
An Easy Route to Aziridine Ketones and Carbinols


Abstract
:1. Introduction
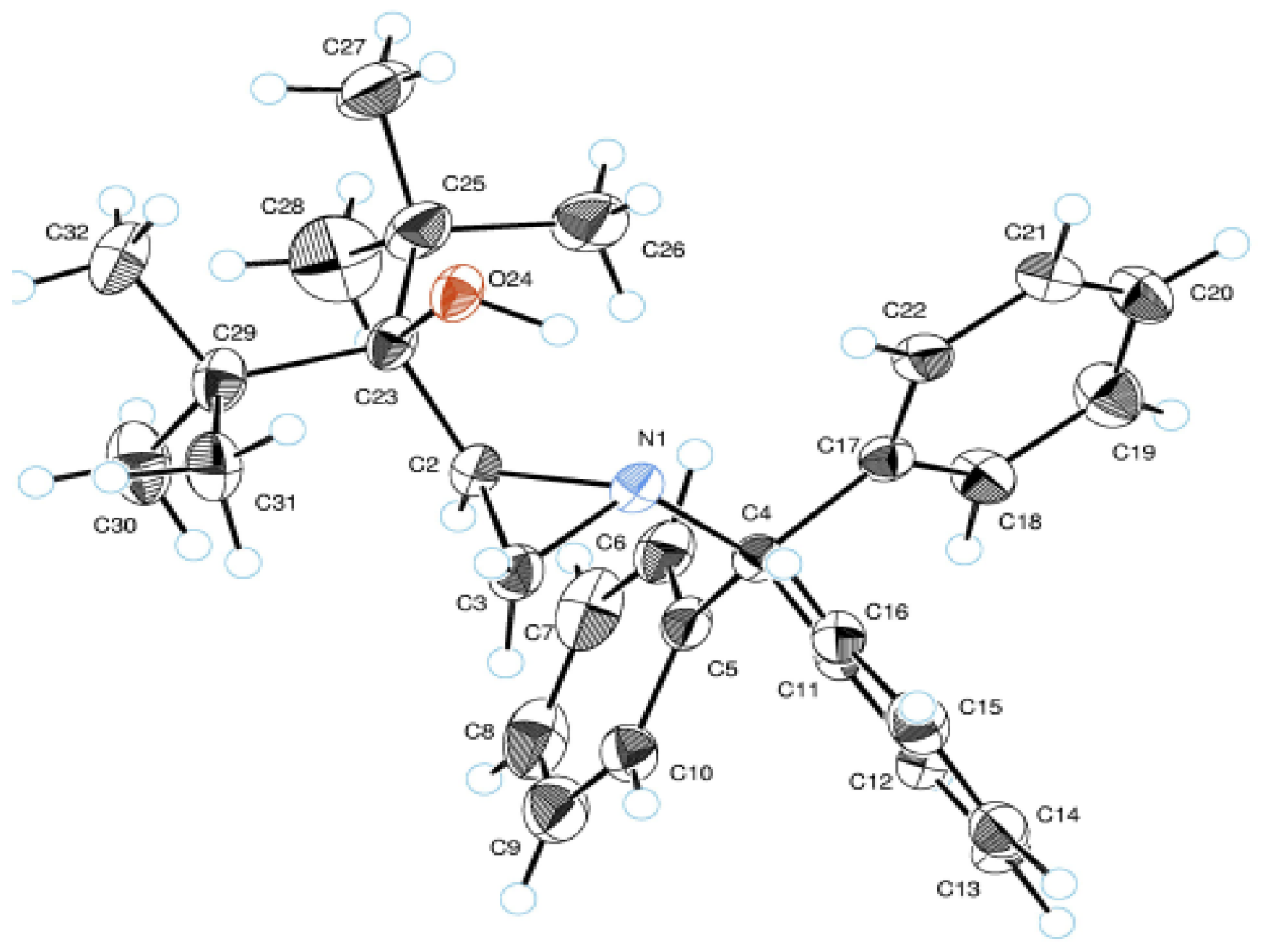
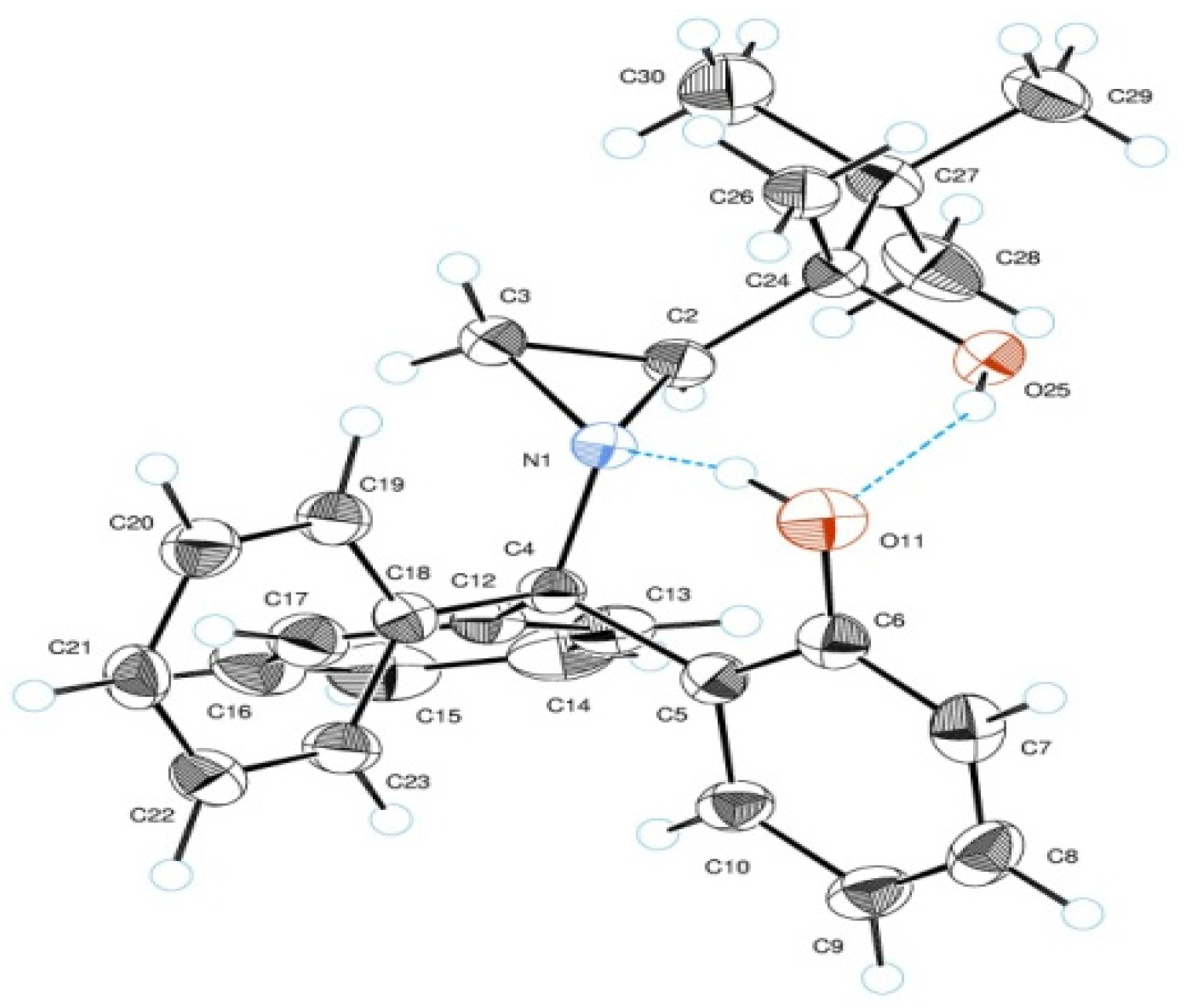
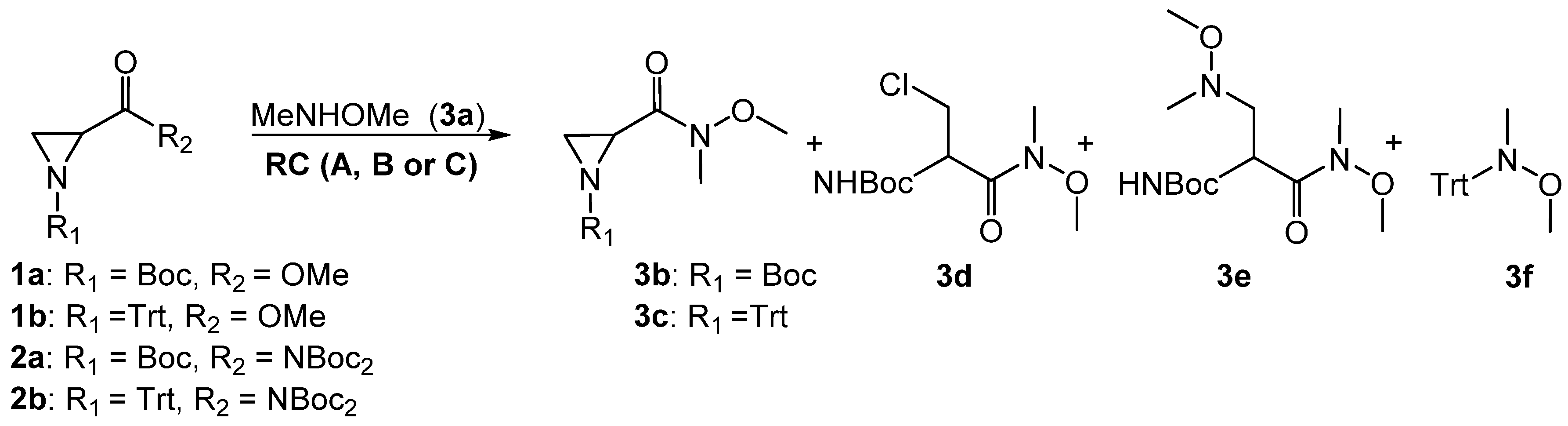
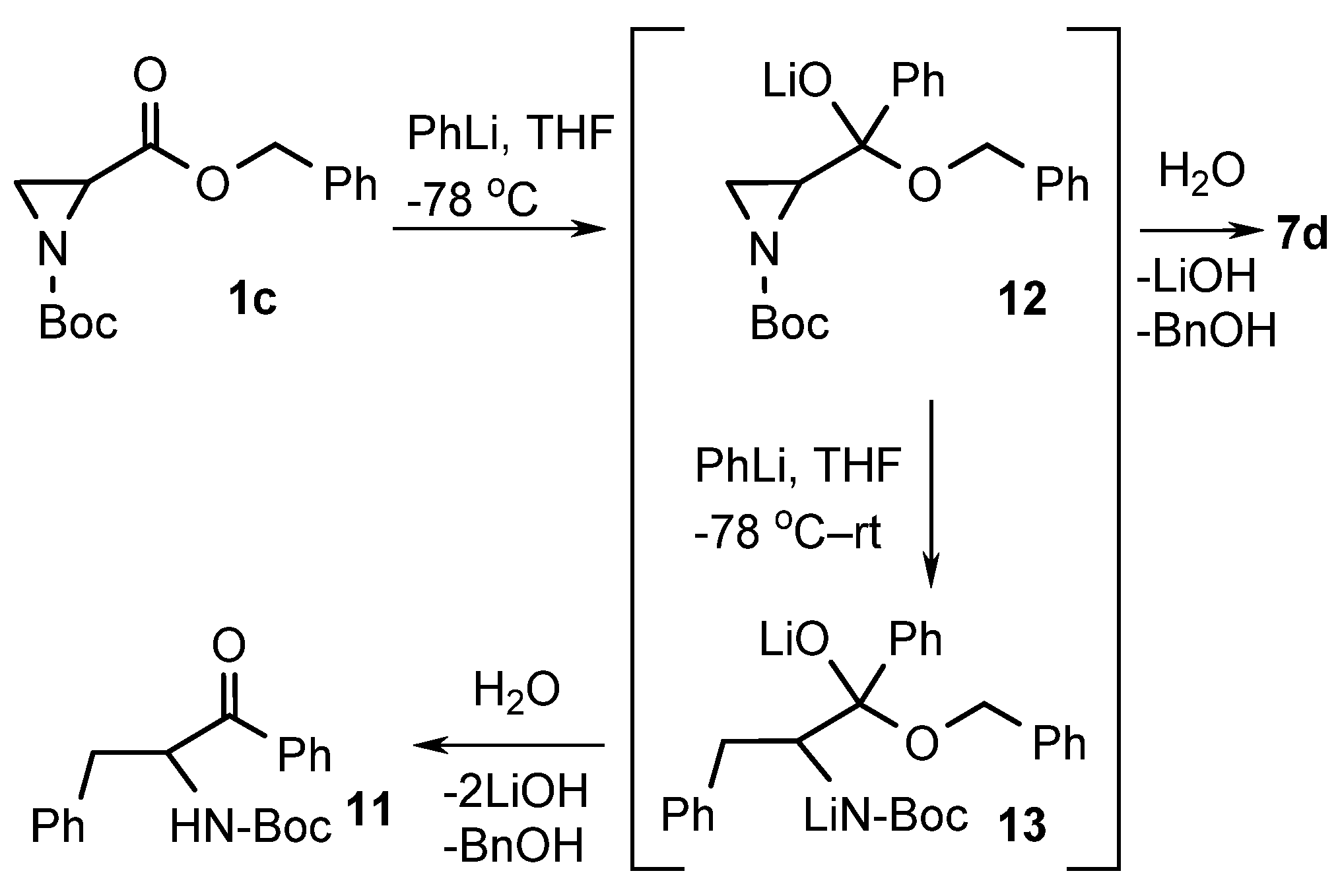
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, G.S. Advances in synthesis and chemistry of aziridines. Adv. Heterocycl. Chem. 2019, 129, 245–335. [Google Scholar] [CrossRef]
- D’Hooghe, M.; Ha, H.-J.; Macha, L. Deployment of Aziridines for the Synthesis of Alkaloids and Their Derivatives. Synthesis 2019, 51, 1491–1515. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Asymmetric Aziridination. Adv. Synth. Catal. 2014, 356, 1899–1935. [Google Scholar] [CrossRef]
- Strumfs, B.; Uljanovs, R.; Velikijs, K.; Trapencieris, P.; Strumfa, I. 3-Arylaziridine-2-carboxylic Acid Derivatives and (3-Arylaziridin-2-yl)ketones: The Aziridination Approaches. Int. J. Mol. Sci. 2021, 22, 9861. [Google Scholar] [CrossRef]
- Zhou, Z.; Kьrti, L. Electrophilic Amination: An Update. Synlett 2019, 30, 1525–1535. [Google Scholar] [CrossRef]
- Degennaro, L.; Trinchera, P.; Luisi, R. Recent Advances in the Stereoselective Synthesis of Aziridines. Chem. Rev. 2014, 114, 7881–7929. [Google Scholar] [CrossRef]
- Yudin, A.K. (Ed.) Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006; p. 492. [Google Scholar]
- Shtrumfs, B.; Hermane, J.; Kalvinsh, I.; Trapencieris, P. Unnatural amino acids. 3. Aziridinyl ketones from esters and amides of aziridine-2-carboxylic acids. Chem. Heterocycl. Compd. 2007, 43, 169–174. [Google Scholar] [CrossRef]
- Adler, M.; Adler, S.; Boche, G. Tetrahedral intermediates in reactions of carboxylic acid derivatives with nucleophiles. J. Phys. Org. Chem. 2005, 18, 193–209. [Google Scholar] [CrossRef]
- Deyrup, J.A.; Moyer, C.L. Aziridinemethanols. J. Org. Chem. 1970, 35, 3424–3428. [Google Scholar] [CrossRef]
- Yu, L.; Kokai, A.; Yudin, A.K. Preparation and Reactivity of Versatile α-Amino Ketones. J. Org. Chem. 2007, 72, 1737–1741. [Google Scholar] [CrossRef]
- Yun, J.M.; Sim, T.B.; Hahm, H.S.; Lee, W.K.; Ha, H.-J. Efficient Synthesis of Enantiomerically Pure 2-Acylaziridines: Facile Syntheses of N-Boc-safingol, N-Boc-d-erythro-sphinganine, and N-Boc-spisulosine from a Common Intermediate. J. Org. Chem. 2003, 68, 7675–7680. [Google Scholar] [CrossRef]
- Woydowski, K.; Liebscher, J. Synthesis of Optically Active 3-Amino-2,3-dihydrobenzopyran-4-ones by Ring Transformation of Aziridinecarboxamides. Synthesis 2000, 2000, 1444–1448. [Google Scholar] [CrossRef]
- Rubin, A.E.; Sharpless, K.B. A Highly Efficient Aminohydroxylation Process. Angew. Chem. Int. Ed. 1997, 36, 2637–2640. [Google Scholar] [CrossRef]
- Molander, G.A.; Stengel, P.J. Reduction of 2-acylaziridines by samarium(II) iodide. An efficient and regioselective route to β-amino carbonyl compounds. Tetrahedron 1997, 53, 8887–8912. [Google Scholar] [CrossRef]
- Sureshbabu, P.; Azeez, S.; Muniyappan, N.; Sabiah, S.; Kandasamy, J. Chemoselective Synthesis of Aryl Ketones from Amides and Grignard Reagents via C(O)–N Bond Cleavage under Catalyst-Free Conditions. J. Org. Chem. 2019, 84, 11823–11838. [Google Scholar] [CrossRef]
- Chan, G.H.; Ong, D.Y.; Yen, Z.; Chiba, S. Reduction of N,N-Dimethylcarboxamides to Aldehydes by Sodium Hydride-Iodide Composite. Helvetica Chim. Acta 2018, 101, e1800049. [Google Scholar] [CrossRef]
- Anthore-Dalion, L.; Benischke, A.D.; Wei, B.; Berionni, G.; Knochel, P. The Halogen–Samarium Exchange Reaction: Synthetic Applications and Kinetics. Angew. Chem. Int. Ed. 2019, 58, 4046–4050. [Google Scholar] [CrossRef]
- Nadano, R.; Fuchibe, K.; Ikeda, M.; Takahashi, H.; Ichikawa, J. Rapid and Slow Generation of 1-Trifluoromethylvinyllithium: Syntheses and Applications of CF3-Containing Allylic Alcohols, Allylic Amines, and Vinyl Ketones. Chem.–Asian J. 2010, 5, 1875–1883. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.-Q.; Zhang, L.; Liao, P.; Song, J.; Bi, X. Silver(I)-Catalyzed Hydroazidation of Ethynyl Carbinols: Synthesis of 2-Azidoallyl Alcohols. Angew. Chem. Int. Ed. 2014, 53, 5305–5309. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, H.; Wittenberg, R.; Egi, M.; Huang, W.; Liebeskind, L.S. Ambient Temperature Synthesis of High Enantiopurity N-Protected Peptidyl Ketones by Peptidyl Thiol Ester−Boronic Acid Cross-Coupling. J. Am. Chem. Soc. 2007, 129, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Strumfs, B.; Hermane, J.; Belyakov, S.; Trapencieris, P. Acyl migration from N to C in aziridine-2-carboxylic esters. Tetrahedron 2013, 70, 355–362. [Google Scholar] [CrossRef]
- Shtrumfs, B.; Chernyak, D.; Kums, I.; Kalvins, I.; Trapencieris, P. Unnatural Amino Acids. 2. Simple Method of Obtaining Esters of Aziridine-2-carboxylic Acids by a Transesterification Reaction. Chem. Heterocycl. Compd. 2004, 40, 725–733. [Google Scholar] [CrossRef]
- Ivanova, J.; Љtrumfs, B.; Ћalubovskis, R. Access to NH-aziridine-2-carboxamides through Davidsen acylimidodicarbonate activation. Comptes Rendus Chim. 2019, 22, 283–293. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
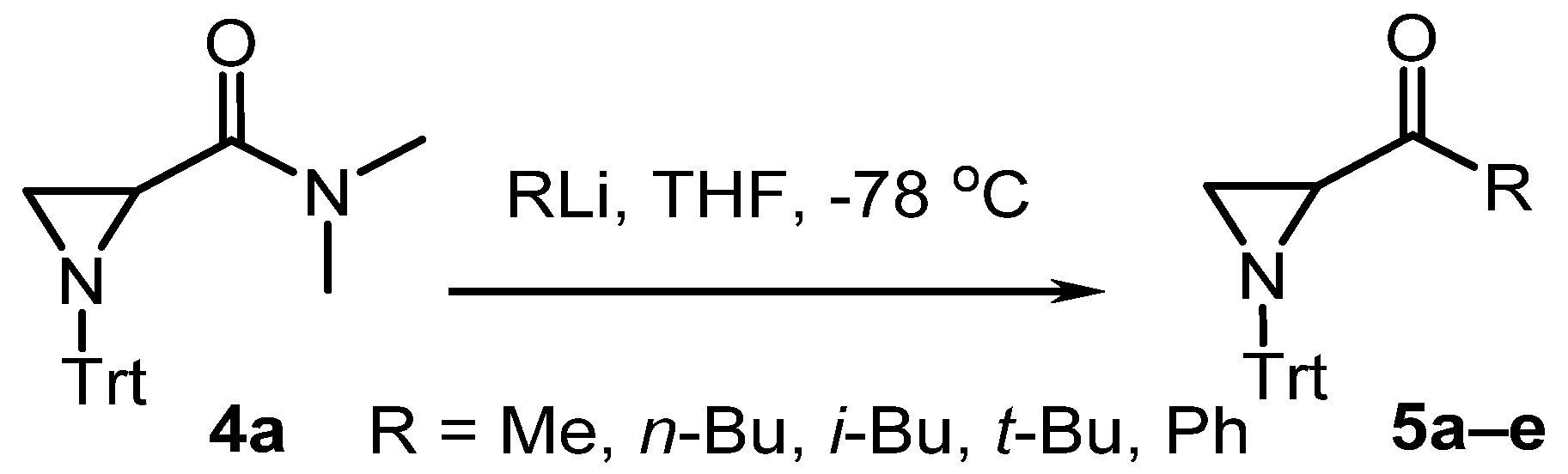{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | SM | RC | Eq. 3a | SM (%) | 3b,c (%) | 3d (%) | 3e (%) | 3f (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | A | 3 | - | - | 55 | 36 | - |
| 2 | 1a | A | 1 | 35 | - | 25 | 20 | - |
| 3 | 1a | B | 3 | 52 | Traces | - | - | - |
| 4 | 1b | A | 3 | - | - | - | - | 62 |
| 5 | 1b | A | 1 | 39 | - | - | - | 42 |
| 6 | 2a | C | 2 | 56 | 31 | - | - | - |
| 7 | 2a | C | 2 | 80 | Traces | - | - | - |
| # | Eq. RLi | Time (h) | R | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1 | 2 | Me | 5a | 77 |
| 2 | 2 | 2 | Me | 5a | 93 |
| 3 | 2 | 24 | Me | 5a | 70 |
| 4 | 1 | 2 | n-Bu | 5b | 39 |
| 5 | 2 | 2 | n-Bu | 5b | 44 |
| 6 | 2 | 24 | n-Bu | 5b | 38 |
| 7 | 1 | 2 | i-Bu | 5c | 41 |
| 8 | 1 | 24 | i-Bu | 5c | 36 |
| 9 | 2 | 2 | i-Bu | 5c | 43 |
| 10 | 2 | 24 | i-Bu | 5c | 47 |
| 11 | 1 | 2 | t-Bu | 5d | 30 |
| 12 | 2 | 2 | t-Bu | 5d | 32 |
| 13 | 2 | 24 | t-Bu | 5d | 29 |
| 14 | 1 | 2 | Ph | 5e | 51 |
| 15 | 1 | 12 | Ph | 5e | 79 |
| 16 | 2 | 2 | Ph | 5e | 50 |
| 17 | 2 | 12 | Ph | 5e | 81 |
| 18 | 4 | 24 | Ph | 5e | 78 |
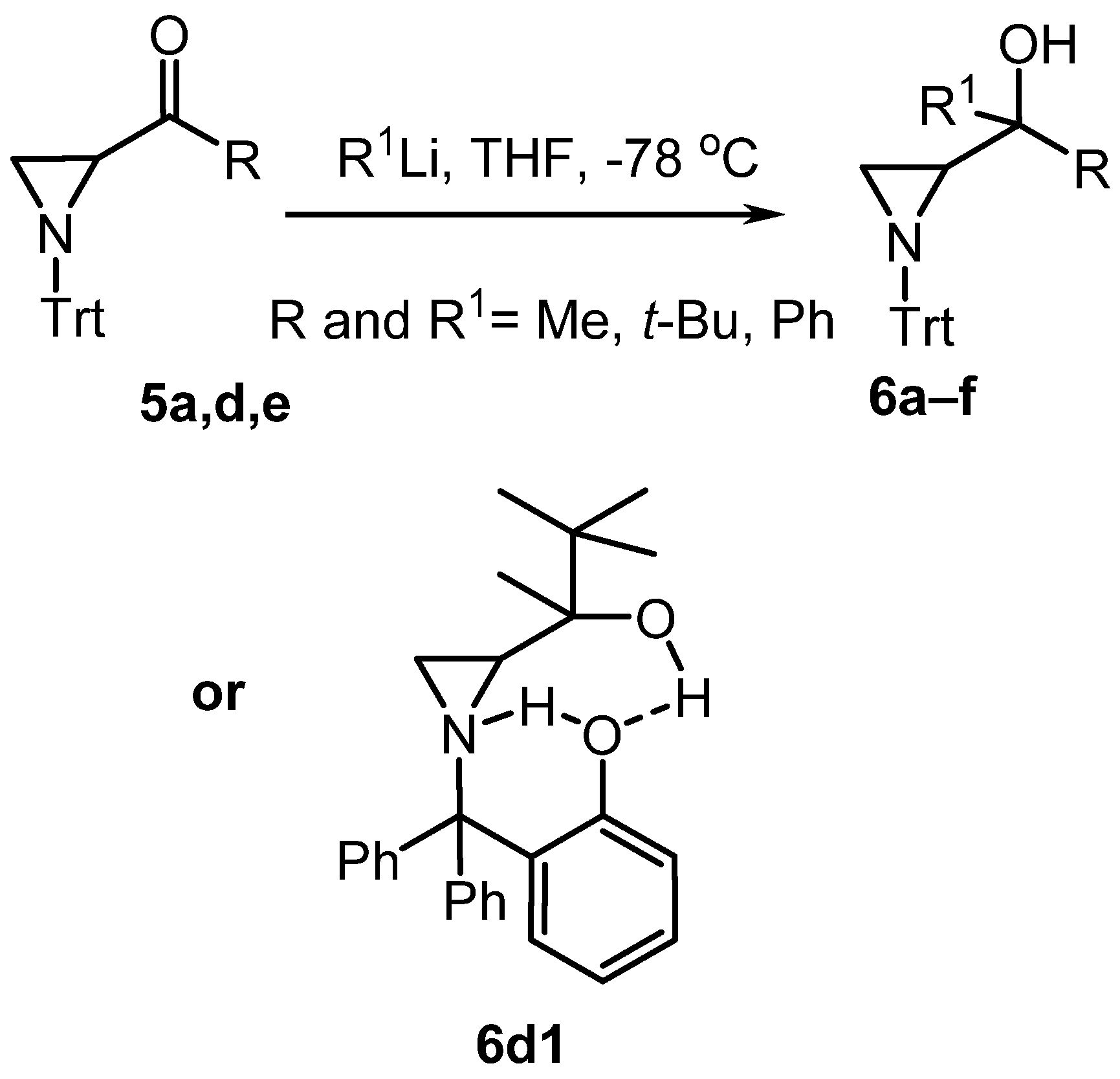
| # | Eq. R1Li | Time (h) | R | R1 | Product | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | 1 | Me | Me | 6a | 33 |
| 2 | 1 | 12 | Me | Me | 6a | 35 |
| 3 | 4 | 1 | Me | Me | 6a | 33 |
| 4 | 4 | 12 | Me | Me | 6a | 32 |
| 5 | 1 | 1 | Ph | Ph | 6b | 82 |
| 6 | 1 | 12 | Ph | Ph | 6b | 85 |
| 7 | 4 | 1 | Ph | Ph | 6b | 79 |
| 8 | 4 | 12 | Ph | Ph | 6b | 99 |
| 9 | 4 | 24 | Ph | Ph | 6b | 98 |
| 10 | 1 | 1 | t-Bu | t-Bu | 6c | 30 |
| 11 | 1 | 12 | t-Bu | t-Bu | 6c | 68 |
| 12 | 4 | 12 | t-Bu | t-Bu | 6c | 70 |
| 13 | 4 | 12 | Me | t-Bu | 6d * | 25 |
| 14 | 4 | 12 | t-Bu | Me | 6d1 | 52 |
| 15 | 4 | 12 | t-Bu | Ph | 6e | 100 |
| 16 | 1 | 12 | Ph | Me | 6f | 88 |
| 17 | 4 | 12 | Ph | Me | 6f | 61 |
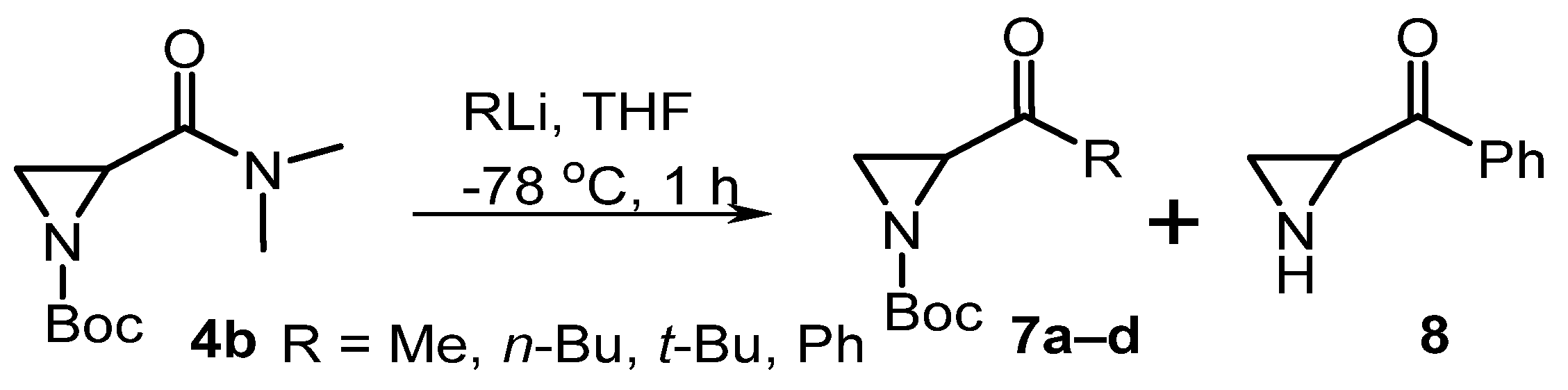
| # | Eq. RLi | R | Product 7 (%) | Product 8 (%) |
|---|---|---|---|---|
| 1 | 1 | Me | 7a (62) | - |
| 2 | 2 | Me | 7a (8) | - |
| 3 | 1 | n-Bu | 7b (75) | - |
| 4 | 1 | t-Bu | 7c (82) | - |
| 5 | 1 | Ph | 7d (60) | - |
| 6 | 4 | Ph | 7d (51) | 35 |
| 7 | 4 * | Ph | - | 66 |
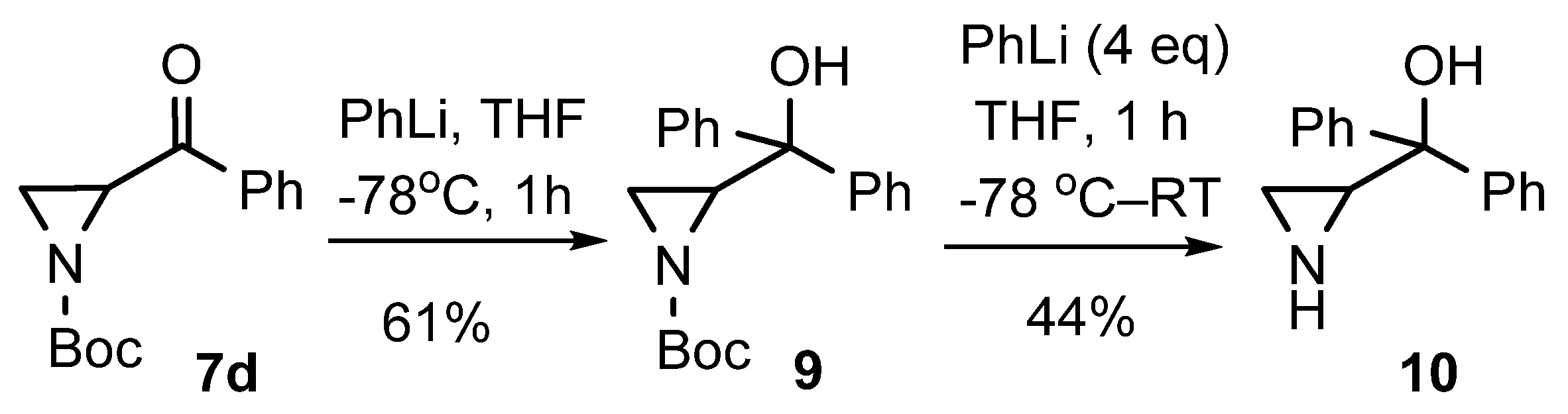
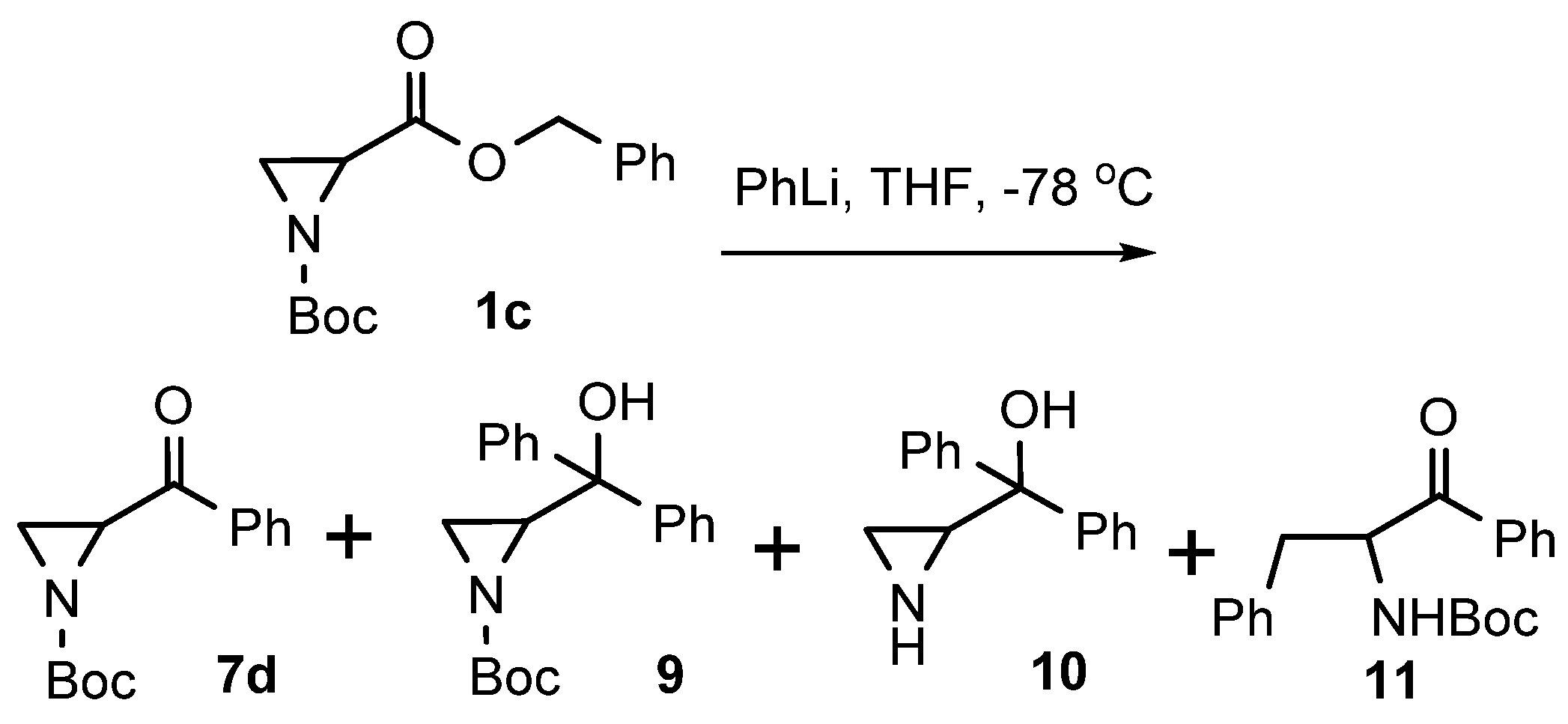
| # | Eq. PhLi | Time (h) | Product 7d (%) | Product 9 (%) | Product 10 (%) | Product 11 (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 26 | 5 | - | - |
| 2 | 2 | 1 | 10 | 40 | - | - |
| 3 | 4 | 1 | 8 | 52 | - | - |
| 4 | 4 | 12 | - | 5 | 22 | - |
| 5 | 4 | 12 * | - | 2 | 15 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strumfs, B.; Hermane, J.; Belyakov, S.; Sobolevs, A.; Velikijs, K.; Uljanovs, R.; Trapencieris, P.; Strumfa, I. An Easy Route to Aziridine Ketones and Carbinols. Int. J. Mol. Sci. 2021, 22, 13145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313145
Strumfs B, Hermane J, Belyakov S, Sobolevs A, Velikijs K, Uljanovs R, Trapencieris P, Strumfa I. An Easy Route to Aziridine Ketones and Carbinols. International Journal of Molecular Sciences. 2021; 22(23):13145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313145
Chicago/Turabian StyleStrumfs, Boriss, Jekaterina Hermane, Sergey Belyakov, Artjoms Sobolevs, Kirils Velikijs, Romans Uljanovs, Peteris Trapencieris, and Ilze Strumfa. 2021. "An Easy Route to Aziridine Ketones and Carbinols" International Journal of Molecular Sciences 22, no. 23: 13145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313145