.; Sobel, E.; Satoh, M.; Reeves, W.H. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007, 56, 3770–3783. [Google Scholar] [CrossRef] [Green Version]
- Thibault, D.L.; Graham, K.L.; Lee, L.Y.; Balboni, I.; Hertzog, P.J.; Utz, P.J. Type I interferon receptor controls B-cell expression of nucleic acid-sensing Toll-like receptors and autoantibody production in a murine model of lupus. Arthritis Res. Ther. 2009, 11, R112. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.Y.; Weinstein, J.S.; Nacionales, D.C.; Scumpia, P.O.; Li, Y.; Butfiloski, E.; van Rooijen, N.; Moldawer, L.; Satoh, M.; Reeves, W.H. A novel type I IFN-producing cell subset in murine lupus. J. Immunol. 2008, 180, 5101–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacionales, D.C.; Weinstein, J.S.; Yan, X.J.; Albesiano, E.; Lee, P.Y.; Kelly-Scumpia, K.M.; Lyons, R.; Satoh, M.; Chiorazzi, N.; Reeves, W.H. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J. Immunol. 2009, 182, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacionales, D.C.; Kelly, K.M.; Lee, P.Y.; Zhuang, H.; Li, Y.; Weinstein, J.S.; Sobel, E.; Kuroda, Y.; Akaogi, J.; Satoh, M.; et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am. J. Pathol. 2006, 168, 1227–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.L.; Dong, X.; Du, S.; Oh, M.; Singh, R.R.; Voskuhl, R.R. A female preponderance for chemically induced lupus in SJL/J mice. Clin. Immunol. 2007, 122, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.R.; Kotenko, S.V.; Kaplan, M.J. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2021, 17, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Acklin, M.; Staeheli, P. Influenza virus resistance of wild mice: Wild-type and mutant Mx alleles occur at comparable frequencies. J. Interferon Res. 1987, 7, 647–656. [Google Scholar] [CrossRef]
- Staeheli, P.; Grob, R.; Meier, E.; Sutcliffe, J.G.; Haller, O. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 1988, 8, 4518–4523. [Google Scholar] [CrossRef] [Green Version]
- Horisberger, M.A.; Staeheli, P.; Haller, O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc. Natl. Acad. Sci. USA 1983, 80, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.L.; Hatesuer, B.; Bergmann, S.; Nedelko, T.; Schughart, K. Protection from Severe Influenza Virus Infections in Mice Carrying the Mx1 Influenza Virus Resistance Gene Strongly Depends on Genetic Background. J. Virol. 2015, 89, 9998–10009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; Staeheli, P.; Hartmann, R.; Jensen, U.B.; Dagnaes-Hansen, F.; Thomsen, A.R.; Chen, Z.; Haugen, H.; et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, R.; Maizes, J.S.; Guinamard, R.; Ono, M.; Takai, T.; Ravetch, J.V. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 1999, 189, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumore, P.M.; Steinman, C.R. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Investig. 1990, 86, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Emlen, W.; Mannik, M. Clearance of circulating DNA-anti-DNA immune complexes in mice. J. Exp. Med. 1982, 155, 1210–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaaji, M.; Mortensen, E.; Jorgensen, L.; Olsen, R.; Rekvig, O.P. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am. J. Pathol. 2006, 168, 1779–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedeke, T.M.; Stockl, F.W.; Weber, R.; Sugisaki, Y.; Batsford, S.R.; Vogt, A. Histones have high affinity for the glomerular basement membrane. Relevance for immune complex formation in lupus nephritis. J. Exp. Med. 1989, 169, 1879–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coritsidis, G.N.; Beers, P.C.; Rumore, P.M. Glomerular uptake of nucleosomes: Evidence for receptor-mediated mesangial cell binding. Kidney Int. 1995, 47, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Termaat, R.M.; Assmann, K.J.; Dijkman, H.B.; van Gompel, F.; Smeenk, R.J.; Berden, J.H. Anti-DNA antibodies can bind to the glomerulus via two distinct mechanisms. Kidney Int. 1992, 42, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Starke, C.; Frey, S.; Wellmann, U.; Urbonaviciute, V.; Herrmann, M.; Amann, K.; Schett, G.; Winkler, T.; Voll, R.E. High frequency of autoantibody-secreting cells and long-lived plasma cells within inflamed kidneys of NZB/W F1 lupus mice. Eur. J. Immunol. 2011, 41, 2107–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espeli, M.; Bokers, S.; Giannico, G.; Dickinson, H.A.; Bardsley, V.; Fogo, A.B.; Smith, K.G. Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol. 2011, 22, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Henderson, S.G.; Brandt, D.; Liu, N.; Guttikonda, R.; Hsieh, C.; Kaverina, N.; Utset, T.O.; Meehan, S.M.; Quigg, R.J.; et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J. Immunol. 2011, 186, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowling, T.K.; Gilkeson, G.S. Mechanisms of tissue injury in lupus nephritis. Arthritis Res. Ther. 2011, 13, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatiades, E.G.; Tremblay, M.E.; Bohm, M.; Crozet, L.; Bisht, K.; Kao, D.; Coelho, C.; Fan, X.; Yewdell, W.T.; Davidson, A.; et al. Immune Monitoring of Trans-endothelial Transport by Kidney-Resident Macrophages. Cell 2016, 166, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervier, B.; Ribon, M.; Tarantino, N.; Mussard, J.; Breckler, M.; Vieillard, V.; Amoura, Z.; Steinle, A.; Klein, R.; Kotter, I.; et al. Increased Concentrations of Circulating Soluble MHC Class I-Related Chain A (sMICA) and sMICB and Modulation of Plasma Membrane MICA Expression: Potential Mechanisms and Correlation With Natural Killer Cell Activity in Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 633658. [Google Scholar] [CrossRef]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef]
- Spada, R.; Rojas, J.M.; Perez-Yague, S.; Mulens, V.; Cannata-Ortiz, P.; Bragado, R.; Barber, D.F. NKG2D ligand overexpression in lupus nephritis correlates with increased NK cell activity and differentiation in kidneys but not in the periphery. J. Leukoc. Biol. 2015, 97, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Schepis, D.; Gunnarsson, I.; Eloranta, M.L.; Lampa, J.; Jacobson, S.H.; Karre, K.; Berg, L. Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology 2009, 126, 140–146. [Google Scholar] [CrossRef]
- Postol, E.; Meyer, A.; Cardillo, F.; de Alencar, R.; Pessina, D.; Nihei, J.; Mariano, M.; Mengel, J. Long-term administration of IgG2a anti-NK1.1 monoclonal antibody ameliorates lupus-like disease in NZB/W mice in spite of an early worsening induced by an IgG2a-dependent BAFF/BLyS production. Immunology 2008, 125, 184–196. [Google Scholar] [CrossRef]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Seillet, C.; Belz, G.T.; Huntington, N.D. Development, Homeostasis, and Heterogeneity of NK Cells and ILC1. Curr. Top Microbiol. Immunol. 2016, 395, 37–61. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Vienne, M.; Tang, L.; Kerdiles, Y.; Etiennot, M.; Escaliere, B.; Galluso, J.; Wei, H.; Sun, R.; Vivier, E.; et al. Liver type 1 innate lymphoid cells develop locally via an interferon-gamma-dependent loop. Science 2021, 371. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Szeto, C.; Han, S.; Yang, L.; Reeves, W.H. Animal Models of Interferon Signature Positive Lupus. Front. Immunol. 2015, 6, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, E.; Laine, E.; Strehler, D.; Staeheli, P. Resistance to influenza virus infection of Mx transgenic mice expressing Mx protein under the control of two constitutive promoters. J. Virol. 1992, 66, 1709–1716. [Google Scholar] [CrossRef] [Green Version]
- Mordstein, M.; Kochs, G.; Dumoutier, L.; Renauld, J.C.; Paludan, S.R.; Klucher, K.; Staeheli, P. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008, 4, e1000151. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aschman, T.; Schaffer, S.; Biniaris Georgallis, S.I.; Triantafyllopoulou, A.; Staeheli, P.; Voll, R.E. Interferon Lambda Regulates Cellular and Humoral Immunity in Pristane-Induced Lupus. Int. J. Mol. Sci. 2021, 22, 11747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111747
Aschman T, Schaffer S, Biniaris Georgallis SI, Triantafyllopoulou A, Staeheli P, Voll RE. Interferon Lambda Regulates Cellular and Humoral Immunity in Pristane-Induced Lupus. International Journal of Molecular Sciences. 2021; 22(21):11747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111747
Chicago/Turabian StyleAschman, Tom, Sandra Schaffer, Stylianos Iason Biniaris Georgallis, Antigoni Triantafyllopoulou, Peter Staeheli, and Reinhard E. Voll. 2021. "Interferon Lambda Regulates Cellular and Humoral Immunity in Pristane-Induced Lupus" International Journal of Molecular Sciences 22, no. 21: 11747. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111747