
Epigenetics of Myotonic Dystrophies: A Minireview

 ,
,  and
and
Abstract
:1. Introduction
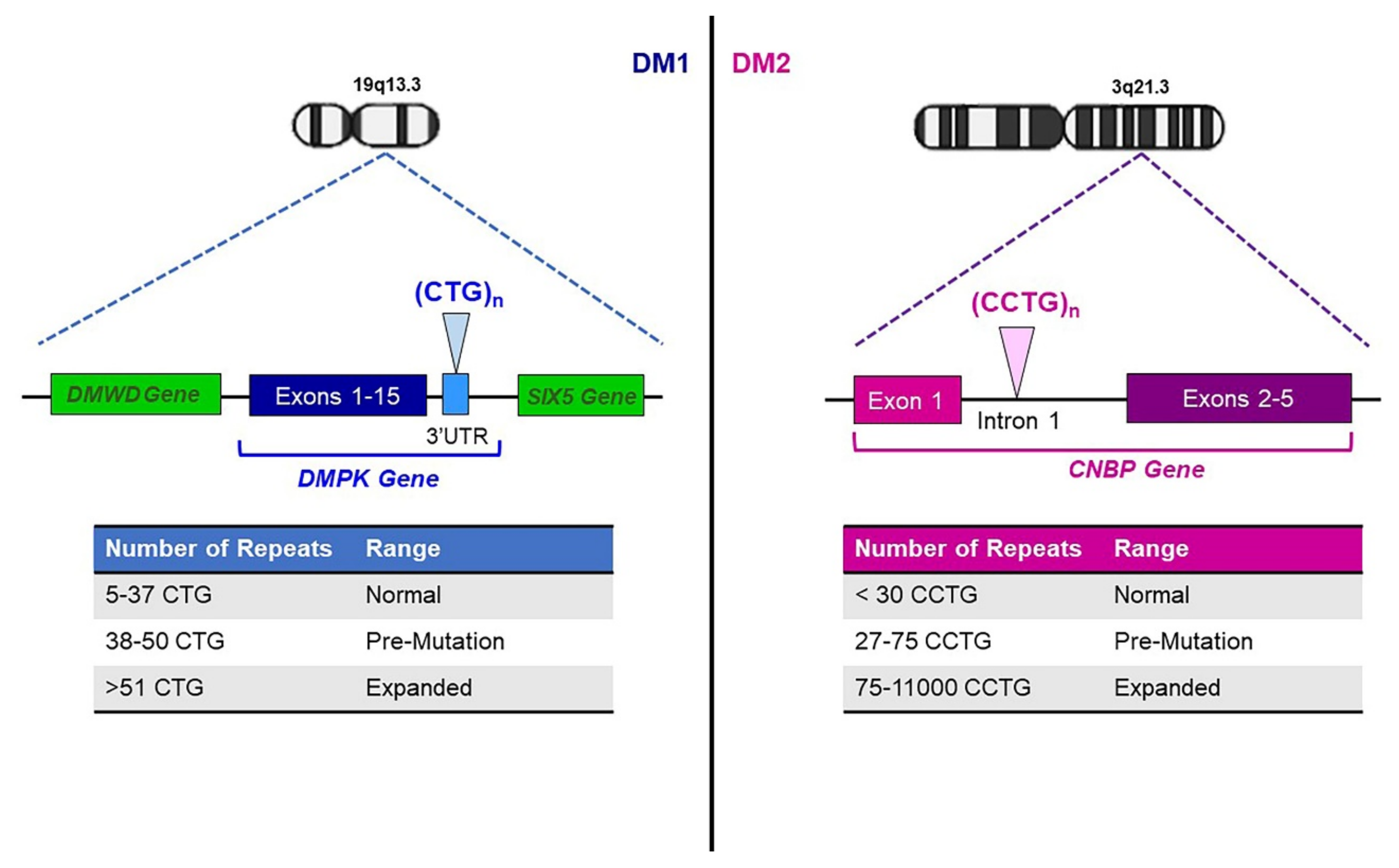
2. Genetics of Myotonic Dystrophies
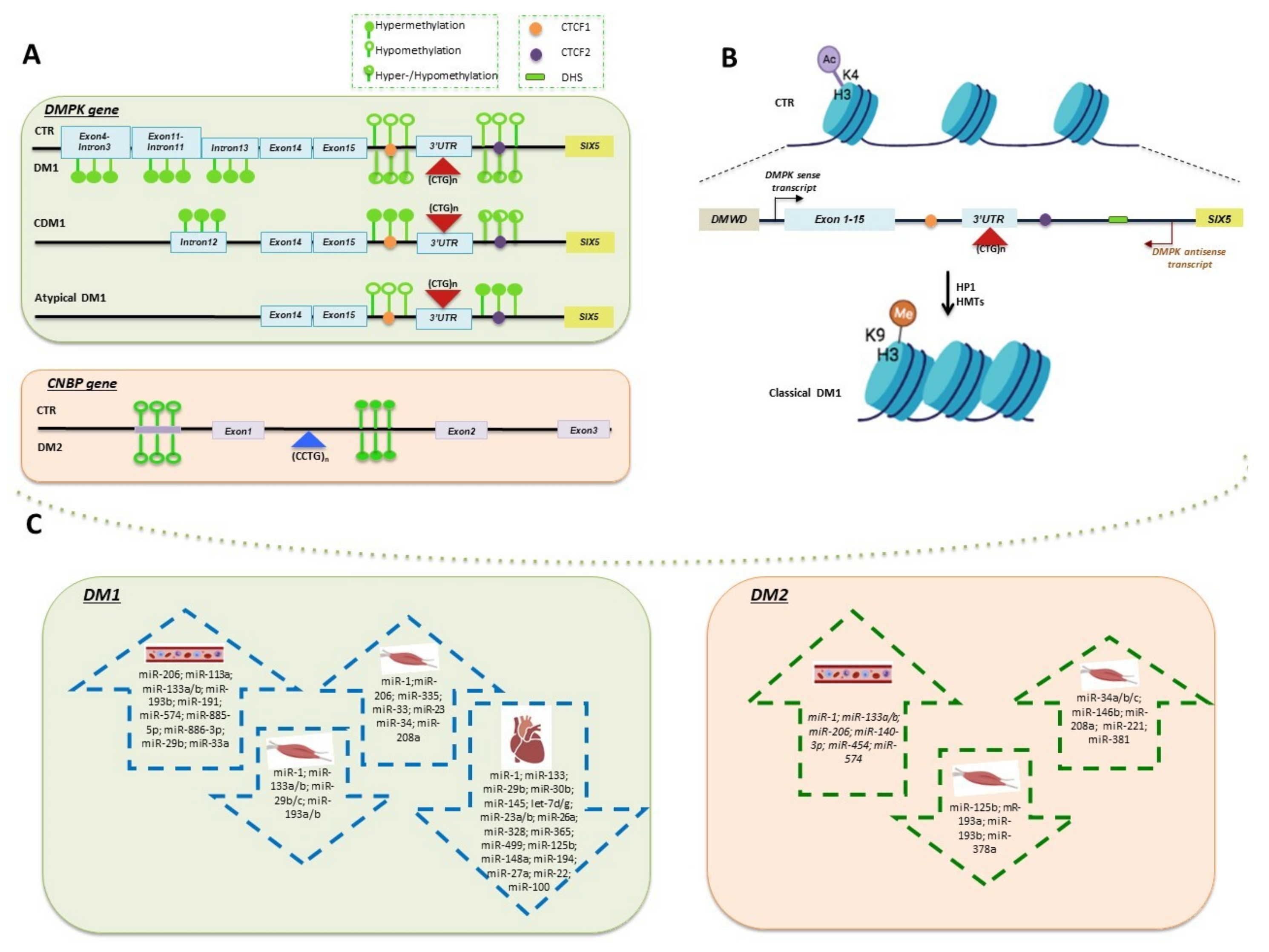
3. Methylation of DMPK and CNBP Genes
4. Chromatin Remodeling of DM1 Locus
5. miRNAs-Based Mechanisms of Epigenetic Regulation in DMs
5.1. Tissue-Specific miRNAs Deregulated in DMs
5.2. Circulating miRNAs Deregulation in DMs
6. Clinical Significance of Epigenetic Signatures in DMs
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Nageshwaran, S.; Festenstein, R. Epigenetics and Triplet-Repeat Neurological Diseases. Front. Neurol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Evans-Galea, M.V.; Hannan, A.J.; Carrodus, N.; Delatycki, M.B.; Saffery, R. Epigenetic modifications in trinucleotide repeat diseases. Trends Mol. Med. 2013, 19, 655–663. [Google Scholar] [CrossRef]
- He, F.; Todd, P.K. Epigenetics in Nucleotide Repeat Expansion Disorders. Semin. Neurol. 2011, 31, 470–483. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F. Epigenetics of neuromuscular disorders. Epigenomics 2020, 12, 2125–2139. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Huguet, A.; Nicole, A.; Munnich, A.; Gourdon, G. Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J. Nucleic Acids 2013, 2013, 567435. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. J. Neuromuscul. Dis. 2015, 2, S59–S71. [Google Scholar] [CrossRef] [Green Version]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic Dystrophy Mutation: An Unstable CTG Repeat in the 3′ Untranslated region of the Gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef]
- Suominen, T.; Bachinski, L.L.; Auvinen, S.; Hackman, P.; Baggerly, K.A.; Angelini, C.; Peltonen, L.; Krahe, R.; Udd, B. Population frequency of myotonic dystrophy: Higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur. J. Hum. Genet. 2011, 19, 776–782. [Google Scholar] [CrossRef]
- Wenninger, S.; Montagnese, F.; Schoser, B. Core Clinical Phenotypes in Myotonic Dystrophies. Front. Neurol. 2018, 9, 303. [Google Scholar] [CrossRef] [Green Version]
- Vanacore, N.; Rastelli, E.; Antonini, G.; Bianchi, M.L.E.; Botta, A.; Bucci, E.; Casali, C.; Costanzi-Porrini, S.; Giacanelli, M.; Gibellini, M.; et al. An Age-Standardized Prevalence Estimate and a Sex and Age Distribution of Myotonic Dystrophy Types 1 and 2 in the Rome Province, Italy. Neuroepidemiology 2016, 46, 191–197. [Google Scholar] [CrossRef]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef]
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.-P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.S.; Swanson, M.; Thornton, C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- Cardani, R.; Bugiardini, E.; Renna, L.V.; Rossi, G.; Colombo, G.; Valaperta, R.; Novelli, G.; Botta, A.; Meola, G. Overexpression of CUGBP1 in Skeletal Muscle from Adult Classic Myotonic Dystrophy Type 1 but Not from Myotonic Dystrophy Type 2. PLoS ONE 2013, 8, e83777. [Google Scholar] [CrossRef] [Green Version]
- Botta, A.; Rossi, G.; Marcaurelio, M.; Fontana, L.; Rosaria D’apice, M.; Brancati, F.; Massa, R.; Monckton, D.G.; Sangiuolo, F.; Novelli, G. Identification and characterization of 5′ CCG interruptions in complex DMPK expanded alleles. Eur. J. Hum. Genet. 2016, 25, 257–261. [Google Scholar] [CrossRef]
- Braida, C.; Stefanatos, R.K.A.; Adam, B.; Mahajan, N.; Smeets, H.J.M.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2010, 19, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. Part A 2009, 149A, 1365–1374. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Bachinski, L.L.; Czernuszewicz, T.; Ramagli, L.S.; Suominen, T.; Shriver, M.D.; Udd, B.; Siciliano, M.J.; Krahe, R. Premutation allele pool in myotonic dystrophy type 2. Neurology 2008, 72, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Mahyera, A.S.; Schneider, T.; Halliger-Keller, B.; Schrooten, K.; Hörner, E.-M.; Rost, S.; Kress, W. Distribution and Structure of DM2 Repeat Tract Alleles in the German Population. Front. Neurol. 2018, 9, 463. [Google Scholar] [CrossRef]
- Botta, A.; Visconti, V.V.; Fontana, L.; Bisceglia, P.; Bengala, M.; Massa, R.; Bagni, I.; Cardani, R.; Sangiuolo, F.; Meola, G.; et al. A 14-Year Italian Experience in DM2 Genetic Testing: Frequency and Distribution of Normal and Premutated CNBP Alleles. Front. Genet. 2021, 12, 668094. [Google Scholar] [CrossRef]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Lanni, S.; Pearson, C.E. Molecular genetics of congenital myotonic dystrophy. Neurobiol. Dis. 2019, 132, 104533. [Google Scholar] [CrossRef]
- Santoro, M.; Fontana, L.; Masciullo, M.; Bianchi, M.L.E.; Rossi, S.; Leoncini, E.; Novelli, G.; Botta, A.; Silvestri, G. Expansion size and presence of CCG/CTC/CGG sequence interruptions in the expanded CTG array are independently associated to hypermethylation at the DMPK locus in myotonic dystrophy type 1 (DM1). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2645–2652. [Google Scholar] [CrossRef]
- Breton, É.; Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.; Mathieu, J.; Gagnon, C.; Richer, L.; Gallais, B.; Bouchard, L. DNA methylation at the DMPK gene locus is associated with cognitive functions in myotonic dystrophy type 1. Epigenomics 2020, 12, 2051–2064. [Google Scholar] [CrossRef]
- Schoser, B.; Timchenko, L. Myotonic Dystrophies 1 and 2: Complex Diseases with Complex Mechanisms. Curr. Genom. 2010, 11, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.N.; van der Plas, E.; Hamilton, M.; Koscik, T.R.; Gutmann, L.; Cumming, S.A.; Monckton, D.G.; Nopoulos, P.C. Variant repeats within the DMPK CTG expansion protect function in myotonic dystrophy type 1. Neurol. Genet. 2020, 6, e504. [Google Scholar] [CrossRef]
- Overend, G.; Légaré, C.; Mathieu, J.; Bouchard, L.; Gagnon, C.; Monckton, D.G. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum. Mol. Genet. 2019, 28, 2245–2254. [Google Scholar] [CrossRef]
- Morales, F.; Vásquez, M.; Cuenca, P.; Campos, D.; Santamaría, C.; Del Valle, G.; Brian, R.; Sittenfeld, M.; Monckton, D.G. Parental age effects, but no evidence for an intrauterine effect in the transmission of myotonic dystrophy type 1. Eur. J. Hum. Genet. 2014, 23, 646–653. [Google Scholar] [CrossRef]
- Cumming, S.A.; Jimenez-Moreno, C.; Okkersen, K.; Wenninger, S.; Daidj, F.; Hogarth, F.; Littleford, R.; Gorman, G.; Bassez, G.; Schoser, B.; et al. Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology 2019, 93, e995–e1009. [Google Scholar] [CrossRef] [Green Version]
- Barbé, L.; Lanni, S.; López-Castel, A.; Franck, S.; Spits, C.; Keymolen, K.; Seneca, S.; Tomé, S.; Miron, I.; Letourneau, J.; et al. CpG Methylation, a Parent-of-Origin Effect for Maternal-Biased Transmission of Congenital Myotonic Dystrophy. Am. J. Hum. Genet. 2017, 100, 488–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbach, P.; Gläser, D.; Vogel, W.; Wolf, M.; Schwemmle, S. The DMPK Gene of Severely Affected Myotonic Dystrophy Patients Is Hypermethylated Proximal to the Largely Expanded CTG Repeat. Am. J. Hum. Genet. 1998, 62, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippova, G.N.; Thienes, C.; Penn, B.H.; Cho, D.H.; Hu, Y.J.; Moore, J.M.; Klesert, T.R.; Lobanenkov, V.V.; Tapscott, S.J. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 2001, 28, 335–343. [Google Scholar] [CrossRef]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense Transcription and Heterochromatin at the DM1 CTG Repeats Are Constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef]
- Yanovsky-Dagan, S.; Avitzour, M.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Mitrani-Rosenbaum, S.; Levy-Lahad, E.; Birnbaum, R.Y.; Gepstein, L.; et al. Uncovering the Role of Hypermethylation by CTG Expansion in Myotonic Dystrophy Type 1 Using Mutant Human Embryonic Stem Cells. Stem Cell Rep. 2015, 5, 221–231. [Google Scholar] [CrossRef] [Green Version]
- López Castel, A.; Nakamori, M.; Tomé, S.; Chitayat, D.; Gourdon, G.; Thornton, C.A.; Pearson, C.E. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 2010, 20, 730–750. [Google Scholar] [CrossRef] [Green Version]
- Buckley, L.; Lacey, M.; Ehrlich, M. Epigenetics of the myotonic dystrophy-associated DMPK gene neighborhood. Epigenomics 2016, 8, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.G.; Mathieu, J.; Gagnon, C.; Bouchard, L. DMPK gene DNA methylation levels are associated with muscular and respiratory profiles in DM1. Neurol. Genet. 2019, 5, e338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Santoro, M.; D’Apice, M.R.; Peluso, F.; Gori, G.; Morrone, A.; Novelli, G.; Dosa, L.; Botta, A. Identification, molecular characterization and segregation analysis of a variant DMPK pre-mutation allele in a three-generation Italian family. Acta Myol. 2020, 39, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Hildonen, M.; Knak, K.L.; Dunø, M.; Vissing, J.; Tümer, Z. Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1. Genes 2020, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Fontana, L.; Maiorca, F.; Centofanti, F.; Massa, R.; Silvestri, G.; Novelli, G.; Botta, A. Expanded [CCTG]n repetitions are not associated with abnormal methylation at the CNBP locus in myotonic dystrophy type 2 (DM2) patients. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1864, 917–924. [Google Scholar] [CrossRef]
- Huang, C.; Sloan, E.A.; Boerkoel, C.F. Chromatin remodeling and human disease. Curr. Opin. Genet. Dev. 2003, 13, 246–252. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone Modifications Depict an Aberrantly Heterochromatinized FMR1 Gene in Fragile X Syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dion, V.; Wilson, J.H. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 2009, 25, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Tomé, S.; Castel, A.L.; Panigrahi, G.B.; Foiry, L.; Hagerman, K.A.; Sroka, H.; Chitayat, D.; Gourdon, G.; Pearson, C.E. Tissue- and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat. Struct. Mol. Biol. 2010, 17, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.W.; Day, J.W. Myotonic Dystrophy: RNA Pathogenesis Comes into Focus. Am. J. Hum. Genet. 2004, 74, 793–804. [Google Scholar] [CrossRef] [Green Version]
- Otten, A.D.; Tapscott, S.J. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc. Natl. Acad. Sci. USA 1995, 92, 5465–5469. [Google Scholar] [CrossRef] [Green Version]
- Frisch, R.; Singleton, K.R.; Moses, P.A.; Gonzalez, I.L.; Carango, P.; Marks, H.G.; Funanage, V.L. Effect of triplet repeat expansion on chromatin structure and expression of dmpk and neighboring genes, six5 and dmwd, in myotonic dystrophy. Mol. Genet. Metab. 2001, 74, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Godde, J.S.; Wolffe, A.P. Nucleosome Assembly on CTG Triplet Repeats. J. Biol. Chem. 1996, 271, 15222–15229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, L.; Huguet-Lachon, A.; Gourdon, G. Sense and Antisense DMPK RNA Foci Accumulate in DM1 Tissues during Development. PLoS ONE 2015, 10, e0137620. [Google Scholar] [CrossRef] [Green Version]
- Sorek, M.; Cohen, L.R.Z.; Meshorer, E. Open chromatin structure in PolyQ disease-related genes: A potential mechanism for CAG repeat expansion in the normal human population. NAR Genom. Bioinform. 2019, 1, e3. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238.e15. [Google Scholar] [CrossRef] [Green Version]
- Buendía, G.A.R.; Leleu, M.; Marzetta, F.; Vanzan, L.; Tan, J.Y.; Ythier, V.; Randall, E.L.; Marques, A.C.; Baubec, T.; Murr, R.; et al. Three-dimensional chromatin interactions remain stable upon CAG/CTG repeat expansion. Sci. Adv. 2020, 6, eaaz4012. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.P.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Turchinovich, A.; Samatov, T.R.; Tonevitsky, A.G.; Burwinkel, B. Circulating miRNAs: Cell-cell communication function? Front. Genet. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Gambardella, S.; Rinaldi, F.; Lepore, S.M.; Viola, A.; Loro, E.; Angelini, C.; Vergani, L.; Novelli, G.; Botta, A. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med. 2010, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. 2011, 21, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Costa, J.M.; Garcia-Lopez, A.; Zuñiga, S.; Fernandez-Pedrosa, V.; Felipo-Benavent, A.; Mata, M.; Jaka, O.; Aiastui, A.; Perez-Alonso, M.; Vilchez, J.J.; et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2012, 22, 704–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritegotto, C.; Ferrati, C.; Pegoraro, V.; Angelini, C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 2017, 38, 619–625. [Google Scholar] [CrossRef]
- Thong, M.-K.; Ambrose, K.K.; Ishak, T.; Lian, L.-H.; Goh, K.-J.; Wong, K.-T.; Ahmad-Annuar, A. Deregulation of microRNAs in blood and skeletal muscles of myotonic dystrophy type 1 patients. Neurol. India 2017, 65, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.-P.; Fischer, M.-C.; Jost, B.; Dembélé, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef]
- Kalsotra, A.; Singh, R.K.; Gurha, P.; Ward, A.J.; Creighton, C.J.; Cooper, T.A. The Mef2 Transcription Network Is Disrupted in Myotonic Dystrophy Heart Tissue, Dramatically Altering miRNA and mRNA Expression. Cell Rep. 2014, 6, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, A.; Greco, S.; Bugiardini, E.; Cardani, R.; Gaia, P.; Gaetano, C.; Meola, G.; Martelli, F. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul. Disord. 2014, 24, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef] [PubMed]
- Koutsoulidou, A.; Photiades, M.; Kyriakides, T.C.; Georgiou, K.; Prokopi, M.; Kapnisis, K.; Łusakowska, A.; Nearchou, M.; Christou, Y.; Papadimas, G.K.; et al. Identification of exosomal muscle-specific miRNAs in serum of myotonic dystrophy patients relating to muscle disease progress. Hum. Mol. Genet. 2017, 26, 3285–3302. [Google Scholar] [CrossRef]
- Pegoraro, V.; Cudia, P.; Baba, A.; Angelini, C. MyomiRNAs and myostatin as physical rehabilitation biomarkers for myotonic dystrophy. Neurol. Sci. 2020, 41, 2953–2960. [Google Scholar] [CrossRef]
- Greco, S.; Perfetti, A.; Fasanaro, P.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Deregulated MicroRNAs in Myotonic Dystrophy Type 2. PLoS ONE 2012, 7, e39732. [Google Scholar] [CrossRef]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2015, 410, 1–13. [Google Scholar] [CrossRef]
- Eisenberg, I.; Alexander, M.S.; Kunkel, L.M. miRNAS in normal and diseased skeletal muscle. J. Cell. Mol. Med. 2008, 13, 2–11. [Google Scholar] [CrossRef]
- Siracusa, J.; Koulmann, N.; Banzet, S. Circulating myomiRs: A new class of biomarkers to monitor skeletal muscle in physiology and medicine. J. Cachex. Sarcopenia Muscle 2017, 9, 20–27. [Google Scholar] [CrossRef]
- André, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Kirby, T.J.; Chaillou, T.; McCarthy, J.J. The role of microRNAs in skeletal muscle health and disease. Front. Biosci. 2015, 20, 37–77. [Google Scholar]
- Krist, B.; Florczyk, U.; Pietraszek-Gremplewicz, K.; Józkowicz, A.; Dulak, J. The Role of miR-378a in Metabolism, Angiogenesis, and Muscle Biology. Int. J. Endocrinol. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenen-Stass, A.M.L.; Betts, C.A.; Lee, Y.F.; Mäger, I.; Turunen, M.P.; El Andaloussi, S.; Morgan, J.E.; Wood, M.J.A.; Roberts, T.C. Selective release of muscle-specific, extracellular microRNAs during myogenic differentiation. Hum. Mol. Genet. 2016, 25, 3960–3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehorst, E.; Ballester-Lopez, A.; Arechavala-Gomeza, V.; Martínez-Piñeiro, A.; Nogales-Gadea, G. The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I. J. Clin. Med. 2020, 9, 3939. [Google Scholar] [CrossRef]
- Koutsoulidou, A.; Phylactou, L.A. Circulating Biomarkers in Muscular Dystrophies: Disease and Therapy Monitoring. Mol. Ther. Methods Clin. Dev. 2020, 18, 230–239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease Form | Tissue | Sample Size | Genomic Context (DMPK Gene) | Method of Analysis | Analysis Outcome | Reference |
|---|---|---|---|---|---|---|
| CDM1; Adult | Dura mater, skeletal muscle, skin biopsies and white blood cells | 30 DM1 | Upstream region of (CTG)n repeats, corresponding to the genomic SacI-HindIII fragment carrying exons 11–15 | Methylation-sensitive restriction enzymes digestion | Hypermethylation in intron 12 at restriction sites of SacII and HhaI in CDM1 patients | [35] |
| DM1 foetuses; DM1 adults; Transgenic DM1 mice | Different source | 13 DM1 vs. 3 CTRs | CTCF binding sites upstream and downstream of the CTG repeats | Bisulphite-sequencing PCR and methylation-sensitive restriction enzymesdigestion | Hypermethylation of upstream sequences in DM1 individuals. In DM1, mice methylation pattern was present up- and down-stream of the CTG array | [40] |
| Childhood-onset; Juvenile/adult-onset; CDM1 with uninterrupted CTG expansions; DM1 “atypical” | Whole blood | 66 DM1 including 9 patients with VRs vs. 30 CTRs | DNA sequences (including CTCF-1 and CTCF-2) in 5′ and in 3′ end regions of the CTG array | MS-HRM | Hypermethylation of upstream sequences in CDM1 and childhood-onset patients with large uninterrupted (CTG)n expansions, significantly associated with maternal transmission. First evidence that DM1 patients with VRs show a distinctive hypermethylation pattern at 3′ end of the CTG array | [27] |
| Premutated DMPK alleles containing VRs | Whole blood | Three-generation Italian family | DNA sequences (including CTCF-1 and CTCF-2) in 5′ and in 3′ end regions of CTG array | Pyrosequencing | Absence of an in cis effect of the (CCG)n interruptions on the methylation of the DM1 locus | [43] |
| DM1-Affected hESC Line Collection | hESCs | 14 DM1 | DNA sequence spanning from exon 11 to the CTG repeats | Pyrosequencing | Marked increase in methylation levels of the expanded allele | [39] |
| Late-onset; Adult; Juvenile; Childhood | Whole blood | 92 DM1 vs. 10 CTRs | Upstream (CTCF1) and downstream (CTCF2) regions | Sanger Sequencing and Massive Parallel Sequencing | DNAme levels of both CTCF sites higher in CDM1 than in non-CDM1 patients | [34] |
| Adult-onset; DM1 “atypical” | Whole blood | 90 DM1 including 8 patients with VRs | CpG sites upstream and downstream of the (CTG)n expansion | Pyrosequencing | DNAme levels upstream of the (CTG)n expansion were correlated with CTG repeat length, and the presence of a VRs was associated with higher DNAme levels compared to pure CTG array | [42] |
| Adult-onset; DM1 “atypical” | Whole blood | 115 DM1 including 12 patients with VRs | Downstream region (no CTCF binding sites) of the (CTG)n repeats | Pyrosequencing | Patients with VRs alleles had distinctive DNAme and cognitive profiles | [28] |
| Noncongenital DM1 | Whole blood | 68 DM1 vs. 73 CTRs | Upstream and downstream regions (no CTCF binding sites) of the (CTG)n repeats | Pyrosequencing | Hypermethylation of both upstream and downstream regions | [44] |
| DM2 | Whole blood | 72 DM2 vs. 50 CTRs | CpG islands in the 5′ promoter region and in the region 3′ of the [CCTG]n repetitions | Pyrosequencing | No significant differences in the methylation profile between DM2 patients and CTRs | [45] |
| Skeletal muscle | 7 DM2 vs. 7 CTRs | |||||
| Skeletal muscle | 7 DM2 vs. 7 CTRs |
| miRNA | Sample Type DM1 | Sample Size | Analysis Outcome (Up or Down Regulation) | Reference |
|---|---|---|---|---|
| miR-206 | Skeletal muscle (vastus lateralis) | 7 DM1 vs. 4 CTR | Up | [64] |
| [miR-1; miR-335 miR-33] | Skeletal muscle (biceps branchii) | 15 DM1 vs. 14 CTR | [Up] | [65] |
| miR-29b/c | Down | |||
| miR-1; miR-7; miR-10 | Skeletal muscle (vastus lateralis, biceps branchii, deltoid) | 5 DM1 vs. 3 CTR | Down | [66] |
| miR-206 | Skeletal muscle (vastus lateralis) | 12 DM1 vs. 6 CTR | Up | [67] |
| [miR-1; miR-133a/b] | [Down] | |||
| miR-1; miR-133a; miR-29c | Skeletal muscle (biceps branchii, deltoid, Gastrocnemium) | 9 DM1 Vs. 9 CTR | Down | [68] |
| miR-1 | Heart | 5 DM1 vs. 8 CTR | Down | [69] |
| miR-1; miR-133; miR-29b; miR-30b; miR-145; let-7d/g; miR-23a/b; miR-26a; miR-328; miR-365; miR-499; miR-125b; miR-148°; miR-194; miR-27a; miR-22; miR-100 | Heart | 8 DM1 vs. 4 CTR | Down | [70] |
| [miR-113a; miR-193b; miR-191; miR-454; miR-574; miR-885-5p; miR-886-3p] | Plasma | 36 DM1 vs. 36 CTR | [Up] | [71] |
| miR-27b | Down | |||
| [miR-1; miR-133a/b; miR-206; miR-140-3p; miR-454; miR-574] | Plasma | 103 DM1 vs. 111 CTR | [Up] | [72] |
| miR-27b | Down | |||
| miR-1; miR-133a/b; miR-206 | Serum | 23 DM1 vs. 23 CTR | Up | [73] |
| miR-1; miR-133a/b; miR-206; miR-113a; miR-193b; miR-191; miR-574; miR-885-5p; miR-886-3p; miR-27b | Serum | 63 DM1 vs. 63 CTR | Up | [74] |
| miR-1; miR-133a/b; miR-206 | Serum | 9 DM1 vs. 7 CTR | Up | [75] |
| miR-133a; miR-29b; miR-33a | Whole blood | 10 DM1 vs. 10 CTR | Up | [68] |
| miR-1 | Heart | 2 DM2 vs. 8 CTR | Down | [69] |
| [miR-34a-5p; miR-34b-3p; miR-34c-5p; miR-146b-5p; miR-208a; miR-221-3p; miR-381] | Skeletal muscle (biceps brachi) | 13 DM2 vs. 13 CTR | [Up] | [76] |
| miR-125b-5p, miR-193a-3p, miR-193b-3p and miR-378a-3p | Down | |||
| miR-1; miR-133a/b; miR-206; miR-140-3p; miR-454; miR-574 | Plasma | 30 DM2 vs. 111 CTR | Up | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visconti, V.V.; Centofanti, F.; Fittipaldi, S.; Macrì, E.; Novelli, G.; Botta, A. Epigenetics of Myotonic Dystrophies: A Minireview. Int. J. Mol. Sci. 2021, 22, 12594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212594
Visconti VV, Centofanti F, Fittipaldi S, Macrì E, Novelli G, Botta A. Epigenetics of Myotonic Dystrophies: A Minireview. International Journal of Molecular Sciences. 2021; 22(22):12594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212594
Chicago/Turabian StyleVisconti, Virginia Veronica, Federica Centofanti, Simona Fittipaldi, Elisa Macrì, Giuseppe Novelli, and Annalisa Botta. 2021. "Epigenetics of Myotonic Dystrophies: A Minireview" International Journal of Molecular Sciences 22, no. 22: 12594. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212594