
Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease



Abstract
:1. Introduction
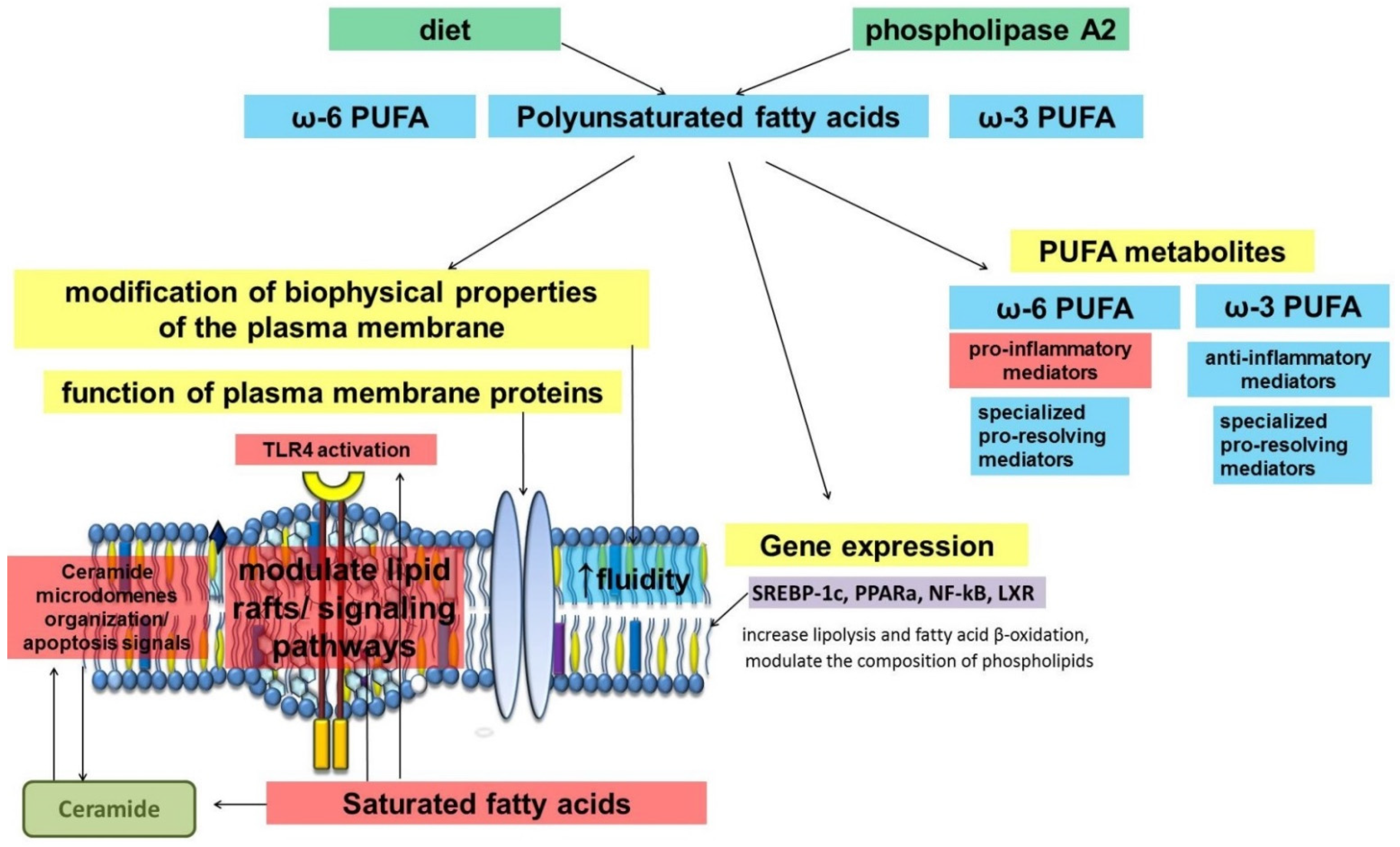
2. Long-Chain Fatty Acids
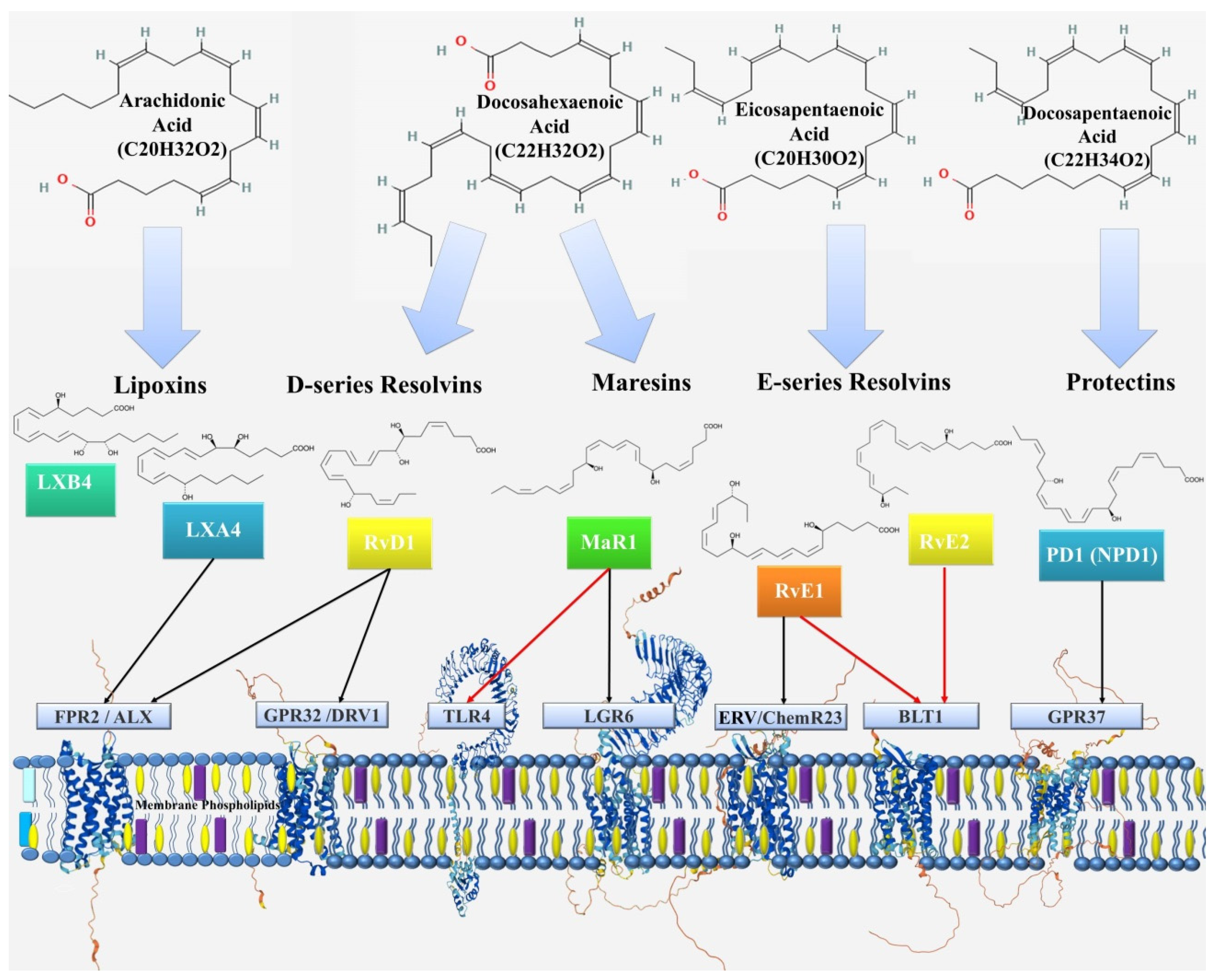
3. Specialized Pro-Resolving Mediators
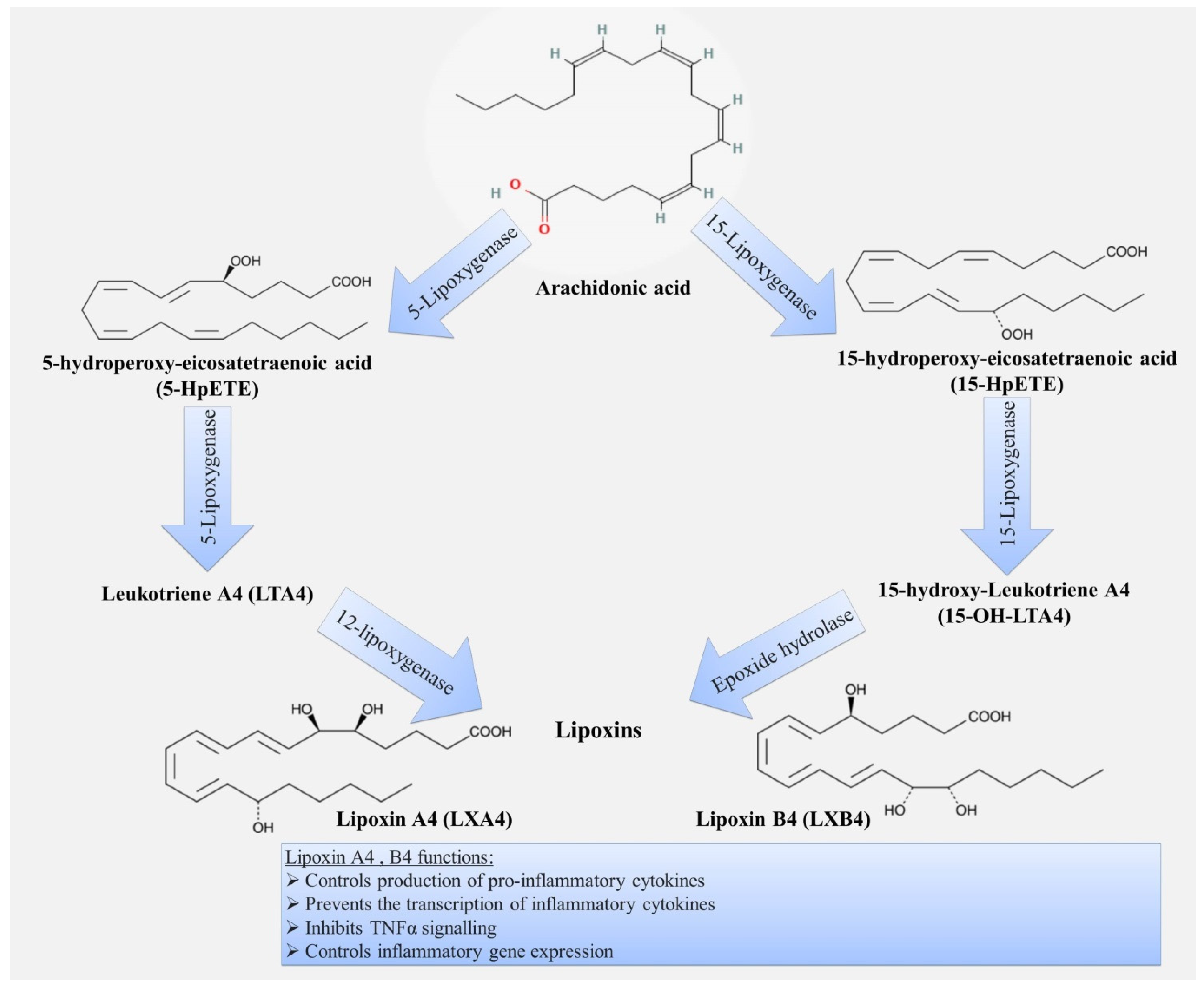
3.1. Lipoxins
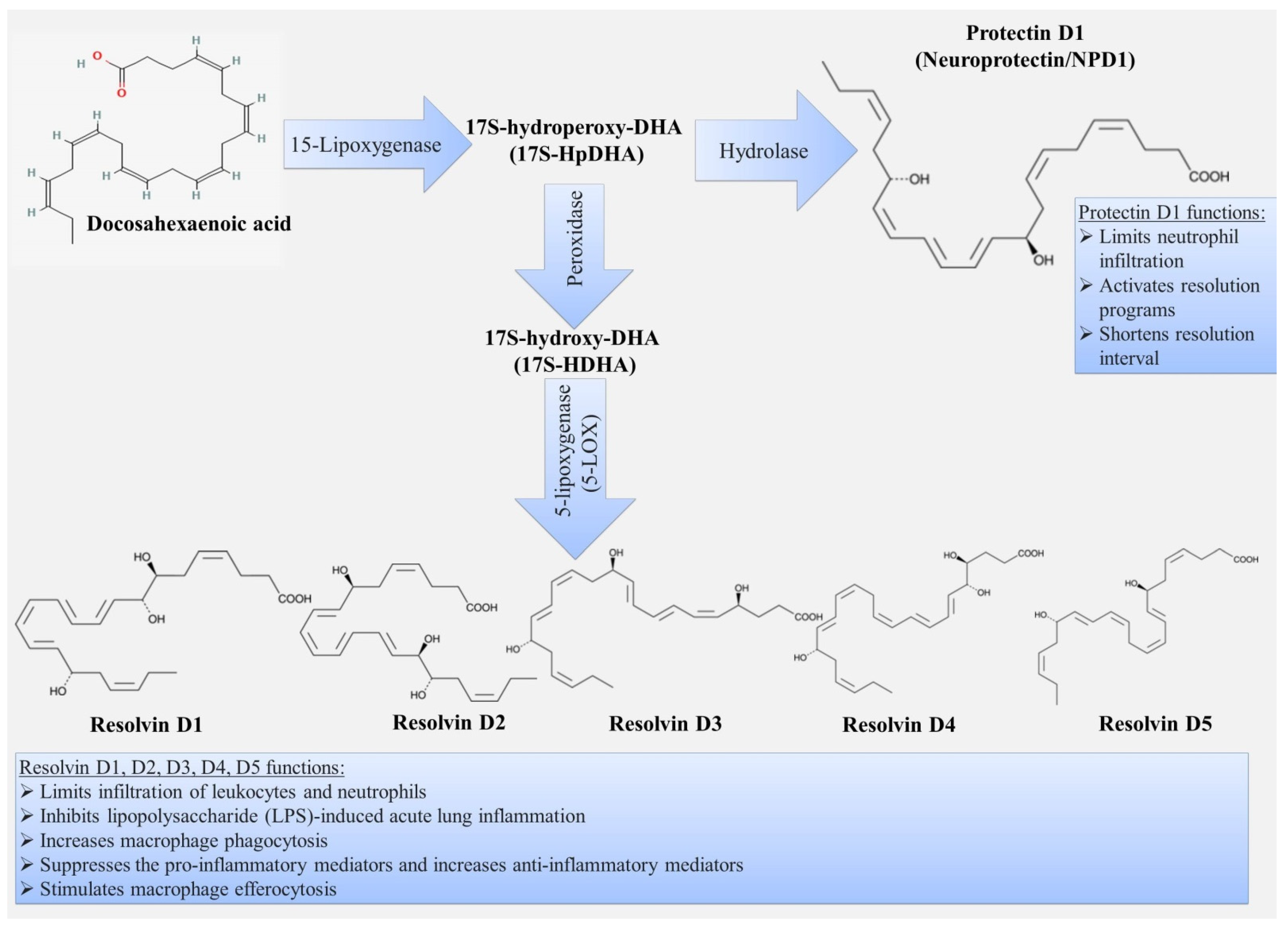
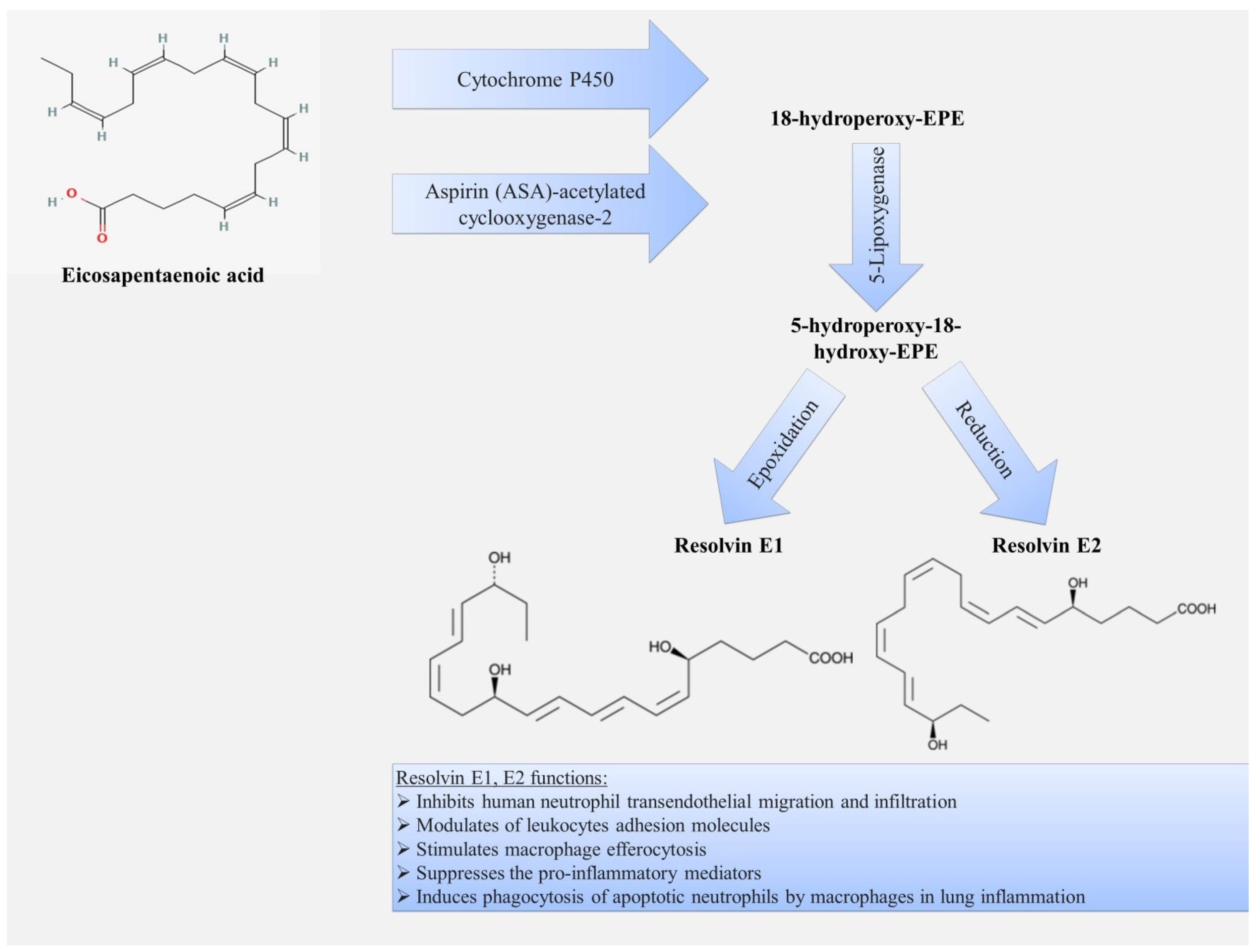
3.2. Resolvins
3.3. Protectins
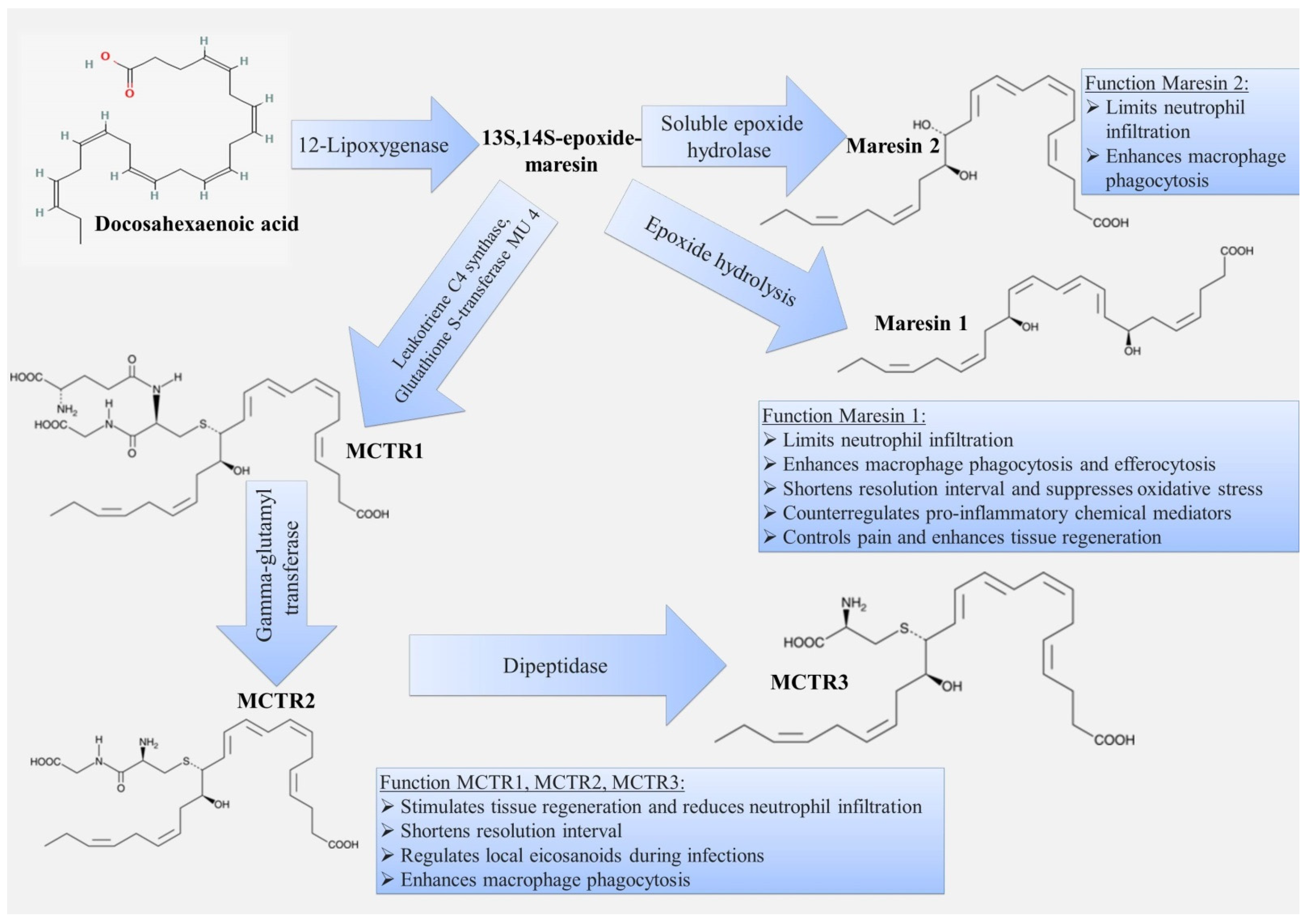
3.4. Maresins
4. Participation of Fatty Acids in Immunometabolic Reprogramming of Macrophages
5. Short-Chain Fatty Acids
6. The Importance of Nutrition in the Progression of COPD
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chronic Obstructive Pulmonary Disease (COPD). Available online: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (accessed on 2 November 2021).
- Mirza, S.; Benzo, R. Chronic Obstructive Pulmonary Disease Phenotypes: Implications for Care. Mayo Clin. Proc. 2017, 92, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkhof, M.; Voorham, J.; Dorinsky, P.; Cabrera, C.; Darken, P.; Kocks, J.W.H.; Sadatsafavi, M.; Sin, D.D.; Carter, V.; Price, D.B. The Long-Term Burden of COPD Exacerbations During Maintenance Therapy and Lung Function Decline. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Viniol, C.; Vogelmeier, C.F. Exacerbations of COPD. Eur. Respir. Rev. 2018, 27, 170103. [Google Scholar] [CrossRef] [Green Version]
- Le Rouzic, O.; Roche, N.; Cortot, A.B.; Tillie-Leblond, I.; Masure, F.; Perez, T.; Boucot, I.; Hamouti, L.; Ostinelli, J.; Pribil, C.; et al. Defining the “Frequent Exacerbator” Phenotype in COPD: A Hypothesis-Free Approach. Chest 2018, 153, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Motavkin, P.A.; Gelzer, B.I. Clinical and Experimental Pathophysiology of the Lungs; Nauka: Moscow, Russia, 1998; p. 366. [Google Scholar]
- Kotlyarov, S.; Kotlyarova, A. Bioinformatic Analysis of ABCA1 Gene Expression in Smoking and Chronic Obstructive Pulmonary Disease. Membranes 2021, 11, 674. [Google Scholar] [CrossRef]
- Boukhenouna, S.; Wilson, M.A.; Bahmed, K.; Kosmider, B. Reactive Oxygen Species in Chronic Obstructive Pulmonary Disease. Oxidative medicine and cellular longevity 2018, 2018, 5730395. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Molecular Mechanisms of Lipid Metabolism Disorders in Infectious Exacerbations of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 7634. [Google Scholar] [CrossRef]
- Yang, A.; Wu, Y.; Yu, G.; Wang, H. Role of specialized pro-resolving lipid mediators in pulmonary inflammation diseases: Mechanisms and development. Respir. Res. 2021, 22, 204. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Newson, J.; Stables, M.; Karra, E.; Arce-Vargas, F.; Quezada, S.; Motwani, M.; Mack, M.; Yona, S.; Audzevich, T.; Gilroy, D.W. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood 2014, 124, 1748–1764. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Targe. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Higgins, A.J.; Lees, P. The acute inflammatory process, arachidonic acid metabolism and the mode of action of anti-inflammatory drugs. Equine Vet. J. 1984, 16, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Ibarguren, M.; López, D.J.; Escribá, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caires, R.; Sierra-Valdez, F.J.; Millet, J.R.M.; Herwig, J.D.; Roan, E.; Vásquez, V.; Cordero-Morales, J.F. Omega-3 Fatty Acids Modulate TRPV4 Function through Plasma Membrane Remodeling. Cell Rep. 2017, 21, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wen, L.; Wang, S.; Zhang, K.; Cui, Y.; Zhang, C.; Feng, L.; Yu, F.; Chen, Y.; Wang, R.; et al. Omega-3 fatty acids improve flow-induced vasodilation by enhancing TRPV4 in arteries from diet-induced obese mice. Cardiovasc. Res. 2020, 117, 2450–2458. [Google Scholar] [CrossRef]
- Weber, J.; Rajan, S.; Schremmer, C.; Chao, Y.-K.; Krasteva-Christ, G.; Kannler, M.; Yildirim, A.Ö.; Brosien, M.; Schredelseker, J.; Weissmann, N.; et al. TRPV4 channels are essential for alveolar epithelial barrier function as protection from lung edema. JCI Insight 2020, 5, e134464. [Google Scholar] [CrossRef]
- Pretorius, E.; du Plooy, J.N.; Soma, P.; Keyser, I.; Buys, A.V. Smoking and fluidity of erythrocyte membranes: A high resolution scanning electron and atomic force microscopy investigation. Nitric Oxide 2013, 35, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Hannan, S.E.; Harris, J.O.; Sheridan, N.P.; Patel, J.M. Cigarette Smoke Alters Plasma Membrane Fluidity of Rat Alveolar Macrophages. Am. Rev. Respir. Dis. 1989, 140, 1668–1673. [Google Scholar] [CrossRef]
- Padmavathi, P.; Reddy, V.D.; Maturu, P.; Varadacharyulu, N. Smoking-Induced Alterations in Platelet Membrane Fluidity and Na+/K+-ATPase Activity in Chronic Cigarette Smokers. J. Atheroscler. Thromb. 2010, 17, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Xu, X.; Qi, M.-Y.; Liu, S.; Song, X.-T.; Zhang, J.-N.; Zhai, Y.-F.; Lu, M.-H.; Han, H.-B.; Lian, Z.-X.; Yao, Y.-C. TLR4 overexpression enhances saturated fatty acid–induced inflammatory cytokine gene expression in sheep. Eur. J. Inflamm. 2018, 16, 2058739218792976. [Google Scholar] [CrossRef] [Green Version]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated Fatty Acid Activates but Polyunsaturated Fatty Acid Inhibits Toll-like Receptor 2 Dimerized with Toll-like Receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harb, H.; Irvine, J.; Amarasekera, M.; Hii, C.S.; Kesper, D.A.; Ma, Y.; D’Vaz, N.; Renz, H.; Potaczek, D.P.; Prescott, S.L.; et al. The role of PKCζ in cord blood T-cell maturation towards Th1 cytokine profile and its epigenetic regulation by fish oil. Biosci. Rep. 2017, 37, BSR20160485. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, N.; Frumento, P.; Harb, H.; Alashkar Alhamwe, B.; Johansson, C.; Eick, L.; Alm, J.; Renz, H.; Scheynius, A.; Potaczek, D.P. Histone Acetylation of Immune Regulatory Genes in Human Placenta in Association with Maternal Intake of Olive Oil and Fish Consumption. Int. J. Mol. Sci. 2019, 20, 1060. [Google Scholar] [CrossRef] [Green Version]
- van der Does, A.M.; Heijink, M.; Persson, L.J.; Kloos, D.-P.; Aanerud, M.; Bakke, P.; Taube, C.; Eagan, T.; Hiemstra, P.S.; Giera, M. Disturbed fatty acid metabolism in airway secretions of patients with Chronic Obstructive Pulmonary Disease. Eur. Respir. J. 2017, 50, PA3913. [Google Scholar] [CrossRef]
- Denisenko, Y.K.; Novgorodtseva, T.P.; Antonyuk, M.V.; Gvozdenko, T.A.; Zhukova, N.V.; Vitkina, T.I.; Gel’tser, B.I. Pathogenesis of immune cell membrane abnormalities in comorbidity of chronic obstructive pulmonary disease and asthma. Pulmonologiya 2018, 28, 647–654. [Google Scholar] [CrossRef]
- Denisenko, Y.K.; Novgorodtseva, T.P.; Vitkina, T.I.; Antonyuk, M.V.; Bocharova, N.V. The fatty acid composition of the mitochondrial membranes of platelets in chronic obstructive pulmonary disease. Klin. Med. 2018, 96, 343–347. [Google Scholar] [CrossRef]
- van der Does, A.M.; Heijink, M.; Mayboroda, O.A.; Persson, L.J.; Aanerud, M.; Bakke, P.; Eagan, T.M.; Hiemstra, P.S.; Giera, M. Dynamic differences in dietary polyunsaturated fatty acid metabolism in sputum of COPD patients and controls. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 224–233. [Google Scholar] [CrossRef]
- Gangopadhyay, S.; Vijayan, V.K.; Bansal, S.K. Lipids of Erythrocyte Membranes of COPD Patients: A Quantitative and Qualitative Study. COPD J. Chronic Obstr. Pulm. Dis. 2012, 9, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Novgorodtseva, T.P.; Denisenko, Y.K.; Zhukova, N.V.; Antonyuk, M.V.; Knyshova, V.V.; Gvozdenko, T.A. Modification of the fatty acid composition of the erythrocyte membrane in patients with chronic respiratory diseases. Lipids Health Dis. 2013, 12, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodtseva, T.P.; Denisenko, Y.K.; Antonyuk, M.V.; Zhukova, N.V. Modification of fatty acid content of cell membranes of erythrocytes at chronic obstructive pulmonary disease. Bull. SB RAMS 2013, 33, 64–69. [Google Scholar]
- Titz, B.; Luettich, K.; Leroy, P.; Boue, S.; Vuillaume, G.; Vihervaara, T.; Ekroos, K.; Martin, F.; Peitsch, M.C.; Hoeng, J. Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD). Int. J. Mol. Sci. 2016, 17, 1583. [Google Scholar] [CrossRef] [Green Version]
- Spiteller, G. Peroxyl radicals: Inductors of neurodegenerative and other inflammatory diseases. Their origin and how they transform cholesterol, phospholipids, plasmalogens, polyunsaturated fatty acids, sugars, and proteins into deleterious products. Free Radic. Biol. Med. 2006, 41, 362–387. [Google Scholar] [CrossRef] [PubMed]
- De Castro, J.; Hernández-Hernández, A.; Rodríguez, M.C.; Sardina, J.L.; Llanillo, M.; Sánchez-Yagüe, J. Comparison of changes in erythrocyte and platelet phospholipid and fatty acid composition and protein oxidation in chronic obstructive pulmonary disease and asthma. Platelets 2007, 18, 43–51. [Google Scholar] [CrossRef]
- Jauregibeitia, I.; Portune, K.; Rica, I.; Tueros, I.; Velasco, O.; Grau, G.; Trebolazabala, N.; Castaño, L.; Larocca, A.V.; Ferreri, C.; et al. Fatty Acid Profile of Mature Red Blood Cell Membranes and Dietary Intake as a New Approach to Characterize Children with Overweight and Obesity. Nutrients 2020, 12, 3446. [Google Scholar] [CrossRef]
- Wada, H.; Goto, H.; Saitoh, E.; Ieki, R.; Okamura, T.; Ota, T.; Hagiwara, S.; Kodaka, T.; Yamamoto, Y. Reduction in plasma free fatty acid in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1465. [Google Scholar] [CrossRef]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am. J. Respir. Cell Mol. Biol. 2014, 51, 284–293. [Google Scholar] [CrossRef]
- Jiang, Z.; Knudsen, N.H.; Wang, G.; Qiu, W.; Naing, Z.Z.C.; Bai, Y.; Ai, X.; Lee, C.-H.; Zhou, X. Genetic Control of Fatty Acid β-Oxidation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2017, 56, 738–748. [Google Scholar] [CrossRef]
- Cornell, K.; Alam, M.; Lyden, E.; Wood, L.; LeVan, T.D.; Nordgren, T.M.; Bailey, K.; Hanson, C. Saturated Fat Intake Is Associated with Lung Function in Individuals with Airflow Obstruction: Results from NHANES 2007–2012. Nutrients 2019, 11, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceco, E.; Celli, D.; Weinberg, S.; Shigemura, M.; Welch, L.C.; Volpe, L.; Chandel, N.S.; Bharat, A.; Lecuona, E.; Sznajder, J.I. Elevated CO(2) Levels Delay Skeletal Muscle Repair by Increasing Fatty Acid Oxidation. Front. Physiol. 2021, 11, 630910. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zhu, Y.; Ma, Y.i.; Xu, Z.; Zao, Y.i.; Wang, J.; Lin, Y.; Comer, G.M. Effect of Supplementing a High-Fat, Low-Carbohydrate Enteral Formula in COPD Patients. Nutrition 2003, 19, 229–232. [Google Scholar] [CrossRef]
- Gong, J.; Zhao, H.; Liu, T.; Li, L.; Cheng, E.; Zhi, S.; Kong, L.; Yao, H.-W.; Li, J. Cigarette Smoke Reduces Fatty Acid Catabolism, Leading to Apoptosis in Lung Endothelial Cells: Implication for Pathogenesis of COPD. Front. Physiol. 2019, 10, 941. [Google Scholar] [CrossRef] [Green Version]
- Pullmannová, P.; Pavlíková, L.; Kováčik, A.; Sochorová, M.; Školová, B.; Slepička, P.; Maixner, J.; Zbytovská, J.; Vávrová, K. Permeability and microstructure of model stratum corneum lipid membranes containing ceramides with long (C16) and very long (C24) acyl chains. Biophys. Chem. 2017, 224, 20–31. [Google Scholar] [CrossRef]
- Sot, J.; Goñi, F.M.; Alonso, A. Molecular associations and surface-active properties of short- and long-N-acyl chain ceramides. Biochim. Biophys. Acta 2005, 1711, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Sot, J.; Aranda, F.J.; Collado, M.I.; Goñi, F.M.; Alonso, A. Different effects of long- and short-chain ceramides on the gel-fluid and lamellar-hexagonal transitions of phospholipids: A calorimetric, NMR, and X-ray diffraction study. Biophys. J. 2005, 88, 3368–3380. [Google Scholar] [CrossRef] [Green Version]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef] [Green Version]
- Goñi, F.M.; Contreras, F.X.; Montes, L.R.; Sot, J.; Alonso, A. Biophysics (and sociology) of ceramides. Biochem. Soc. Symp. 2005, 72, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Balte, P.; Hoffman, E.A.; Oelsner, E.; Pistenmaa, C.L.; Michos, E.; Watson, K.; Laine, A.; Angelini, E.; Wysoczanski, A.; Stukovsky, K.D.H.; et al. Associations of Plasma Omega-3 Fatty Acid Levels with Longitudinal Change in Percent Emphysema, Spirometry, and Chronic Lower Respiratory Disease Events: The Mesa Lung Study. In C15. PREDICTING OUTCOMES IN COPD; American Thoracic Society: New York, NY, USA, 2020; p. A4489. [Google Scholar]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. “Cell Membrane Theory of Senescence” and the Role of Bioactive Lipids in Aging, and Aging Associated Diseases and Their Therapeutic Implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef]
- Das, U.N. Arachidonic acid and lipoxin A4 as possible endogenous anti-diabetic molecules. Prostaglandins Leukot Essent Fatty Acids 2013, 88, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic acid and lipoxin A4 attenuate alloxan-induced cytotoxicity to RIN5F cells in vitro and type 1 diabetes mellitus in vivo. Biofactors 2017, 43, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Arachidonic acid and lipoxinA4 attenuate streptozotocin-induced cytotoxicity to RIN5 F cells in vitro and type 1 and type 2 diabetes mellitus in vivo. Nutrition 2017, 35, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Rutting, S.; Papanicolaou, M.; Xenaki, D.; Wood, L.G.; Mullin, A.M.; Hansbro, P.M.; Oliver, B.G. Dietary ω-6 polyunsaturated fatty acid arachidonic acid increases inflammation, but inhibits ECM protein expression in COPD. Respir. Res. 2018, 19, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Leuti, A.; Maccarrone, M.; Chiurchiù, V. Proresolving Lipid Mediators: Endogenous Modulators of Oxidative Stress. Oxidative medicine and cellular longevity 2019, 2019, 8107265. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Samuelsson, B.; Dahlén, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef]
- Malawista, S.E.; de Boisfleury Chevance, A.; van Damme, J.; Serhan, C.N. Tonic inhibition of chemotaxis in human plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 17949–17954. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Christman, B.W.; Christman, J.W.; Dworski, R.; Blair, I.A.; Prakash, C. Prostaglandin E2 limits arachidonic acid availability and inhibits leukotriene B4 synthesis in rat alveolar macrophages by a nonphospholipase A2 mechanism. J. Immunol. 1993, 151, 2096–2104. [Google Scholar] [PubMed]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef]
- Duvall, M.G.; Bruggemann, T.R.; Levy, B.D. Bronchoprotective mechanisms for specialized pro-resolving mediators in the resolution of lung inflammation. Mol. Aspects Med. 2017, 58, 44–56. [Google Scholar] [CrossRef]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 2013, 5, 174ra126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Fukunaga, K.; Levy, M.A.; Levy, B.D. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am. J. Pathol. 2006, 168, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Serhan, C.N.; Dahlén, S.E.; Drazen, J.M.; Hay, D.W.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Maddox, J.F.; Perez, H.D.; Serhan, C.N. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 1994, 180, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.; Jin, Y.; Chen, Y.; Inomata, T.; Lee, H.; Chauhan, S.K.; Petasis, N.A.; Serhan, C.N.; Dana, R. The resolvin D1 analogue controls maturation of dendritic cells and suppresses alloimmunity in corneal transplantation. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5944–5951. [Google Scholar] [CrossRef]
- Maddox, J.F.; Hachicha, M.; Takano, T.; Petasis, N.A.; Fokin, V.V.; Serhan, C.N. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 1997, 272, 6972–6978. [Google Scholar] [CrossRef] [Green Version]
- Gastardelo, T.S.; Cunha, B.R.; Raposo, L.S.; Maniglia, J.V.; Cury, P.M.; Lisoni, F.C.; Tajara, E.H.; Oliani, S.M. Inflammation and cancer: Role of annexin A1 and FPR2/ALX in proliferation and metastasis in human laryngeal squamous cell carcinoma. PLoS ONE 2014, 9, e111317. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Maddox, J.F.; Petasis, N.A.; Akritopoulou-Zanze, I.; Papayianni, A.; Brady, H.R.; Colgan, S.P.; Madara, J.L. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 1995, 34, 14609–14615. [Google Scholar] [CrossRef]
- Papayianni, A.; Serhan, C.N.; Brady, H.R. Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J. Immunol. 1996, 156, 2264–2272. [Google Scholar]
- Colgan, S.P.; Serhan, C.N.; Parkos, C.A.; Delp-Archer, C.; Madara, J.L. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J. Clin. Investig. 1993, 92, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Maddox, J.F.; Serhan, C.N. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: Selective inactivation by dehydrogenation and reduction. J. Exp. Med. 1996, 183, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.; Mitchell, S.; Brady, H.R.; Godson, C. Lipoxins: Revelations on resolution. Trends Pharmacol. Sci. 2001, 22, 391–395. [Google Scholar] [CrossRef]
- Higgins, G.; Ringholz, F.; Buchanan, P.; McNally, P.; Urbach, V. Physiological impact of abnormal lipoxin A₄ production on cystic fibrosis airway epithelium and therapeutic potential. Biomed. Res. Int. 2015, 2015, 781087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenchegowda, S.; Bazan, N.G.; Bazan, H.E. EGF stimulates lipoxin A4 synthesis and modulates repair in corneal epithelial cells through ERK and p38 activation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2240–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, G.; Buchanan, P.; Perriere, M.; Al-Alawi, M.; Costello, R.W.; Verriere, V.; McNally, P.; Harvey, B.J.; Urbach, V. Activation of P2RY11 and ATP release by lipoxin A4 restores the airway surface liquid layer and epithelial repair in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 178–190. [Google Scholar] [CrossRef] [PubMed]
- József, L.; Zouki, C.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 13266–13271. [Google Scholar] [CrossRef] [Green Version]
- Bonnans, C.; Levy, B.D. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am. J. Respir. Cell Mol. Biol. 2007, 36, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, A.T.; McCormick, B.; Neish, A.S.; Petasis, N.A.; Gronert, K.; Serhan, C.N.; Madara, J.L. Pathogen-induced chemokine secretion from model intestinal epithelium is inhibited by lipoxin A4 analogs. J. Clin. Investig. 1998, 101, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Verrière, V.; Grumbach, Y.; Chiron, R.; Urbach, V. Lxa4 effect On intracellular Ca2+, Cl-secretion, tight junction formation and IL-8 production in normal and cf airway epithelium: 174. Pediatr. Pulmonol. 2006, 41, 268–269. [Google Scholar]
- Fierro, I.M.; Kutok, J.L.; Serhan, C.N. Novel Lipid Mediator Regulators of Endothelial Cell Proliferation and Migration: Aspirin-Triggered-15R-Lipoxin A4 and Lipoxin A4. J. Pharmacol. Exp. Ther. 2002, 300, 385–392. [Google Scholar] [CrossRef]
- El Kebir, D.; József, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Balode, L.; Strazda, G.; Jurka, N.; Kopeika, U.; Kislina, A.; Beinare, M.; Bukovskis, M.; Taivans, I. LSC 2011 Abstract: The role of lipoxin A4 in the chronic obstructive pulmonary disease. Eur. Respir. J. 2011, 38, p414. [Google Scholar]
- Balode, L.; Isajeva, D.; Kislina, A.; Isajevs, S.; Strazda, G.; Jurka, N.; Kopeika, U.; Bukovskis, M.; Taivans, I. Chronic obstructive pulmonary disease is characterized with suppressed lipoxin A4 and increased lipoxin receptor expression in lungs. Eur. Respir. J. 2012, 40, P4588. [Google Scholar]
- Fritscher, L.G.; Post, M.; Rodrigues, M.T.; Silverman, F.; Balter, M.; Chapman, K.R.; Zamel, N. Profile of eicosanoids in breath condensate in asthma and COPD. J. Breath Res. 2012, 6, 026001. [Google Scholar] [CrossRef]
- Balode, L.; Isajevs, S.; Svirina, D.; Kopeika, U.; Strazda, G.; Taivans, I. Lipoxin A4 receptor expression in smokers with and without COPD. Eur. Respir. J. 2011, 38, p3900. [Google Scholar]
- Sha, Y.-H.; Hu, Y.-W.; Gao, J.-J.; Wang, Y.-C.; Ma, X.; Qiu, Y.-R.; Li, S.-F.; Zhao, J.-Y.; Huang, C.; Zhao, J.-J.; et al. Lipoxin A4 promotes ABCA1 expression and cholesterol efflux through the LXRα signaling pathway in THP-1 macrophage-derived foam cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6708–6715. [Google Scholar] [PubMed]
- Demetz, E.; Schroll, A.; Auer, K.; Heim, C.; Patsch, J.R.; Eller, P.; Theurl, M.; Theurl, I.; Theurl, M.; Seifert, M.; et al. The Arachidonic Acid Metabolome Serves as a Conserved Regulator of Cholesterol Metabolism. Cell Metab. 2014, 20, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Xu, Z.; Yin, X.; Zheng, F.; Lin, X.; Pan, Q.; Li, H. Inverse Relationship between Serum Lipoxin A4 Level and the Risk of Metabolic Syndrome in a Middle-Aged Chinese Population. PLoS ONE 2015, 10, e0142848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef]
- Libreros, S.; Shay, A.E.; Nshimiyimana, R.; Fichtner, D.; Martin, M.J.; Wourms, N.; Serhan, C.N. A New E-Series Resolvin: RvE4 Stereochemistry and Function in Efferocytosis of Inflammation-Resolution. Front. Immunol. 2021, 11, 631319. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. UAS 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Norling, L.V.; Dalli, J.; Flower, R.J.; Serhan, C.N.; Perretti, M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: Receptor-dependent actions. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Gemperle, C.; Rimann, N.; Hersberger, M. Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32. J. Immunol. 2016, 196, 3429–3437. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.Y.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Fredman, G.; Serhan, C.N. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 2012, 180, 2018–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuga, K.; Yang, R.; Porter, T.F.; Agrawal, N.; Petasis, N.A.; Irimia, D.; Toner, M.; Serhan, C.N. Rapid Appearance of Resolvin Precursors in Inflammatory Exudates: Novel Mechanisms in Resolution. J. Immunol. 2008, 181, 8677–8687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Hong, S.; Vaidya, V.S.; Lu, Y.; Fredman, G.; Serhan, C.N.; Bonventre, J.V. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006, 177, 5902–5911. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.-L. Resolvins a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Gong, X.; Wan, J.Y.; Zhang, L.; Zhang, Z.; Li, H.Z.; Min, S. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm. Pharmacol. Ther. 2011, 24, 434–441. [Google Scholar] [CrossRef]
- Croasdell, A.; Thatcher, T.H.; Kottmann, R.M.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Sime, P.J.; Phipps, R.P. Resolvins attenuate inflammation and promote resolution in cigarette smoke-exposed human macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L888–L901. [Google Scholar] [CrossRef] [Green Version]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: Evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [Green Version]
- Rogerio, A.P.; Haworth, O.; Croze, R.; Oh, S.F.; Uddin, M.; Carlo, T.; Pfeffer, M.A.; Priluck, R.; Serhan, C.N.; Levy, B.D. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J. Immunol. 2012, 189, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Abdulnour, R.E.; Sham, H.P.; Douda, D.N.; Colas, R.A.; Dalli, J.; Bai, Y.; Ai, X.; Serhan, C.N.; Levy, B.D. Aspirin-triggered resolvin D1 is produced during self-resolving gram-negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol. 2016, 9, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.M.; Thatcher, T.H.; Colas, R.A.; Serhan, C.N.; Phipps, R.P.; Sime, P.J. Resolvin D1 Reduces Emphysema and Chronic Inflammation. Am. J. Pathol. 2015, 185, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.M.; Sapinoro, R.E.; Thatcher, T.H.; Croasdell, A.; Levy, E.P.; Fulton, R.A.; Olsen, K.C.; Pollock, S.J.; Serhan, C.N.; Phipps, R.P.; et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS ONE 2013, 8, e58258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posso, S.V.; Quesnot, N.; Moraes, J.A.; Brito-Gitirana, L.; Kennedy-Feitosa, E.; Barroso, M.V.; Porto, L.C.; Lanzetti, M.; Valença, S.S. AT-RVD1 repairs mouse lung after cigarette smoke-induced emphysema via downregulation of oxidative stress by NRF2/KEAP1 pathway. Int. Immunopharmacol. 2018, 56, 330–338. [Google Scholar] [CrossRef]
- Xie, W.; Wang, H.; Liu, Q.; Li, Y.; Wang, J.; Yao, S.; Wu, Q. ResolvinD1 reduces apoptosis and inflammation in primary human alveolar epithelial type 2 cells. Lab. Investig. 2016, 96, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 2018, 11, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Pena, K.B.; Ramos, C.O.; Soares, N.P.; da Silva, P.F.; Bandeira, A.C.; Costa, G.P.; Cangussú, S.D.; Talvani, A.; Bezerra, F.S. The administration of a high refined carbohydrate diet promoted an increase in pulmonary inflammation and oxidative stress in mice exposed to cigarette smoke. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 3207–3217. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.F.; Dona, M.; Fredman, G.; Krishnamoorthy, S.; Irimia, D.; Serhan, C.N. Resolvin E2 formation and impact in inflammation resolution. J. Immunol. 2012, 188, 4527–4534. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.L.; Louis, N.A.; Tomassetti, S.E.; Canny, G.O.; Arita, M.; Serhan, C.N.; Colgan, S.P. Resolvin E1 promotes mucosal surface clearance of neutrophils: A new paradigm for inflammatory resolution. FASEB J. 2007, 21, 3162–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cash, J.L.; Bena, S.; Headland, S.E.; McArthur, S.; Brancaleone, V.; Perretti, M. Chemerin15 inhibits neutrophil-mediated vascular inflammation and myocardial ischemia-reperfusion injury through ChemR23. EMBO Rep. 2013, 14, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.L.; Hart, R.; Russ, A.; Dixon, J.P.; Colledge, W.H.; Doran, J.; Hendrick, A.G.; Carlton, M.B.; Greaves, D.R. Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 2008, 205, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.Y.; Leung, L.L. Proteolytic regulatory mechanism of chemerin bioactivity. Acta Biochim. Biophys. Sin. 2009, 41, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Herová, M.; Schmid, M.; Gemperle, C.; Hersberger, M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 2015, 194, 2330–2337. [Google Scholar] [CrossRef] [Green Version]
- Parolini, S.; Santoro, A.; Marcenaro, E.; Luini, W.; Massardi, L.; Facchetti, F.; Communi, D.; Parmentier, M.; Majorana, A.; Sironi, M.; et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007, 109, 3625–3632. [Google Scholar] [CrossRef]
- Samson, M.; Edinger, A.L.; Stordeur, P.; Rucker, J.; Verhasselt, V.; Sharron, M.; Govaerts, C.; Mollereau, C.; Vassart, G.; Doms, R.W.; et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 1998, 28, 1689–1700. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and anti-inflammatory actions: Pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem. Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Yoshida, M.; Hong, S.; Tjonahen, E.; Glickman, J.N.; Petasis, N.A.; Blumberg, R.S.; Serhan, C.N. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA 2005, 102, 7671–7676. [Google Scholar] [CrossRef] [Green Version]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, H.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M.; Ohira, T.; Sun, Y.P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461. [Google Scholar] [CrossRef] [Green Version]
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Uppin, V.; Acharya, P.; Ravichandra Talahalli, R. Modulatory Potentials of n-3 Polyunsaturated Fatty Acids in Inflammatory Diseases. In Apolipoproteins, Triglycerides and Cholesterol; IntechOpen: London, UK, 2020. [Google Scholar]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [Green Version]
- Cash, J.L.; Norling, L.V.; Perretti, M. Resolution of inflammation: Targeting GPCRs that interact with lipids and peptides. Drug Discov. Today 2014, 19, 1186–1192. [Google Scholar] [CrossRef] [Green Version]
- Isobe, Y.; Arita, M.; Matsueda, S.; Iwamoto, R.; Fujihara, T.; Nakanishi, H.; Taguchi, R.; Masuda, K.; Sasaki, K.; Urabe, D.; et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J Biol. Chem. 2012, 287, 10525–10534. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Aoki-Saito, H.; Fukuda, H.; Ikeda, H.; Koga, Y.; Yatomi, M.; Tsurumaki, H.; Maeno, T.; Saito, T.; Nakakura, T.; et al. Resolvin E3 attenuates allergic airway inflammation via the interleukin-23-interleukin-17A pathway. FASEB J. 2019, 33, 12750–12759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, P.C.; Libreros, S.; Serhan, C.N. Resolution metabolomes activated by hypoxic environment. Sci. Adv. 2019, 5, eaax4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The Protectin Family of Specialized Pro-resolving Mediators: Potent Immunoresolvents Enabling Innovative Approaches to Target Obesity and Diabetes. Front. Pharmacol. 2019, 9, 1582. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Vik, A.; Ramon, S.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Synthesis and anti-inflammatory and pro-resolving activities of 22-OH-PD1, a monohydroxylated metabolite of protectin D1. J. Nat. Prod. 2014, 77, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Marcheselli, V.L.; Serhan, C.N.; Bazan, N.G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA 2004, 101, 8491–8496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, N.G. Neuroprotectin D1 (NPD1): A DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005, 15, 159–166. [Google Scholar] [CrossRef]
- Serhan, C.N.; Fredman, G.; Yang, R.; Karamnov, S.; Belayev, L.S.; Bazan, N.G.; Zhu, M.; Winkler, J.W.; Petasis, N.A. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011, 18, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariel, A.; Li, P.L.; Wang, W.; Tang, W.X.; Fredman, G.; Hong, S.; Gotlinger, K.H.; Serhan, C.N. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 2005, 280, 43079–43086. [Google Scholar] [CrossRef] [Green Version]
- Ariel, A.; Fredman, G.; Sun, Y.P.; Kantarci, A.; Van Dyke, T.E.; Luster, A.D.; Serhan, C.N. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 2006, 7, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Wei, J.; Gronert, K. The role of pro-resolving lipid mediators in ocular diseases. Mol. Aspects Med. 2017, 58, 37. [Google Scholar] [CrossRef]
- Kraft, J.D.; Blomgran, R.; Lundgaard, I.; Quiding-Järbrink, M.; Bromberg, J.S.; Börgeson, E. Specialized Pro-Resolving Mediators and the Lymphatic System. Int. J. Mol. Sci. 2021, 22, 2750. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.R.; Spur, B.W. Total synthesis of the macrophage derived anti-inflammatory lipid mediator Maresin 1. Tetrahedron Lett. 2012, 53, 4169–4172. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Spur, B.W. First total synthesis of the macrophage derived anti-inflammatory and pro-resolving lipid mediator Maresin 2. Tetrahedron Lett. 2015, 56, 256–259. [Google Scholar] [CrossRef]
- Tang, S.; Wan, M.; Huang, W.; Stanton, R.C.; Xu, Y. Maresins: Specialized Proresolving Lipid Mediators and Their Potential Role in Inflammatory-Related Diseases. Mediat. Inflamm. 2018, 2018, 2380319. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, Y.; Ma, Z.; Ma, M.; Wang, D.; Xie, G.; Yin, Y.; Zhang, P.; Tao, K. Maresin 1 Mitigates Inflammatory Response and Protects Mice from Sepsis. Mediat. Inflamm. 2016, 2016, 3798465. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Spur, B.W. First total synthesis of pro-resolving and tissue-regenerative Maresin sulfido-conjugates. Tetrahedron Lett. 2015, 56, 3936–3940. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 397–413. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dalli, J.; Chiang, N.; Baron, R.M.; Quintana, C.; Serhan, C.N. Plasticity of Leukocytic Exudates in Resolving Acute Inflammation Is Regulated by MicroRNA and Proresolving Mediators. Immunity 2013, 39, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Wu, Y.; Zhao, F.; Wang, J. Maresin 1 Ameliorates Lung Ischemia/Reperfusion Injury by Suppressing Oxidative Stress via Activation of the Nrf-2-Mediated HO-1 Signaling Pathway. Oxidative Med. Cell. Longev. 2017, 2017, 9634803. [Google Scholar] [CrossRef]
- Chiang, N.; Libreros, S.; Norris, P.C.; de la Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.F.; Hao, H.; Tu, W.S.; Guo, N.; Zhou, X.Y. Maresins: Anti-inflammatory pro-resolving mediators with therapeutic potential. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7442–7453. [Google Scholar] [CrossRef]
- Qiu, S.; Li, P.; Zhao, H.; Li, X. Maresin 1 alleviates dextran sulfate sodium-induced ulcerative colitis by regulating NRF2 and TLR4/NF-kB signaling pathway. Int. Immunopharmacol. 2020, 78, 106018. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.-W.; Mei, H.-X.; Ye, Y.; Xu, H.-R.; Xiang, S.-Y.; Yang, Q.; Zheng, S.-X.; Smith, F.-G.; Jin, S.-W. MCTR1 enhances the resolution of lipopolysaccharide-induced lung injury through STAT6-mediated resident M2 alveolar macrophage polarization in mice. J. Cell. Mol. Med. 2020, 24, 9646–9657. [Google Scholar] [CrossRef]
- Li, H.; Hao, Y.; Yang, L.L.; Wang, X.Y.; Li, X.Y.; Bhandari, S.; Han, J.; Liu, Y.J.; Gong, Y.Q.; Scott, A.; et al. MCTR1 alleviates lipopolysaccharide-induced acute lung injury by protecting lung endothelial glycocalyx. J. Cell. Physiol. 2020, 235, 7283–7294. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef]
- Saradna, A.; Do, D.C.; Kumar, S.; Fu, Q.L.; Gao, P. Macrophage polarization and allergic asthma. Transl. Res. 2018, 191, 1–14. [Google Scholar] [CrossRef]
- Abdelaziz, M.H.; Abdelwahab, S.F.; Wan, J.; Cai, W.; Huixuan, W.; Jianjun, C.; Kumar, K.D.; Vasudevan, A.; Sadek, A.; Su, Z.; et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J. Transl. Med. 2020, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Chawla, A. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 275–297. [Google Scholar] [CrossRef] [Green Version]
- Namgaladze, D.; Brüne, B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1796–1807. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2009, 457, 318–321. [Google Scholar] [CrossRef]
- Posokhova, E.; Khoshchenko, O.; Chasovskikh, M.; Pivovarova, E.; Dushkin, M. Lipid synthesis in macrophages during inflammation in vivo: Effect of agonists of peroxisome proliferator activated receptors α and γ and of retinoid X receptors. Biochemistry 2008, 73, 296–304. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Xu, B.; Hui, C.K.M.; Chung, K.F.; Adcock, I.M. Role of Metabolic Reprogramming in Pulmonary Innate Immunity and Its Impact on Lung Diseases. J. Innate Immun. 2020, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovich, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Vaughan, A.; Frazer, Z.A.; Hansbro, P.M.; Yang, I.A. COPD and the gut-lung axis: The therapeutic potential of fibre. J. Thorac. Dis. 2019, 11, S2173–S2180. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.A.; Macfarlane, G.T. Dissimilatory amino Acid metabolism in human colonic bacteria. Anaerobe 1997, 3, 327–337. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, P.B.; Clausen, M.R. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand. J. Gastroenterol. Suppl. 1996, 216, 132–148. [Google Scholar] [CrossRef]
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [CrossRef]
- Roy, C.C.; Kien, C.L.; Bouthillier, L.; Levy, E. Short-chain fatty acids: Ready for prime time? Nutr. Clin. Pract. 2006, 21, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Wiltrout, D.W.; Satter, L.D. Contribution of propionate to glucose synthesis in the lactating and nonlactating cow. J. Dairy Sci. 1972, 55, 307–317. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Layden, B.T.; Angueira, A.R.; Brodsky, M.; Durai, V.; Lowe, W.L., Jr. Short chain fatty acids and their receptors: New metabolic targets. Transl. Res. 2013, 161, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Kaneyuki, T.; Tagawa, K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim. Biophys. Acta 2001, 1532, 79–87. [Google Scholar] [CrossRef]
- Bell-Parikh, L.C.; Guengerich, F.P. Kinetics of cytochrome P450 2E1-catalyzed oxidation of ethanol to acetic acid via acetaldehyde. J. Biol. Chem. 1999, 274, 23833–23840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merezhinskaya, N.; Ogunwuyi, S.A.; Mullick, F.G.; Fishbein, W.N. Presence and localization of three lactic acid transporters (MCT1, -2, and -4) in separated human granulocytes, lymphocytes, and monocytes. J. Histochem. Cytochem. 2004, 52, 1483–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, E.M.; Knuplez, E.; Marsche, G. Role of Short Chain Fatty Acids and Apolipoproteins in the Regulation of Eosinophilia-Associated Diseases. Int. J. Mol. Sci. 2021, 22, 4377. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, N.E.; Kotarsky, K.; Owman, C.; Olde, B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun. 2003, 303, 1047–1052. [Google Scholar] [CrossRef]
- Ulven, T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front. Endocrinol. 2012, 3, 111. [Google Scholar] [CrossRef] [Green Version]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nature Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, Y.W.; Chen, C.T.; Ho, C.M.; Su, W.H.; Jou, Y.S. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Tao, J.; Chen, P.; Chen, L.; Sharma, S.; Wang, G.; Dong, Q. Sodium butyrate inhibits colorectal cancer cell migration by downregulating Bmi-1 through enhanced miR-200c expression. Mol. Nutr. Food Res. 2018, 62, 1700844. [Google Scholar] [CrossRef]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J. Inflamm. 2010, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal Transduct. Target. Ther. 2019, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Jardou, M.; Lawson, R. Supportive therapy during COVID-19: The proposed mechanism of short-chain fatty acids to prevent cytokine storm and multi-organ failure. Med. Hypotheses 2021, 154, 110661. [Google Scholar] [CrossRef]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X.; et al. Short-Chain Fatty Acids and Their Association with Signalling Pathways in Inflammation, Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Yoon, H.J.; Park, M.K.; Lee, H.; Park, T.S.; Park, D.W.; Moon, J.-Y.; Kim, T.H.; Sohn, J.W.; Kim, S.-H.; Yoon, H.-R. Effects of respiratory short-chain fatty acids on bronchial inflammation in asthma. World Allergy Organ. J. 2020, 13. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c(-) Patrolling Monocyte Hematopoiesis and CD8(+) T Cell Metabolism. Immunity 2018, 48, 992–1005.e1008. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.A.; Jackson, J.; Stanton, M.; Rojas-Triana, A.; Bober, L.; Laverty, M.; Yang, X.; Zhu, F.; Liu, J.; Wang, S.; et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J. Gastroenterol. 2009, 15, 5549–5557. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e285. [Google Scholar] [CrossRef]
- Ghorbani, P.; Santhakumar, P.; Hu, Q.; Djiadeu, P.; Wolever, T.M.; Palaniyar, N.; Grasemann, H. Short-chain fatty acids affect cystic fibrosis airway inflammation and bacterial growth. Eur. Respir. J. 2015, 46, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tian, X.; Maruyama, D.; Arjomandi, M.; Prakash, A. Lung immune tone via gut-lung axis: Gut-derived LPS and short-chain fatty acids’ immunometabolic regulation of lung IL-1β, FFAR2, and FFAR3 expression. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L65–L78. [Google Scholar] [CrossRef]
- Richards, L.B.; Li, M.; Folkerts, G.; Henricks, P.A.J.; Garssen, J.; van Esch, B.C.A.M. Butyrate and Propionate Restore the Cytokine and House Dust Mite Compromised Barrier Function of Human Bronchial Airway Epithelial Cells. Int. J. Mol. Sci. 2020, 22, 65. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Mise, N.; Jia, J.; Genoyer, E.; Yildirim, A.; Meiners, S.; Eickelberg, O. Cigarette smoke-induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-β. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, M.; Kan-o, K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef] [PubMed]
- Rutting, S.; Xenaki, D.; Malouf, M.; Horvat, J.C.; Wood, L.G.; Hansbro, P.M.; Oliver, B.G. Short-chain fatty acids increase TNFα-induced inflammation in primary human lung mesenchymal cells through the activation of p38 MAPK. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L157–L174. [Google Scholar] [CrossRef] [Green Version]
- Bailón, E.; Cueto-Sola, M.; Utrilla, P.; Rodríguez-Cabezas, M.E.; Garrido-Mesa, N.; Zarzuelo, A.; Xaus, J.; Gálvez, J.; Comalada, M. Butyrate in vitro immune-modulatory effects might be mediated through a proliferation-related induction of apoptosis. Immunobiology 2010, 215, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Hogg, J.C.; Sin, D.D. Bacterial microbiome of lungs in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2014, 9, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Li, N.; Yang, Z.; Liao, B.; Pan, T.; Pu, J.; Hao, B.; Fu, Z.; Cao, W.; Zhou, Y.; He, F.; et al. Chronic exposure to ambient particulate matter induces gut microbial dysbiosis in a rat COPD model. Respir. Res. 2020, 21, 271. [Google Scholar] [CrossRef]
- Reiss, A.; Jacobi, M.; Rusch, K.; Schwiertz, A. Association of dietary type with fecal microbiota and short chain fatty acids in vegans and omnivores. J. Int. Soc. Microbiota 2016, 1, 1. [Google Scholar]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Alonso, V.R.; Guarner, F. Intestinal microbiota composition in adults. In Probiotic Bacteria and Their Effect on Human Health and Well-Being; Karger Publishers: Basel, Switzerland, 2013; Volume 107, pp. 17–24. [Google Scholar]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Madan, J.C.; Koestler, D.C.; Stanton, B.A.; Davidson, L.; Moulton, L.A.; Housman, M.L.; Moore, J.H.; Guill, M.F.; Morrison, H.G.; Sogin, M.L.; et al. Serial Analysis of the Gut and Respiratory Microbiome in Cystic Fibrosis in Infancy: Interaction between Intestinal and Respiratory Tracts and Impact of Nutritional Exposures. mBio 2012, 3, e00251-12. [Google Scholar] [CrossRef] [Green Version]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Marsland, B.J.; Trompette, A.; Gollwitzer, E.S. The Gut-Lung Axis in Respiratory Disease. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 2), S150–S156. [Google Scholar] [CrossRef]
- Sze, M.A.; Dimitriu, P.A.; Hayashi, S.; Elliott, W.M.; McDonough, J.E.; Gosselink, J.V.; Cooper, J.; Sin, D.D.; Mohn, W.W.; Hogg, J.C. The lung tissue microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.O.; Kim, O.-H.; Kim, S.J.; Lee, S.H.; Yun, S.; Lim, S.E.; Yoo, H.J.; Shin, Y.; Lee, S.W. High-fiber diets attenuate emphysema development via modulation of gut microbiota and metabolism. Sci. Rep. 2021, 11, 7008. [Google Scholar] [CrossRef]
- Woodmansey, E.J. Intestinal bacteria and ageing. J. Appl. Microbiol. 2007, 102, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Woodmansey, E.J.; McMurdo, M.E.; Macfarlane, G.T.; Macfarlane, S. Comparison of compositions and metabolic activities of fecal microbiotas in young adults and in antibiotic-treated and non-antibiotic-treated elderly subjects. Appl. Environ. Microbiol. 2004, 70, 6113–6122. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill–Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724.e1712. [Google Scholar] [CrossRef] [Green Version]
- Senghor, B.; Sokhna, C.; Ruimy, R.; Lagier, J.-C. Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 2018, 7–8, 1–9. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-6/omega-3 essential fatty acids: Biological effects. World Rev. Nutr. Diet 2009, 99, 1–16. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Importance of the omega-6/omega-3 balance in health and disease: Evolutionary aspects of diet. World Rev. Nutr. Diet 2011, 102, 10–21. [Google Scholar] [CrossRef]
- Wood, L.G.; Scott, H.A.; Garg, M.L.; Gibson, P.G. Innate immune mechanisms linking non-esterified fatty acids and respiratory disease. Prog. Lipid Res. 2009, 48, 27–43. [Google Scholar] [CrossRef]
- Shahar, E.; Boland, L.L.; Folsom, A.R.; Tockman, M.S.; McGovern, P.G.; Eckfeldt, J.H. Docosahexaenoic acid and smoking-related chronic obstructive pulmonary disease. The Atherosclerosis Risk in Communities Study Investigators. Am. J. Respir. Crit. Care Med. 1999, 159, 1780–1785. [Google Scholar] [CrossRef]
- Hirayama, F.; Lee, A.H.; Binns, C.W.; Hiramatsu, N.; Mori, M.; Nishimura, K. Dietary intake of isoflavones and polyunsaturated fatty acids associated with lung function, breathlessness and the prevalence of chronic obstructive pulmonary disease: Possible protective effect of traditional Japanese diet. Mol. Nutr. Food Res. 2010, 54, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, X.; Lei, T.; Zhang, C.; Zhang, M.; Wang, Y.; Zhu, L.; Liu, J. Effect of Omega-3 Fatty Acids on Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 2677–2686. [Google Scholar] [CrossRef]
- Mickleborough, T.D.; Lindley, M.R.; Ionescu, A.A.; Fly, A.D. Protective effect of fish oil supplementation on exercise-induced bronchoconstriction in asthma. Chest 2006, 129, 39–49. [Google Scholar] [CrossRef]
- Mickleborough, T.D.; Murray, R.L.; Ionescu, A.A.; Lindley, M.R. Fish oil supplementation reduces severity of exercise-induced bronchoconstriction in elite athletes. Am. J. Respir. Crit. Care Med. 2003, 168, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Lopata, A.L.; Smuts, C.M.; Baatjies, R.; Jeebhay, M.F. Relationship between Serum Omega-3 Fatty Acid and Asthma Endpoints. Int. J. Environ. Res. Public Health 2018, 16, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, S.C.M.; Brigham, E.P.; Woo, H.; Hanson, C.K.; McCormack, M.C.; Koch, A.; Putcha, N.; Hansel, N.N. Omega-3 fatty acid intake and prevalent respiratory symptoms among U.S. adults with COPD. BMC Pulm. Med. 2019, 19, 97. [Google Scholar] [CrossRef] [Green Version]
- Shahar, E.; Folsom, A.R.; Melnick, S.L.; Tockman, M.S.; Comstock, G.W.; Gennaro, V.; Higgins, M.W.; Sorlie, P.D.; Ko, W.-J.; Szklo, M. Dietary n-3 Polyunsaturated Fatty Acids and Smoking-Related Chronic Obstructive Pulmonary Disease. New Engl. J. Med. 1994, 331, 228–233. [Google Scholar] [CrossRef]
- Shaheen, S.O.; Jameson, K.A.; Syddall, H.E.; Aihie Sayer, A.; Dennison, E.M.; Cooper, C.; Robinson, S.M.; Group, T.H.C.S. The relationship of dietary patterns with adult lung function and COPD. Eur. Respir. J. 2010, 36, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaluza, J.; Harris, H.; Wallin, A.; Linden, A.; Wolk, A. Dietary Fiber Intake and Risk of Chronic Obstructive Pulmonary Disease: A Prospective Cohort Study of Men. Epidemiology 2018, 29, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, J.; Larsson, S.C.; Orsini, N.; Linden, A.; Wolk, A. Fruit and vegetable consumption and risk of COPD: A prospective cohort study of men. Thorax 2017, 72, 500–509. [Google Scholar] [CrossRef]
- Kaluza, J.; Harris, H.R.; Linden, A.; Wolk, A. Long-term consumption of fruits and vegetables and risk of chronic obstructive pulmonary disease: A prospective cohort study of women. Int. J. Epidemiol. 2018, 47, 1897–1909. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, S.; Kotlyarova, A. Atherosclerosis as a risk factor in the prognosis of the survival of patients with COPD. Eur. Heart J. Acute Cardiovasc. Care 2021, 10. [Google Scholar] [CrossRef]
- Xue, M.; Cai, C.; Guan, L.; Xu, Y.; Lin, J.; Zeng, Y.; Hu, H.; Chen, R.; Wang, H.; Zhou, L.; et al. Exploration of n-6 and n-3 Polyunsaturated Fatty Acids Metabolites Associated with Nutritional Levels in Patients with Severe Stable Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, R.; Wouters, E.F.; Creutzberg, E.C.; Weling-Scheepers, C.A.; Schols, A.M. Polyunsaturated fatty acids improve exercise capacity in chronic obstructive pulmonary disease. Thorax 2005, 60, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelakis, E.; Armougom, F.; Million, M.; Raoult, D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012, 7, 91–109. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Howard, A.G.; Meyer, K.A.; Tsilimigras, M.C.B.; Avery, C.L.; Sha, W.; Sun, S.; Zhang, J.; Su, C.; et al. Circulating Short-Chain Fatty Acids Are Positively Associated with Adiposity Measures in Chinese Adults. Nutrients 2020, 12, 2127. [Google Scholar] [CrossRef] [PubMed]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Louis, S.; Rings, A.; Messner, S.; Camarinha-Silva, A.; Seifert, J.; Bischoff, S.C.; et al. Intestinal Microbiota and Microbial Metabolites Are Changed in a Pig Model Fed a High-Fat/Low-Fiber or a Low-Fat/High-Fiber Diet. PLoS ONE 2016, 11, e0154329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.K.; Abbaspour, A.; Bulik, C.M.; Carroll, I.M. The intestinal microbiota and anorexia nervosa: Cause or consequence of nutrient deprivation. Curr. Opin. Endocr. Metab. Res. 2021, 19, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Mack, I.; Cuntz, U.; Grämer, C.; Niedermaier, S.; Pohl, C.; Schwiertz, A.; Zimmermann, K.; Zipfel, S.; Enck, P.; Penders, J. Weight gain in anorexia nervosa does not ameliorate the faecal microbiota, branched chain fatty acid profiles and gastrointestinal complaints. Sci. Rep. 2016, 6, 26752. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Keshavarzian, A.; Rose, D.J. Impact of dietary fiber fermentation from cereal grains on metabolite production by the fecal microbiota from normal weight and obese individuals. J. Med. Food 2013, 16, 862–867. [Google Scholar] [CrossRef]
- Baxter, N.T.; Schmidt, A.W.; Venkataraman, A.; Kim, K.S.; Waldron, C.; Schmidt, T.M. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 2019, 10, e02566-18. [Google Scholar] [CrossRef] [Green Version]
- Rutting, S.; Xenaki, D.; Lau, E.; Horvat, J.; Wood, L.G.; Hansbro, P.M.; Oliver, B.G. Dietary omega-6, but not omega-3, polyunsaturated or saturated fatty acids increase inflammation in primary lung mesenchymal cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L922–L935. [Google Scholar] [CrossRef]
- McKeever, T.M.; Lewis, S.A.; Cassano, P.A.; Ocké, M.; Burney, P.; Britton, J.; Smit, H.A. The relation between dietary intake of individual fatty acids, FEV1 and respiratory disease in Dutch adults. Thorax 2008, 63, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Atlantis, E.; Cochrane, B. The association of dietary intake and supplementation of specific polyunsaturated fatty acids with inflammation and functional capacity in chronic obstructive pulmonary disease: A systematic review. Int. J. Evid. Based Healthc. 2016, 14, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Fulton, A.S.; Hill, A.M.; Williams, M.T.; Howe, P.R.; Coates, A.M. Paucity of evidence for a relationship between long-chain omega-3 fatty acid intake and chronic obstructive pulmonary disease: A systematic review. Nutr. Rev. 2015, 73, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, K.; Lavrynenko, O.; Titz, B.; Pater, C.; Hoeng, J.; Ivanov, N.V. Lipid-based biomarkers for CVD, COPD, and aging–A translational perspective. Prog. Lipid Res. 2020, 78, 101030. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Meister, M.; Zhang, S.; Vong, C.-I.; Wang, S.; Fang, R.; Li, L.; Wang, P.G.; Massion, P.; Ji, X. Identification of lipid biomarker from serum in patients with chronic obstructive pulmonary disease. Respir. Res. 2020, 21, 242. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipids | Anti-Inflammatory Mechanisms | Changes in COPD | References |
|---|---|---|---|
| Long-chain polyunsaturated fatty acids | modulation of biophysical properties of plasma membranes (lipid ordering, fluidity, lipid rafts); regulation of membrane proteins function; gene expression (NF-kB, SREBP); substrate for synthesis of specialized pro-resolving mediators. | modification of the fatty acid composition of phospholipids of plasma membranes; lipid peroxidation; changes in the composition of free fatty acids; increased utilization of fatty acids. | [18,19,20,21,22,25,28,29,31,32,33,35,36,37,53,54,55,56,57] |
| Short-chain fatty acids | cell metabolism; cell differentiation; HDAC inhibition; modulation of mucosal inflammation; epithelial cell proliferation; junctional permeability. | changes in the composition of the intestinal microflora; eating disorders. | [204,209,210,211,212,213,214,221,222,223,224,225,229,230,231,232,233,236,237,238,239,240,241,244,245,246,247,248,249,254] |
| Specialized pro-resolving mediators | inhibition of neutrophil chemotaxis; inhibition of transendothelial and transepithelial migration of neutrophils; stimulation of phagocytosis and efferocytosis by macrophages; inhibition of cytokine production; influence on the proliferation of epithelial cells; participation in cholesterol homeostasis. | decreased production of pro-resolving mediators leads to: persistence of inflammation in the bronchi; development of emphysema; provides comorbid relationship with metabolic syndrome and atherosclerosis. | [9,10,60,67,68,69,70,71,72,73,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,98,99,100,101,103,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,129,130,131,132,133,134,135,136,137,138,141,142,143,150,151,153,154,155,156,157,158,159,162,163,164,165,167,168,170,171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S.; Kotlyarova, A. Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 12803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312803
Kotlyarov S, Kotlyarova A. Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2021; 22(23):12803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312803
Chicago/Turabian StyleKotlyarov, Stanislav, and Anna Kotlyarova. 2021. "Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 22, no. 23: 12803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312803