
Interaction of KRSR Peptide with Titanium Dioxide Anatase (100) Surface: A Molecular Dynamics Simulation Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
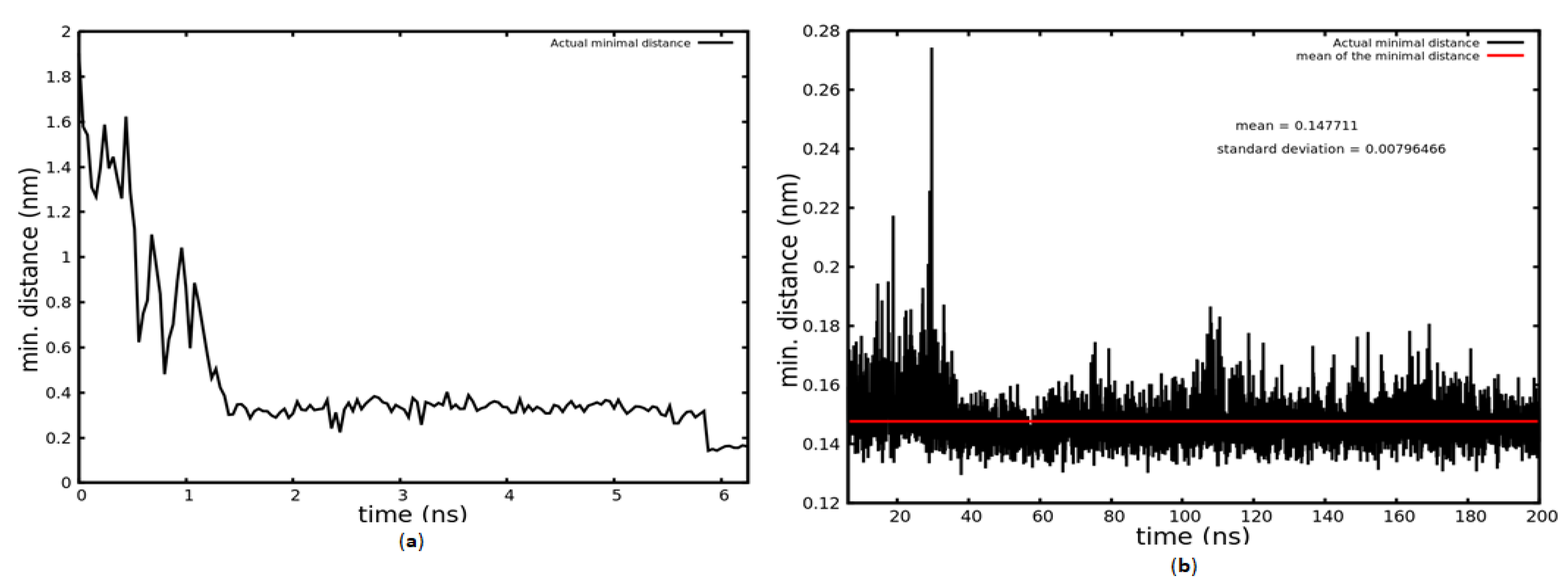
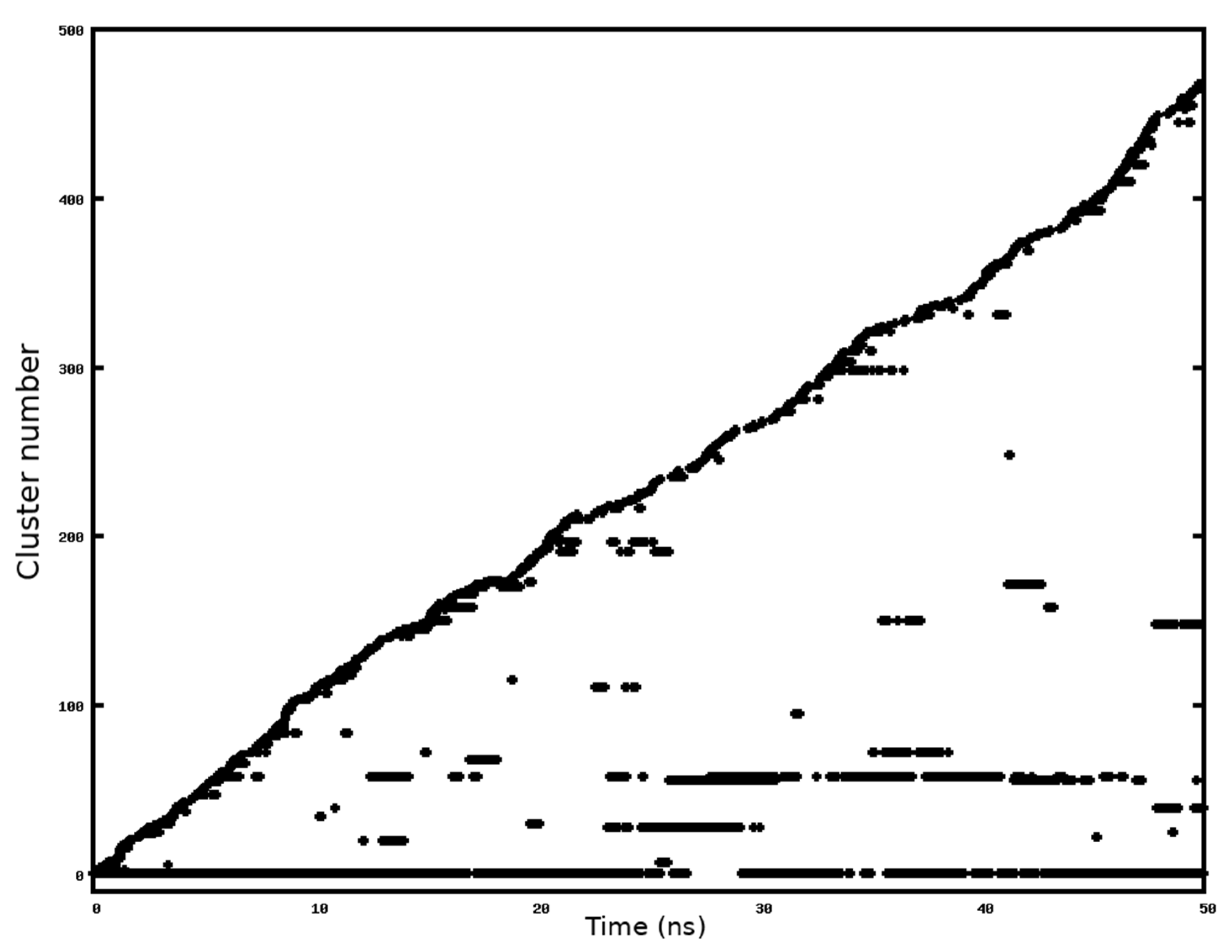
2.1. MD Simulation
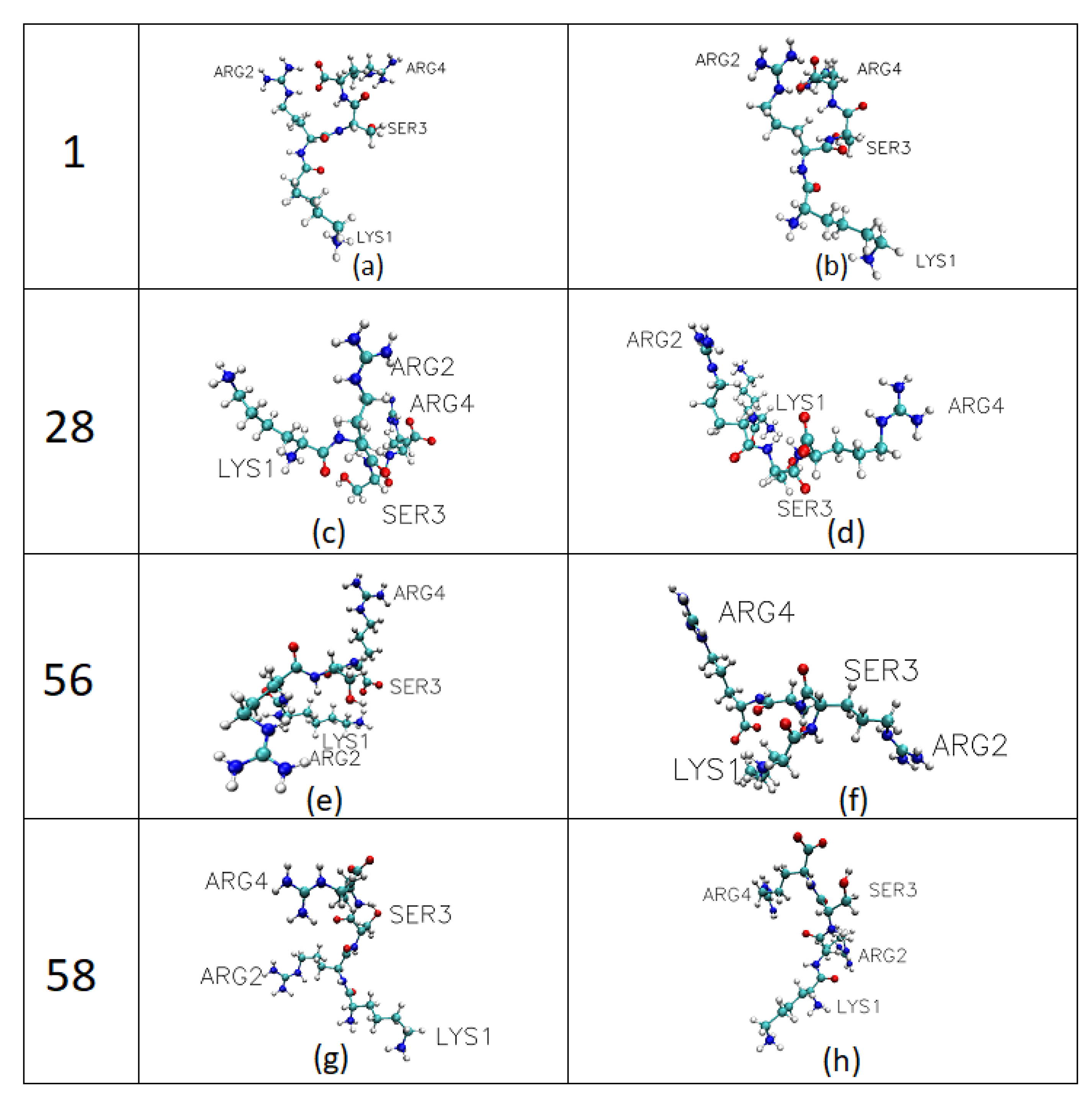
2.2. Determination of Adhered Peptide Conformations by Replica Exchange Molecular Dynamic Simulation (REMD)
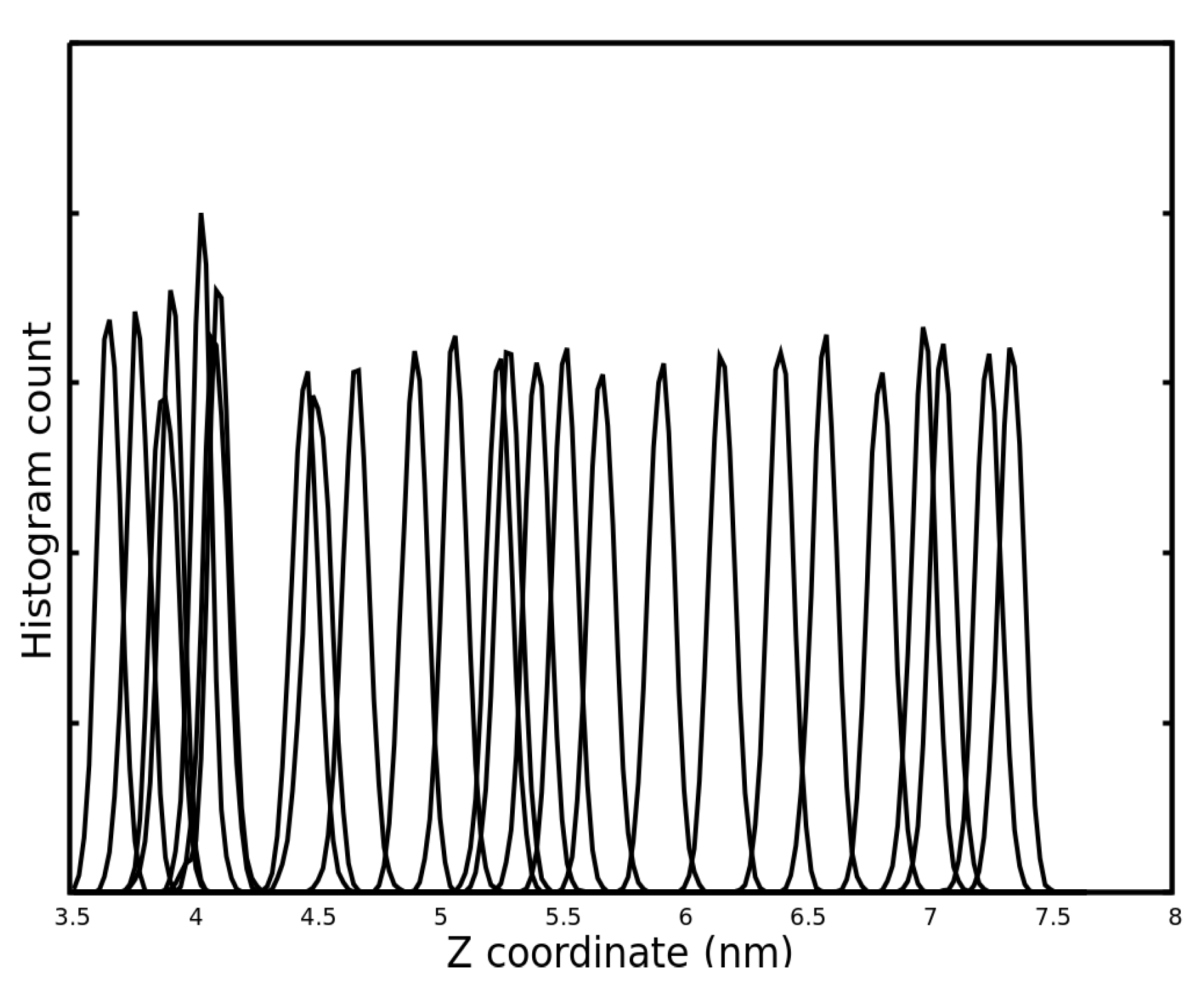
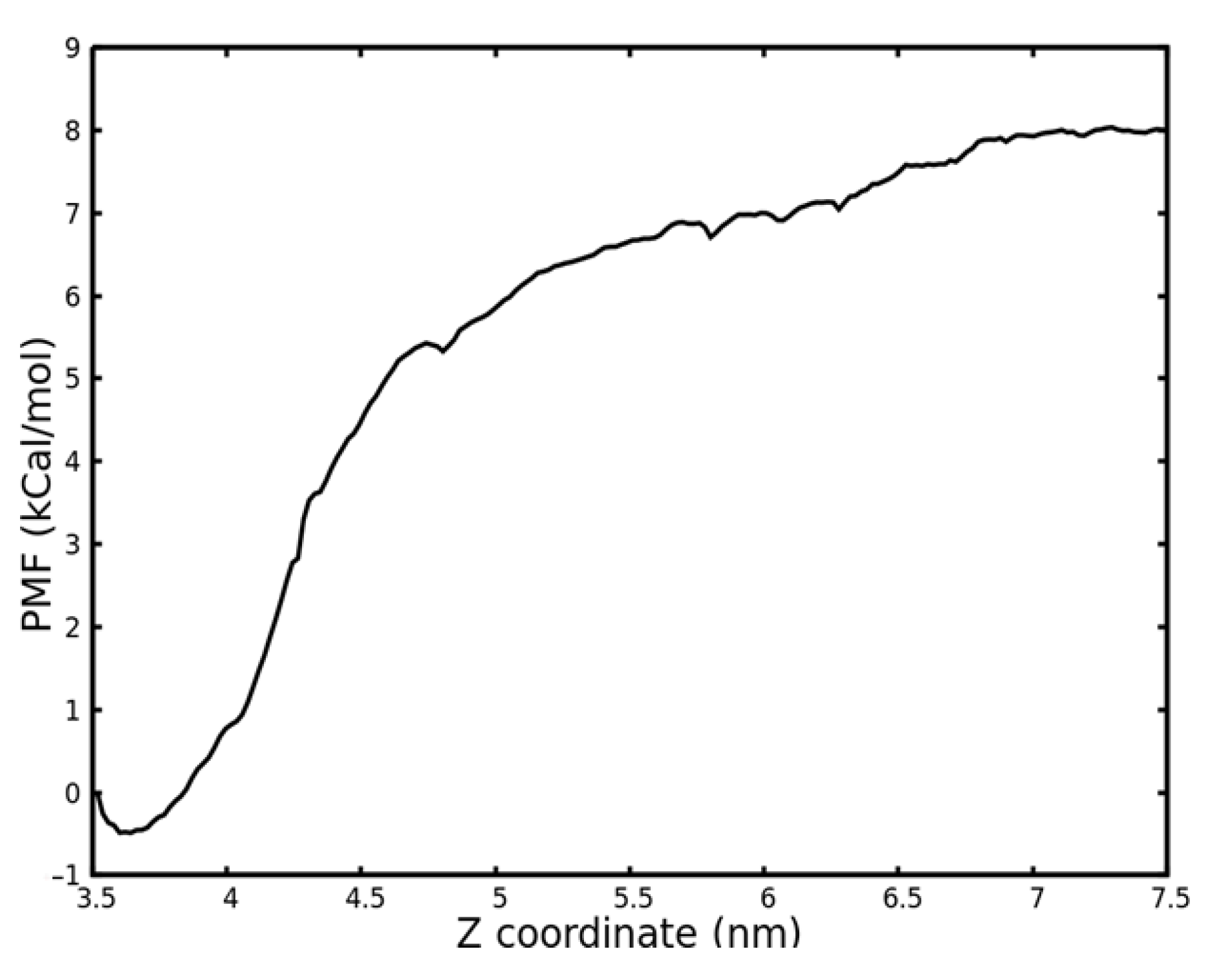
2.3. Binding Free Energy Calculation by Potential of Mean Force (PMF) Calculation
2.4. Discussion
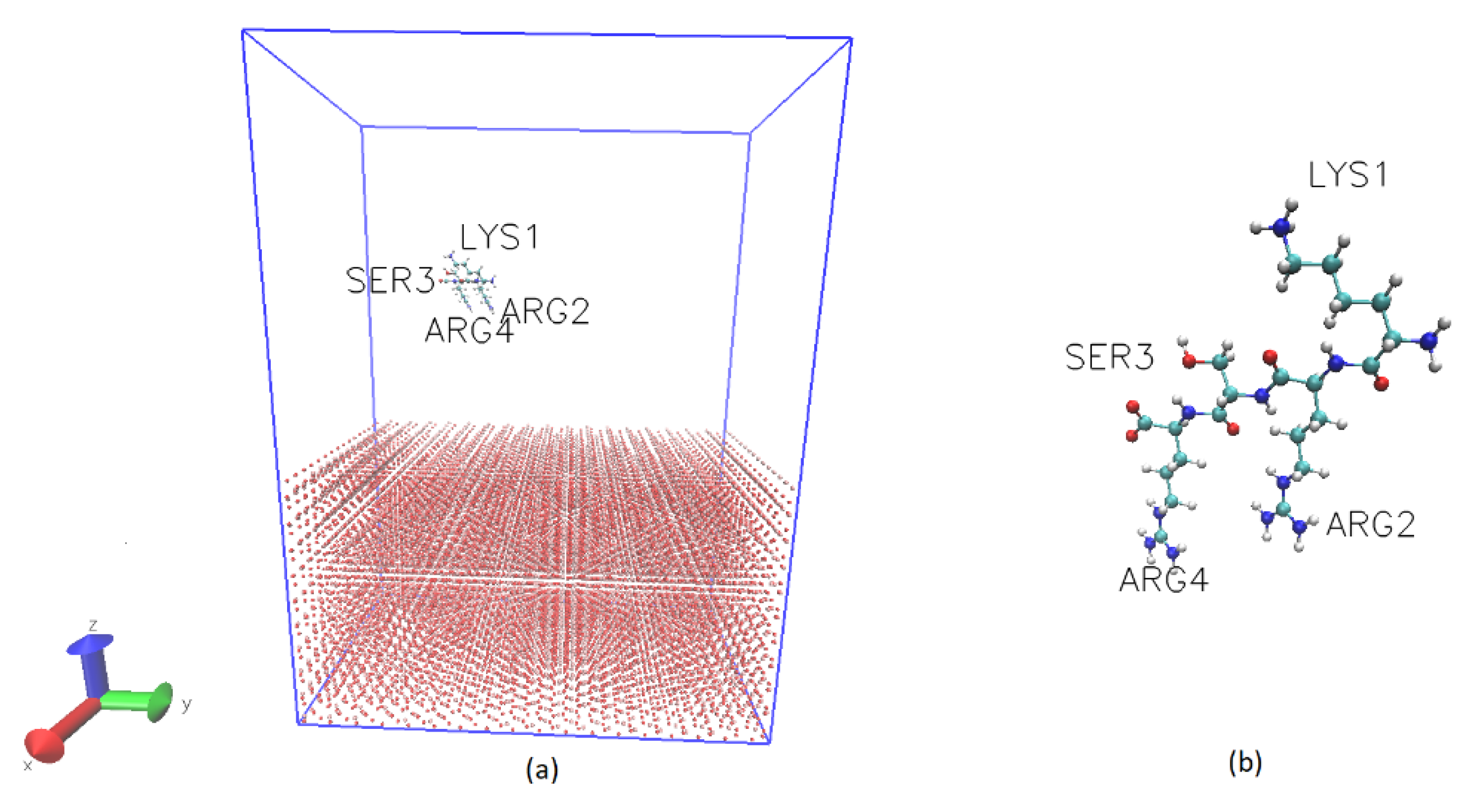
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yvoni, K.; Margarita, S.; Maria-Eleni, D.; Bennani, V.; Bakopoulou, A.; Tsouknidas, A.; Michailidis, M.; Michalakis, K. New Ti-Alloys and Surface Modifications to Improve the Mechanical Properties and the Biological Response to Orthopedic and Dental Implants: A Review. BioMed Res. Int. 2016, 2016, 2908570. [Google Scholar]
- Collet, T.; Atanasiu, J.P.; de Cussac, J.B.; Oufroukhi, K.; Bothorel, H.; Saffarini, M.; Badatcheff, F. Midterm outcomes of titanium modular femoral necks in total hip arthroplasty. Ann. Transl. Med. 2017, 5, 395. [Google Scholar] [CrossRef] [Green Version]
- Kruyt, I.J.; Nelissen, R.C.; Mylanus, E.A.M.; Hol, M.K.S. Three-year Outcomes of a Randomized Controlled Trial Comparing a 4.5-mm-Wide to a 3.75-mm-Wide Titanium Implant for Bone Conduction Hearing. Otol. Neurotol. 2018, 39, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.E.; Pjetursson, B.E.; Glauser, R.; Zembic, A.; Zwahlen, M.; Lang, N.P. A systematic review of the 5-year survival and complication rates of implant-supported single crowns. Clin. Oral Implant. Res. 2008, 19, 119–130. [Google Scholar] [CrossRef]
- Li, L.H.; Kong, Y.M.; Kim, H.W.; Kim, Y.W.; Kim, H.E.; Heo, S.J.; Koak, J.Y. Improved biological performance of Ti implants due to surface modification by micro-arc oxidation. Biomaterials 2004, 25, 2867–2875. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wang, Z.; Yin, H.; Wang, S.; Yan, P.; Huang, B.; Wang, X.; Li, R.; Zong, X.; Han, H.; et al. Understanding the anatase–rutile phase junction in charge separation and transfer in a TiO2 electrode for photoelectrochemical water splitting. Chem. Sci. 2016, 7, 6076–6082. [Google Scholar] [CrossRef] [Green Version]
- Ong, J.; Lucas, L.C.; Raikar, G.N.; Connatser, R.; Gregory, J.C. Spectroscopic characterization of passivated titanium in a physiologic solution. J. Mater. Sci. Mater. Electron. 1995, 6, 113–119. [Google Scholar] [CrossRef]
- Lavenus, S.; Louarn, G.; Layrolle, P. Nanotechnology and Dental Implants. Int. J. Biomater. 2010, 2010, 915327. [Google Scholar] [CrossRef]
- Miller, R.; Guo, Z.; Vogler, E.A.; Siedlecki, C.A. Plasma coagulation response to surfaces with nanoscale chemical heterogeneity. Biomaterials 2006, 27, 208–215. [Google Scholar] [CrossRef]
- Ferris, D.M.; Moodie, G.D.; Dimond, P.M.; Gioranni, C.W.; Ehrlich, M.G.; Valentini, R.F. RGD-coated titanium implants stimulate increased bone formation in vivo. Biomaterials 1999, 20, 2323–2331. [Google Scholar] [CrossRef]
- Chen, S.; Guo, Y.; Liu, R.; Wu, S.; Fang, J.; Huang, B.; Li, Z.; Chen, Z.; Chen, Z. Tuning surface properties of bone biomaterials to manipulate osteoblastic cell adhesion and the signaling pathways for the enhancement of early osseointegration. Colloids Surf. B Biointerfaces 2018, 1, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Rammelt, S.; Illert, T.; Bierbaum, S.; Scharnweber, D.; Zwipp, H.; Schneiders, W. Coating of titanium implants with collagen, RGD peptide and chondroitin sulfate. Biomaterials 2006, 27, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nat. Cell Biol. 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Control of cell motility and tumour invasion by extracellular matrix interactions. Br. J. Cancer 1992, 66, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Dee, K.C.; Andersen, T.T.; Bizios, R. Design and function of novel osteoblast-adhesive peptides for chemical modification of biomaterials. J. Biomed. Mater. Res. 1998, 5, 371–377. [Google Scholar] [CrossRef]
- Hasenbein, M.E.; Andersen, T.T.; Bizios, R. Micropatterned surfaces modified with select peptides promote exclusive interactions with osteoblasts. Biomaterials 2002, 23, 3937–3942. [Google Scholar] [CrossRef]
- Balasundaram, G.; Webster, T.J. Increased osteoblast adhesion on nanograined Ti modified with KRSR. J. Biomed. Mater. Res. A 2007, 1, 602–611. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Sul, Y.T.; Johansson, C.B.; Petronis, S.; Krozer, A.; Jeong, Y.; Wennerberg, A.; Albrektsson, T. Characteristics of the surface oxides on turned and electrochemically oxidized pure titanium implants up to dielectric breakdown: The oxide thickness, micropore configurations, surface roughness, crystal structure and chemical composition. Biomaterials 2002, 23, 491–501. [Google Scholar] [CrossRef]
- Zhang, H.P.; Lu, X.; Leng, Y.; Watari, F.; Weng, J.; Feng, B.; Qu, S. Effects of aqueous environment and surface defects on Arg-Gly-Asp peptide adsorption on titanium oxide surfaces investigated by molecular dynamics simulation. J. Biomed. Mater. Res. A 2011, 96, 466–476. [Google Scholar] [CrossRef]
- Rajh, T.; Dimitrijevic, N.M.; Bissonnette, M.; Koritarov, T.; Konda, V. Titanium dioxide in the service of the biomedical revolution. Chem. Rev. 2014, 114, 10177–10216. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, K.; Chen, X.; Liang, X.; Zheng, Y.; Li, L. Amino acid adsorption on anatase (101) surface at vacuum and aqueous solution: A density functional study. J. Mol. Model. 2018, 24, 107. [Google Scholar] [CrossRef]
- Cucchi, A.; Molè, F.; Rinaldi, L.; Marchetti, C.; Corinaldesi, G. The Efficacy of an Anatase-Coated Collar Surface in Inhibiting the Bacterial Colonization of Oral Implants: A Pilot Prospective Study in Humans. Int. J. Oral Maxillofac. Implants 2018, 33, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Chen, M.; Guo, C.; Zhao, X.; Yuan, C. Peptide-TiO2 interaction in aqueous solution: Conformational dynamics of RGD using different water models. J. Phys. Chem. B 2010, 114, 4692–4701. [Google Scholar] [CrossRef]
- Song, D.P.; Chen, M.J.; Liang, Y.C.; Bai, Q.S.; Chen, J.X.; Zheng, X.F.Y. Adsorption of tripeptide RGD on rutile TiO2 nanotopography surface in aqueous solution. Acta Biomater. 2010, 6, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, M.H.; Wang, K.F.; Liu, Y.; Zhang, H.P.; Lu, X.; Zhang, X.D. Computer simulation of biomolecule-biomaterial interactions at surfaces and interfaces. Biomed. Mater. 2015, 10, 032001. [Google Scholar] [CrossRef] [Green Version]
- Sowmiya, M.; Senthilkumar, K. Adsorption of proline, hydroxyproline and glycine on anatase (001) surface: A first-principle study. Theor. Chem. Acc. 2016, 135, 12. [Google Scholar] [CrossRef]
- Sowmiya, M.; Senthilkumar, K. Adsorption of RGD tripeptide on anatase (001) surface—A first principle study. Comput. Mater. Sci. 2015, 104, 124–129. [Google Scholar] [CrossRef]
- YazdanYar, A.; Aschauer, U.; Bowen, P. Interaction of biologically relevant ions and organic molecules with titanium oxide (rutile) surfaces: A review on molecular dynamics studies. Colloids Surf. B Biointerfaces 2018, 161, 563–577. [Google Scholar] [CrossRef]
- Luan, B.; Huynh, T.; Zhou, R. Simplified TiO2 force fields for studies of its interaction with biomolecules. J. Chem. Phys. 2015, 142, 234102. [Google Scholar] [CrossRef]
- Yang, W.; Xi, X.; Shen, X.; Liu, P.; Hu, Y.; Cai, K. Titania nanotubes dimensions-dependent protein adsorption and its effect on the growth of osteoblasts. J. Biomed. Mater. Res. Part A 2014, 102, 3598–3608. [Google Scholar] [CrossRef]
- Hoyos-Nogués, M.; Falgueras-Batlle, E.; Ginebra, M.P.; Manero, J.M.; Gil, J.; Mas-Moruno, C. A Dual Molecular Biointerface Combining RGD and KRSR Sequences Improves Osteoblastic Functions by Synergizing Integrin and Cell-Membrane Proteoglycan Binding. Int. J. Mol. Sci. 2019, 20, 1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, M.J.; Mijajlovic, M.; Biggs, M.J. Molecular-level understanding of protein adsorption at the interface between water and a strongly interacting uncharged solid surface. J. Am. Chem. Soc. 2014, 136, 5323–5331. [Google Scholar] [CrossRef]
- Polimeni, M.; Petridis, L.; Smith, J.C.; Arcangeli, C. Dynamics at a peptide–TiO2 anatase (101) interface. J. Phys. Chem. B 2017, 121, 8869–8877. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. Method J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Beutner, R.; Michael, J.; Schwenzer, B.; Scharnweber, D. Biological nano-functionalization of titanium-based biomaterial surfaces: A flexible toolbox. J. R. Soc. Interface 2010, 7 (Suppl. S1), S93–S105. [Google Scholar] [CrossRef] [Green Version]
- Valentin, A.H.; Weber, J. Receptor technology--cell binding to P-15: A new method of regenerating bone quickly and safely-preliminary histomorphometrical and mechanical results in sinus floor augmentations. Keio J. Med. 2004, 53, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, B.F.; Schuler, M.; Tosatti, S.; Textor, M.; Schwartz, Z.; Boyan, B.D. Osteoblast response to titanium surfaces functionalized with extracellular matrix peptide biomimetics. Clin. Oral Implant. Res. 2011, 22, 865–872. [Google Scholar] [CrossRef] [Green Version]
- García, A.; Reyes, C. Bio-adhesive Surfaces to Promote Osteoblast Differentiation and Bone Formation. J. Dent. Res. 2005, 84, 407–413. [Google Scholar] [CrossRef]
- Agosta, L.; Zollo, G.; Arcangeli, C.; Buonocore, F.; Gala, F.; Celino, M. Water driven adsorption of amino acids on the (101) anatase TiO2 surface: An ab initio study. Phys. Chem. Chem. Phys. 2015, 17, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Ciacchi, L.C. Specific Material Recognition by Small Peptides Mediated by the Interfacial Solvent Structure. J. Am. Chem. Soc. 2012, 134, 2407–2413. [Google Scholar] [CrossRef]
- Sumita, M.; Hu, C.; Tateyama, Y. Interface Water on TiO2 Anatase (101) and (001) Surfaces: First-Principles Study with TiO2 Slabs Dipped in Bulk Water. J. Phys. Chem. C 2010, 114, 18529–18537. [Google Scholar] [CrossRef]
- Sultan, A.M.; Hughes, Z.E.; Walsh, T.R. Binding Affinities of Amino Acid Analogues at the Charged Aqueous Titania Interface: Implications for Titania-Binding Peptides. Langmuir 2014, 30, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.; Lyubartsev, A. Molecular Dynamics Simulations of Adsorption of Amino Acid Side Chain Analogues and a Titanium Binding Peptide on the TiO2 (100) Surface. J. Phys. Chem. C 2015, 119, 18126–18139. [Google Scholar] [CrossRef]
- Arcangeli, C.; Borriello, I.; Gianese, G.; Celino, M.; Morales, P. Organic Functionalization of Metal Oxide Surfaces: An Atomic Scale Modeling Approach. Nanosci. Nanotechnol. Lett. 2013, 5, 1147–1154. [Google Scholar] [CrossRef]
- Lemkul, J.; Bevan, D. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Muir, J.; Costa, D.; Idriss, H. DFT computational study of the RGD peptide interaction with the rutile TiO2 (110) surface. Surf. Sci. 2014, 624, 8–14. [Google Scholar] [CrossRef]
- Schneider, J.; Ciacchi, L.C. A Classical Potential to Model the Adsorption of Biological Molecules on Oxidized Titanium Surfaces. J. Chem. Theory Comput. 2010, 7, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-N.; Lu, X.; Zhang, H.-P.; Weng, J.; Watari, F.; Leng, Y. DFT Study of the Adsorption of Aspartic Acid on Pure, N-Doped, and Ca-Doped Rutile (110) Surfaces. J. Phys. Chem. C 2011, 115, 18572–18581. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; van Drunen, R.; van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M.K.R. Gromacs: A Parallel Computer for Molecular Dynamics Simulations; Physics Computing 92; de Groot, R.A., Nadrchal, J., Eds.; World Scientific: Singapore, 1993. [Google Scholar]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simula-tion and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulati ons with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Springer International Publishing: London, UK; Cham, Switzerland, 2015; Volume 8759, pp. 3–27. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.L.D.R.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, G.N.M.; De Groot, B.L.; Grubmüller, S., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, C.J.; Sabine, T.M.; Dickson, F. Structural and thermal parameters for rutile and anatase. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Allouche, A.-R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymer 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; Awasthi, N. Probing a Continuous Polar Defect: A Reaction Coordinate for Pore Formation in Lipid Membranes. J. Chem. Theory Comput. 2017, 13, 2352–2366. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarjányi, T.; Bogár, F.; Minarovits, J.; Gajdács, M.; Tóth, Z. Interaction of KRSR Peptide with Titanium Dioxide Anatase (100) Surface: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2021, 22, 13251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413251
Tarjányi T, Bogár F, Minarovits J, Gajdács M, Tóth Z. Interaction of KRSR Peptide with Titanium Dioxide Anatase (100) Surface: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences. 2021; 22(24):13251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413251
Chicago/Turabian StyleTarjányi, Tamás, Ferenc Bogár, Janos Minarovits, Márió Gajdács, and Zsolt Tóth. 2021. "Interaction of KRSR Peptide with Titanium Dioxide Anatase (100) Surface: A Molecular Dynamics Simulation Study" International Journal of Molecular Sciences 22, no. 24: 13251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413251