Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges



1
Department of Neurology, University Medical Center Göttingen, 37075 Göttingen, Germany
2
German Center for Neurodegenerative Diseases (DZNE), 37075 Göttingen, Germany
3
Biomedical Engineering and Sciences Department, School of Mechanical and Manufacturing Engineering (SMME), National University of Sciences and Technology (NUST), Bolan Road, H-12, 44000 Islamabad, Pakistan
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(3), 1085; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031085
Submission received: 30 December 2020
/
Revised: 18 January 2021
/
Accepted: 19 January 2021
/
Published: 22 January 2021
(This article belongs to the Special Issue Amyloids, Prions and Related Phenomena)
Abstract
:Proteinopathy refers to a group of disorders defined by depositions of amyloids within living tissue. Neurodegenerative proteinopathies, including Alzheimer’s disease, Parkinson’s disease, Creutzfeldt–Jakob disease, and others, constitute a large fraction of these disorders. Amyloids are highly insoluble, ordered, stable, beta-sheet rich proteins. The emerging theory about the pathophysiology of neurodegenerative proteinopathies suggests that the primary amyloid-forming proteins, also known as the prion-like proteins, may exist as multiple proteoforms that contribute differentially towards the disease prognosis. It is therefore necessary to resolve these disorders on the level of proteoforms rather than the proteome. The transient and hydrophobic nature of amyloid-forming proteins and the minor post-translational alterations that lead to the formation of proteoforms require the use of highly sensitive and specialized techniques. Several conventional techniques, like gel electrophoresis and conventional mass spectrometry, have been modified to accommodate the proteoform theory and prion-like proteins. Several new ones, like imaging mass spectrometry, have also emerged. This review aims to discuss the proteoform theory of neurodegenerative disorders along with the utility of these proteomic techniques for the study of highly insoluble proteins and their associated proteoforms.
1. Introduction
Proteinopathies, also known as protein conformational diseases or amyloidosis, are a group of diseases associated with the deposition of misaggregated proteins in various organs [1]. These atypical protein conformations, known as amyloids, are β-sheet rich, insoluble, fibrillar assemblies that retain a uniform structure containing β-pleated sheets running perpendicular to the fiber axis [2]. With the aid of amyloid assemblies of thirty proteins, this phenomenon is reported to be the cause of almost fifty disorders [3]. Although the pathophysiology and symptoms of proteinopathies vary with the nature of misaggregated proteins and affected populations of cells, the primary cascade leading to the formation and deposition of misaggregated proteins is highly similar. Their propagation closely resembles the replication of scrapie isoform of cellular prion protein (PrPSc), the first infectious protein to be discovered in conjunction with human diseases, leading to the use of the terms ‘prion proteins’ and ‘prion-like proteins’ to describe amyloid-forming proteins [4].
The conversion of physiological proteins to pathological amyloid fibrils is an intriguing, albeit partially understood, process initiated by molecular stressors that collapse the native structure by breaking the backbone of the helix and prompting interactions between side chains of resident amino acids [5]. The peptide then refolds into a compact β-sheet rich secondary structure that is stabilized by the presence of electrostatic interactions. The conversion of native helical structure to a thermodynamically favorable β-sheet rich conformation is also known as ‘monomer activation’. These misfolded units can self-replicate by interacting with their physiological counterparts and altering their conformation via deformed templating [6]. The combination of these altered structures, or primary nucleation, leads to the formation of an aggregate that can seed the formation of amyloid fibrils [7]. These seeds undergo a repetitive cycle that involves the assembly of multiple toxic oligomeric species leading to the formation of various multimers, protofibrils (2.5 to 3 nm in diameter) and fibrils (a combination of two strands with a diameter of 6 to 10 nm; [8]). The primary event of nucleation and fibril formation is relatively slow and is referred to as the lag phase of growth. The intertwining of protofibrils and fibrils leads to the formation of mature fibrils that are 60–120 nm in diameter (Figure 1; [2]). X-ray diffraction and nuclear magnetic resonance analysis shows a spacing of approximately 10 Å between the layers of beta-sheets and approximately 4.7 Å between multiple β-strands, depicting a uniform and stable assembly [7]. The addition of monomers to fibrils changes their conformation so that it matches the residues present in the aggregates, leading to the growth of amyloid fibrils, a step referred to as ‘secondary nucleation’ [9]. At this point, the growth of amyloid fibrils reaches an exponential phase causing rapid accumulation of aggregates (Figure 1). Exosome-mediated transport can then spread the oligomeric species to other parts of the affected organ, spreading the pathology [10,11]. Depending on the post-translational processing and three-dimensional folding, each prion-like protein can generate diverse variants (proteoforms) that may contribute differentially towards disease prognosis.
Owing to the distinct, possibly optimal, redox and biochemical profile of the nervous tissue, a major fraction of proteinopathies in humans are associated with the central nervous system, making them a key problem for neuroscientists. In most cases, minuscule variations in protein sequences can lead to structural variations in their three-dimensional conformations, generate amyloids, and alter their seeding and infectious capabilities and the phenotype of associated disease [12,13]. The key to solving the riddle of these proteinopathies lies in a thorough investigation of their proteinaceous culprits. However, a lack of appropriate techniques to purify, identify, and characterize remains a major hurdle despite a collective effort of several research groups around the globe.
Strides in expression, structural and functional proteomics, to accommodate highly fibrillar and polymorphic prion-like proteins, are the need of the hour. The current review aims to highlight variants of amyloidogenic proteins involved in proteinopathies and describe the utility and limitations of proteomic tools in their study.
2. Neurodegenerative Proteinopathies
Some of the most common neurodegenerative disorders—Alzheimer’s disease (AD), Parkinson’s disease (PD), Creutzfeldt–Jacob disease (CJD), Dementia with Lewy bodies (DLB), Huntington disease (HD), and Amyloid Lateral Sclerosis (ALS) —are proteinopathies. Together, these diseases affect millions of lives around the world and have devastating economic implications. AD, the most frequently diagnosed among these listed diseases, affects almost one-tenth of the population above 65 years of age [14]. The number of people suffering from these diseases is increasing rapidly with an increase in life expectancy and it is predicted that by 2050, 135.46 million people will be living with various types of neurodegenerative dementias [15]. Despite the attention of the scientific community, these disorders are far from resolved. The patients can be treated to alleviate the symptoms, but the lack of a cure still means that such a diagnosis can seal their fate.
Although proteinopathies present similarities in their pathological mechanisms, the psychological and physiological symptoms of all these disorders vary and depend on the region of the brain affected. A summary of age at onset, primary sites of pathology, and common symptoms of major neurodegenerative proteinopathies is presented in Table 1. These variations, in turn, are dictated by the proteins that are involved in amyloid formation (Table 2).
In addition to similarities in the mechanism of propagation, prion-like proteins have also adapted another interesting aspect of PrPSc biology. PrPSc can give rise to several clinical variants of prion diseases. This heterogeneity has been attributed to the existence of distinct PrP strains. Strains are defined as conformers of a specific amyloidogenic protein, in this case PrPSc, that differ with respect to their transmission, brain-lesion profiles, incubation periods, and disease phenotypes along with certain biochemical characteristics like Post-translational modifications, sensitivity to proteinase-K, and electrophoretic mobility. The distinct conformational characteristics of each PrP strain are transmitted into the host where it propagates and causes distinct phenotypes [40]. Codon 129 polymorphism gives rise to at least three known strains of PrP in humans [41].
Strain theory is now applicable to most prion-like proteins (Figure 2) [42,43]. α-Synuclein, for example, has been known to be the culprit behind characteristically distinct pathologies, i.e., PD, DLB, and multiple system atrophy, while microtubule-associated protein tau is involved in multiple different tauopathies either as the primary cause or as a co-pathology [44,45]. In the case of Aβ, it has been known for several years that different proteoforms vary in their capability to form amyloids, seeding proficiencies, three-dimensional conformations, transport mechanisms and toxicities [46,47]. Each proteoform can adopt and propagate in multiple conformations [48]. These conformers do not only possess distinct biochemical signatures but also have different stabilities, distribution and morphology in the brain [49]. Moreover, accumulating evidence shows that many neurodegenerative proteinopathies can exist as rapidly progressive and other clinically distinct variants even though the underlying prion-like proteins and mechanisms are the same [50,51]. The capability of one protein to give rise to clinically distinct disorders and alter the progression of a disease has further complicated the characterization of neurodegenerative proteinopathies.
The study of prion-like proteins now encompasses the study of all variants/proteoforms rather than focusing on one parent entity. The existence of proteins as different functional variants is a known fact. These functional variants dictate the localization, uptake, recycling, and biological functions of a protein. In the case of prion-like proteins, the presence of distinct variants involved in neurodegenerative proteinopathies has been verified by several groups over the past two decades [26,28,52]. Although several different terms have been previously used in the literature to classify these variations, any prion-like protein can have:
- Genetic variants (based on mutations).
- Isoforms (based on differences in post-transcriptional modifications).
- Proteoforms (based on differences in post-translation processing and three-dimensional conformation).
- Strains (based on differences in infectivity and incubation periods).
With the acceptance of the notion that different isoforms, proteoforms, or strains of prion-like proteins may differ with respect to their molecular insult mechanisms and dictate the prognosis of associated pathology, the availability of high-resolution data about the sequence and structure has become the key in characterizing, diagnosing, and treating neurodegenerative proteinopathies [53,54,55,56,57,58]. It is therefore mandatory to establish tools that can provide insight into minor changes within the sequence, post-translational processing, and structure of a protein in its undigested form or native conformations.
3. Utilizing Proteomic Platforms to Understand Neurodegenerative Proteinopathies
The identification and characterization of proteoforms and strains require high-resolution proteomic techniques that can analyze proteins in their intact native state. As the focus of the scientific community is shifting now from proteome to proteoforms, several such techniques are being refined and developed. Two-dimensional gel electrophoresis (2D-GE) is becoming popular again as an important method to visualize the presence of proteoforms of the target protein [59]. Based on the preliminary evidence from gel-based techniques, the proteoforms can then be sequenced directly in their native forms using top-down mass spectrometry [60,61]. The recently developed imaging mass spectrometry (IMS) can localize proteoforms in the tissues directly, diminishing the need for immunofluorescence microscopy and proteoform-specific sensitive antibodies [62,63]. In addition, conformational variation between related proteoforms can be understood by utilizing hydrogen/deuterium exchange mass spectrometry [64].
Theoretically, these techniques should identify and characterize any protein and its associated proteoforms isolated from a range of biological specimens. However, the attributes of prion-like proteins make them especially challenging. Biochemically, these proteins are highly insoluble, fibrillar, heterogenous, and transient in nature. Therefore, minor changes in detergents, chaotropes, solvents, pH, plasticware, and storage conditions can result in the formation of multimers and fibrils and/or loss of proteins. The results require careful analysis to rule out any technical artifacts. In some cases, like for AD-associated Aβ peptides, their quantity is very low, therefore requiring a large amount of brain samples or enrichment techniques. However, careful planning and execution of experiments can provide useful insights into the molecular mechanisms involved in neurodegenerative proteinopathies.
In the following sections the application, and challenges, of proteomic techniques to expand our knowledge from the proteome of neurodegenerative proteinopathies to the proteoforms of associated prion-like proteins will be discussed.
3.1. Two-Dimensional Gel Electrophoresis (2D-GE)
Gel electrophoresis, coupled with immunoblotting and mass spectrometry, is an integral tool for proteomic labs and provides valuable insights into the physiological and pathological functions of proteins [65]. Even the most straight-forward gel electrophoresis experiments allow the visualization of the proteome and quantification of various proteins in comparison to others. The basic principle of this technique relies on the efficient separation of proteins based on their biochemical characteristics. One-dimensional gel electrophoresis performs the separation of proteins based on their respective molecular weight [66]. On the other hand, its two-dimensional counterpart utilizes the differences in isoelectric points in addition to molecular weights to separate proteins enabling the visualization of thousands of proteins [67]. The latter tool is ideal for separating the intact proteoforms of any protein even if they have minor differences in biochemical properties in response to differential cleavage and post-translational modifications [68]. This attribute of 2D-GE has increased its popularity over the last few years and its applicability towards the study of proteoforms of prion-like proteins is becoming increasingly evident [69].
The analysis of proteins through gel-based techniques requires the proteins to be in a soluble, denatured, and monomeric state as well as the retention of epitopes essential for the immunoblotting or pull-down assays. In addition, for 2D-GE, the proteins should retain their native charges. Bringing fibrillar assemblies of prion-like proteins into this state requires an understanding about the type of aggregates, their constituent proteins and their amount in the tissue [70]. Prion-like proteins are not soluble in most commonly used detergents and chaotropes. In fact, some of these agents, including Triton X-100, Tween-20, CHAPS, and sodium dodecyl sulphate (low concentrations), enhance the insolubility in certain cases and increase the formation of multimers in the process. Furthermore, the solubility of different conformations, i.e., monomers, multimers, and plaques, of the same target protein is different, thus requiring a sequential fractionation-based approach to isolate all aggregates from a sample [71]. Therefore, the cocktail of detergents, chaotropic agents, and reducing agents, and the pH of the system must be tailored according to the target protein and its conformation. If improperly solubilized, they fail to enter the gel, compromising the analysis [72]. Proteins become highly insoluble as they reach their isoelectric points (pI) and therefore their ability to form aggregates increases [73]. The selected rehydration buffer for 2D-GE must also ensure that the proteins stay soluble throughout the running period.
Mild agents, like phosphate buffered saline (PBS), have been used for the isolation of smaller assemblies, i.e., monomers and oligomers [71]. Larger fibrillar assemblies need stronger ionic detergents, mostly sodium dodecyl sulphate (SDS) or sodium N-lauroylsarcosinate (Sarkosyl), for effective solubilization [72]. However, the use of ionic detergents disturbs the native charges that are required for 2D-GE and interferes with mass spectrometric analysis [74,75]. High concentrations of chaotropic agents, including urea and guanidine hydrochloride, are also very effective for the solubilization of fibrillar assemblies [76,77,78]. Amongst the chaotropes, uncharged urea is most commonly used for the extraction of proteins for subsequent 2D-GE. Its combination with low concentrations of CHAPS, a zwitterionic detergent, preserves the native charges of proteins and prevents the formation of aggregates [79]. In addition, formic acid, trifluoracetic acid and other organic acids can also be employed for the solubilization of fibrillar assemblies [70]. These solvents can be exchanged or neutralized prior to analysis.
Once the proteins are solubilized with their native charges intact, they can be subjected to isoelectric focusing using a range of commercially available IPG strips. The separation on the second dimension using polyacrylamide gels requires additional considerations. The presence of SDS in polyacrylamide gel electrophoresis has drastic effects on the mobility of some prion-like proteins. In the case of Aβ, this phenomenon has been reported previously [80]. Supplementation of polyacrylamide gels with urea nullifies the effects of SDS and improves the resolution of proteoforms even if the differences among them are based on single amino acid only [79,81]. The gels can then be subjected to immunoblotting. Using 2D-GE, in comparison to 1D-GE, would not require specific antibodies against all targeted proteoforms since they are already resolved into specific spots instead of a single band. One antibody against the parent proteoform would detect all its associated proteoforms. The virtual 2D-GE based immunoblots for Aβ are shown in Figure 3.
The optimization of 2D-GE for prion-like proteins has offered a straightforward and economical preliminary approach to detect the presence of proteoforms. It can be employed to obtain information about glycosylation, sialylation, presence/absence of GPI-anchor, and differences in post-PK cleavage pattern for PrPSc from brains and cerebrospinal fluid of patients [82,83,84]. Similarly, in addition to splice-variants, phosphorylation, acetylation, O-GlcNAcylation of tau proteins have been studied using 2D-GE [85,86,87]. Combined with mass spectrometry, 2D-GE is a powerful tool for the identification and the analysis of known and novel prion-like proteins.
3.2. ESI and MALDI Based Top-Down Mass Spectrometry
Top-down proteomics is a relatively new addition to the field of mass spectrometry. Although still in the development stages, this technique is ideal for the study of proteoforms. Unlike bottom-up proteomics, it does not involve the digestion of proteins to peptides before the analysis and thus no information about the sequence and its post-translational modifications is lost. The proteins can be confidently annotated as the complete sequence is available. The analysis, however, is much more complicated than bottom-up proteomics as the digested peptides are not only easier to solubilize and fragment, they also have ample bioinformatic support. In addition, the number of total proteoforms is far greater than the total number of functional gene products initially expected to exist. Hence top-down proteomics requires more sensitive and high-resolution mass spectrometric techniques [88]. This technique has been used for the study of prokaryotic proteoforms for over two decades now and is rapidly advancing to accommodate the complicated world of eukaryotic proteoforms [89,90].
Prior to analysis using this technique, proteins need to be extracted and purified in solvents compatible with mass spectrometry (extraction and purification discussed in the previous section). The introduction of complex mixtures into columns and analyzers is performed by combining two fractionation techniques or two-dimensional fractionation. The differential size, isoelectric pH, molecular weight, or solubility of proteins can be targeted. Reverse-phase liquid chromatography can be coupled with size exclusion chromatography, hydrophobic interaction chromatography, capillary isoelectric focusing, or capillary zone gel electrophoresis to achieve this goal. The intact proteins are converted into intact ions using soft ionization methods like electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI). They are then fragmented, and the MS/MS data are utilized for identification. Mostly ESI coupled with Fourier-transform ion cyclotron resonance (FT-ICR) analyzer is utilized for the analysis of intact proteins because of their ability to generate multiple charged fragments and high-resolution capabilities, respectively. Known sequences from parent proteoforms can be used to comprehend the differences within associated proteoforms [91].
An additional advantage of top-down mass spectrometry is the quantification of native proteoforms. As the measurements involve complete sequence coverage of intact analytes, results from top-down mass spectrometry can be directly used to quantify proteoforms [92]. This approach, after refinement, can be potentially used as a specific and sensitive substitute to enzyme-linked immunosorbent assays. Moreover, the top-down approach in combination with hydrogen/deuterium exchange mass spectrometry, discussed in Section 3.4, can be employed to study conformational variations among associated proteoforms in their native form thereby providing valuable insights into functionally-relevant structural differences [93].
Top-down proteomics is becoming increasingly applicable for the study of eukaryotic proteome and proteoforms [92,94]. It is also being used for the study of native prion-like proteins. α-Synuclein has been subjected to this technique to identify its metal-binding domains that contribute towards the pathology of Parkinson’s disease [95]. Similarly, the native structure of tau and its interactions with aggregation inhibitors have also been targeted [96]. The heterogeneity of Aβ proteoforms in AD has also been established using a top-down approach [26]. The number of publications in this field is increasing every year and soon, it may become the primary tool for the study of proteinopathies.
3.3. MALDI Imaging
The spatial distribution of proteins in tissues, in addition to their identification and expression, is necessary for its complete characterization in physiological and pathological conditions. For several decades, immunostaining and immunofluorescence have fulfilled this aim [97]. However, they require specific and sensitive antibodies for each target. In the case of proteoforms, this condition cannot always be met as the proteoforms of the same proteins can vary because of slightly altered post-translational modifications and the antibodies might not be specific enough to detect these alterations. Secondly, antibody-based methods require prior knowledge about the target, and the discovery of novel proteins is not possible.
The alternative to these conventional techniques is MALDI imaging mass spectrometry (MALDI-IMS). It involves the MS-based detection of analytes on frozen or paraffin-embedded tissue samples directly. It was first reported in the 1990s and is becoming popular for its ability to profile proteins, lipids, metabolites, glycoproteins, and nucleotides in academic and clinical settings [62,98]. It can be used for the analysis of proteins through both bottom-up and top-down approaches. The protocols, equipment, and software for MALDI-IMS are advancing rapidly and have already been developed to accommodate the analysis of proteoforms and prion-like proteins [99,100,101].
The preparation of samples for MALDI-IMS is relatively straightforward. It involves sectioning of tissues and their mounting on indium-tin oxide coated slides. Ethanol fixation is employed to limit the delocalization of proteins. Impurities, like salts and lipids, are removed using a combination of organic solvents, and the tissue is coated with trypsin (if digestion of the analyte is required). The matrix is selected based on the size and biochemical attributes of the target analyte and uniformly coated on the tissue using automated sprayers followed by analysis. Simultaneously, the same tissue can be stained using cresyl violet, hematoxylin and eosin, methylene blue, or immunohistochemistry to visualize the cellular population. These stained images can be overlaid with MALDI-IMS images to assign each protein to a certain cellular population or region of interest. There are several reviews and methodology papers available for fine-tuning the protocols according to the requirements of each protein [100,102].
The samples can be analyzed using time of flight (TOF) or the more sensitive and accurate FT-ICR analyzers [100]. The m/z values from the detected peaks can be compared against protein databases or, in the case of novel proteoforms, in silico fragmentation results to annotate proteins. The results can be validated by analysis of proteins from the target region or whole tissue extract using Liquid chromatography-electrospray ionization-Tandem mass spectrometry LC-ESI-MS/MS to obtain sequences of target peptides. As the MALDI-based approach results in the generation of singly charged species, the targeted analysis of the m/z ratio is less complicated, especially in the top-down approach for smaller proteins and can be utilized to understand differences among associated proteoforms. However, the analysis of larger proteins in their native states is still challenging and requires the integration of various related mass spectrometry-based methods to detect and confidently annotate proteins [103].
In the context of the neurodegenerative proteinopathies, this technique has been especially useful for the AD-associated Aβ peptide and has been employed to identify the signature of Aβ proteoforms and distribution in AD (Figure 4) [104,105]. Aβ is a small peptide (4kDa) and is conveniently detected directly on the tissue if the quality of tissue is good and impurities have been removed effectively. Other prion-like proteins, including tau, have also been studied using MALDI-IMS [106]. It has also been utilized to unveil aberrations in brain architecture caused by prion-like pathologies [107].
In addition to providing useful insights into the pathological roles of prion-like proteins and their proteoforms, this technique can also be used to detect the localization of pharmacologically active substances and their effects on pathological proteoforms [98]. Although still under active research, this attribute of MALDI-IMS will be useful for understanding and treating neurodegenerative proteinopathies.
3.4. Hydrogen/Deuterium Exchange Mass Spectrometry
The alterations in sequence and post-translational modifications of proteins mainly impart functional changes by modulating the three-dimensional structure of proteins. Structural alterations in fibrillar aggregates formed by variants of prion-like proteins are an important source of variability in their toxicity and are frequently used as an explanation of heterogeneity in the clinical presentation of proteinopathies. Slight alterations in structure translate to modified biochemical properties, incubation periods, propagation, and toxicities. Common structural techniques, including electron microscopy and atomic force microscopy, have been popular for obtaining low-resolution images of amyloid morphology whereas X-ray fiber diffraction, nuclear magnetic resonance and Fourier-transform infrared spectroscopy have been used to provide detailed information about secondary structures and 3D organization [108]. However, the prerequisite of purified and highly concentrated amyloids in most of these techniques is a major drawback. With the advances in hydrogen/deuterium exchange mass spectrometry, proteomics has stepped into the field of structural biology and is providing critical knowledge about prion-like proteins.
In contrast to low resolution imaging and infrared spectroscopy techniques, hydrogen/deuterium exchange mass spectrometry (HDX-MS) is based on the ease of isotopic exchange on the peptide chain. The rate of this exchange is dependent on the 3D folded state of the protein, the chemical properties of amino acid residues, their intrinsic bonding, and the aggregation status, and can therefore depict the physiological states effectively [109]. This technique is becoming increasingly useful for assessing the conformational diversity of prions and associating them with the clinical heterogeneity of neurodegenerative disorders [110,111,112,113].
4. Conclusions
The heterogeneity of neurodegenerative proteinopathies has rapidly expanded the study of this group of diseases from the proteome to proteoforms and the availability of high-throughput and sensitive proteomic techniques has become the need of the hour. Although some conventional techniques, like 2D-GE, are already usable for this purpose, several top-down and structural mass spectrometric techniques are being optimized to enhance our knowledge of this field (Table 3). The utility of these tools for the analysis of prion-like proteins associated with neurodegenerative proteinopathies will dictate the advancements in understanding, diagnosing, and treating these disorders.
Author Contributions
Literature review and manuscript preparation, A.N. and S.Z.; Conceptualization and Proof-reading, I.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a seed grant of the Physics-to-Medicine Initiative Göttingen (LM der Niedersächsischen Vorab).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; A Tennent, G.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils11Edited by F. E. Cohen. J. Mol. Biol. 2000, 300, 1033–1039. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Fraser, P.E. Prions and Prion-like Proteins. J. Biol. Chem. 2014, 289, 19839–19840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Johansson, J.; Stromberg, R.; Nilsson, L. Unfolding of the Amyloid β-Peptide Central Helix: Mechanistic Insights from Molecular Dynamics Simulations. PLoS ONE 2011, 6, e17587. [Google Scholar] [CrossRef] [Green Version]
- Spagnolli, G.; Rigoli, M.; Inverardi, G.N.; Codeseira, Y.B.; Biasini, E.; Requena, J.R. Modeling PrPSc Generation Through Deformed Templating. Front. Bioeng. Biotechnol. 2020, 8, 590501. [Google Scholar] [CrossRef] [PubMed]
- Gillam, J.E.; Macphee, C.E. Modelling amyloid fibril formation kinetics: Mechanisms of nucleation and growth. J. Phys. Condens. Matter 2013, 25, 373101. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Ionescu-Zanetti, C.; Pope, M.; Li, J.; Nielson, L.; Ramírez-Alvarado, M.; Regan, L.; Fink, A.L.; Carter, S.A. A General Model for Amyloid Fibril Assembly Based on Morphological Studies Using Atomic Force Microscopy. Biophys. J. 2003, 85, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Theint, T.; Nadaud, P.S.; Aucoin, D.; Helmus, J.J.; Pondaven, S.P.; Surewicz, K.; Surewicz, W.K.; Jaroniec, C.P. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Deng, J.; Ma, S.; Zhao, W.; Chang, Z.; Yu, K.; Yang, J. Structural Mechanism of Barriers to Interspecies Seeding Transmissibility of Full-Length Prion Protein Amyloid. ChemBioChem 2019, 20, 2757–2766. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Prince, M.; Guerchet, M.; Prina, M. The global impact of dementia 2013–2050. Alzheimer’s Dis. Int. 2013, 8, 23. [Google Scholar]
- Johnson, S.C.; Schmitz, T.; Moritz, C.; Meyerand, M.; Rowley, H.; Alexander, A.; Hansen, K.; Gleason, C.; Carlsson, C.; Ries, M.; et al. Activation of brain regions vulnerable to Alzheimer’s disease: The effect of mild cognitive impairment. Neurobiol. Aging 2006, 27, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Manix, M.; Kalakoti, P.; Henry, M.; Thakur, J.D.; Menger, R.; Guthikonda, B.; Nanda, A. Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg. Focus 2015, 39, E2. [Google Scholar] [CrossRef]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch. Aerzteblatt Online 2016, 113, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.H. Huntington’s disease genetics. NeuroRX 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.L.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govoni, V.; Della-Coletta, E.; Cesnik, E.; Casetta, I.; Granieri, E. Can the age at onset give a clue to the pathogenesis of ALS? Acta Neurol. Belg. 2016, 117, 221–227. [Google Scholar] [CrossRef]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Wallon, D.; Goupil, C.; Richard, A.-C.; Pottier, C.; Dorval, V.; Sarov-Rivière, M.; Riant, F.; Hervé, D.; Amouyel, P.; et al. Mutation in the 3′untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 2016, 24, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildburger, N.C.; Esparza, T.J.; LeDuc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M.-Y. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ. 1998, 5, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Westaway, D. Current ReviewInfectious and Genetic Manifestations of Prion Diseases. Mol. Plant. Microbe Interact. 1991, 4, 226–233. [Google Scholar] [CrossRef]
- Zafar, S.; Younas, N.; Shafiq, M.; Zerr, I. Prion Protein Strain Diversity and Disease Pathology. In Prions—Some Physiological and Pathophysiological Aspects; IntechOpen: London, UK, 2019. [Google Scholar]
- Mompeán, M.; Baralle, M.; Buratti, E.; Laurents, D.V. An Amyloid-Like Pathological Conformation of TDP-43 Is Stabilized by Hypercooperative Hydrogen Bonds. Front. Mol. Neurosci. 2016, 9, 125. [Google Scholar] [CrossRef]
- di Donato, M.; Craig, L.; Huff, M.E.; Thayer, M.M.; Cardoso, R.M.; Kassmann, C.J.; Lo, T.P.; Bruns, C.K.; Powers, E.T.; Kelly, J.W.; et al. ALS Mutants of Human Superoxide Dismutase Form Fibrous Aggregates Via Framework Destabilization. J. Mol. Biol. 2003, 332, 601–615. [Google Scholar] [CrossRef]
- Molnar, K.S.; Karabacak, N.M.; Johnson, J.L.; Wang, Q.; Tiwari, A.; Hayward, L.J.; Coales, S.J.; Hamuro, Y.; Agar, J.N. A Common Property of Amyotrophic Lateral Sclerosis-associated Variants. J. Biol. Chem. 2009, 284, 30965–30973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 240–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Lee, J.H.; Jeon, J.H.; Lee, M.J. Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum. Mutat. 2001, 17, 493–503. [Google Scholar] [CrossRef]
- Pont, L.; Poturcu, K.; Benavente, F.; Barbosa, J.; Sanz-Nebot, V. Comparison of capillary electrophoresis and capillary liquid chromatography coupled to mass spectrometry for the analysis of transthyretin in human serum. J. Chromatogr. A 2016, 1444, 145–153. [Google Scholar] [CrossRef]
- di Figlia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of hun-tingtin in Huntington disease. Neuroscientist 2011, 17, 475–492. [Google Scholar] [CrossRef]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.A.; Tattum, M.H.; Jones, S.; Bhelt, D.; Batchelor, M.; Clarke, A.R.; Collinge, J.; Jackson, G.S. Codon 129 polymorphism of the human prion protein influences the kinetics of amyloid formation. J. Gen. Virol. 2006, 87, 2443–2449. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. Transmissible human proteopathies: An expanding field. Diagn. Histopathol. 2019, 25, 16–22. [Google Scholar] [CrossRef]
- Petersen, C.; Nolan, A.L.; Resende, E.D.P.F.; Miller, Z.; Ehrenberg, A.J.; Gorno-Tempini, M.L.; Rosen, H.J.; Kramer, J.H.; Spina, S.; Rabinovici, G.D.; et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 2019, 138, 597–612. [Google Scholar] [CrossRef]
- Tolosa, E.; Campdelacreu, J. Clinical overview of the synucleinopathies. Mov. Disord. 2003, 18, 21–27. [Google Scholar] [CrossRef]
- Irwin, D. Tauopathies as clinicopathological entities. Park. Relat. Disord. 2016, 22, S29–S33. [Google Scholar] [CrossRef] [Green Version]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 1992, 1, 546–554. [Google Scholar] [CrossRef]
- Martel, C.L.; Mackic, J.B.; McComb, J.; Ghiso, J.; Zlokovic, B.V. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer’s amyloid β in guinea pigs. Neurosci. Lett. 1996, 206, 157–160. [Google Scholar] [CrossRef]
- Chakraborty, S.; Das, P. Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Sci. Rep. 2017, 7, 9941. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, J.; Jucker, M.; Walker, L.C. Aβ seeds and prions: How close the fit? Prion 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Schmidt, C.; Redyk, K.; Meissner, B.; Krack, L.; von Ahsen, N.; Roeber, S.; Kretzschmar, H.A.; Zerr, I. Clinical Features of Rapidly Progressive Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2010, 29, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Esparcia, P.; Sideris-Lampretsas, G.; Hernandez-Ortega, K.; Grau-Rivera, O.; Sklaviadis, T.; Gelpi, E.; & Ferrer, I. Al-tered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am. J. Neurodegener. Dis. 2017, 6, 15. [Google Scholar]
- Espinoza, M.; de Silva, R.; Dickson, D.W.; Davies, P. Differential Incorporation of Tau Isoforms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Boban, M.; Šarac, H.; Mimica, N.; Mladinov, M.; Süßmair, C.; Ackl, N.; Šimić, G. CSF tau proteins in differential di-agnosis of dementia. Transl. Neurosci. 2010, 1, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Galante, D.; Corsaro, A.; Florio, T.; Vella, S.; Pagano, A.; Sbrana, F.; Vassalli, M.; Perico, A.; D’Arrigo, C. Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1-42 and Aβpy3-42 beta-amyloids. Int. J. Biochem. Cell Biol. 2012, 44, 2085–2093. [Google Scholar] [CrossRef]
- Stein, K.C.; True, H.L. Prion Strains and Amyloid Polymorphism Influence Phenotypic Variation. PLoS Pathog. 2014, 10, e1004328. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimer’s Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nat. Cell Biol. 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; Lograsso, P.; Lasmézas, C.I. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Zhou, T. Application of Two-Dimensional Gel Electrophoresis in Combination with Mass Spectrometry in the Study of Hormone Proteoforms. In Mass Spectrometry—Future Perceptions and Applications; IntechOpen: London, UK, 2019. [Google Scholar]
- Silva, C.J. Applying the tools of chemistry (mass spectrometry and covalent modification by small molecule reagents) to the detection of prions and the study of their structure. Prion 2014, 8, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Patrie, S.M. Top-Down Mass Spectrometry: Proteomics to Proteoforms. In Taurine 6; Springer Nature: Berlin/Heidelberg, Germany, 2016; Volume 919, pp. 171–200. [Google Scholar]
- Stoeckli, M.; Chaurand, P.; Hallahan, D.E.; Caprioli, R.M. Imaging mass spectrometry: A new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001, 7, 493–496. [Google Scholar] [CrossRef]
- di Lillo, M.; Ait-Belkacem, R.; Esteve, C.; Pellegrini, D.; Nicolardi, S.; Costa, M.; Vannini, E.; de Graaf, E.L.; Caleo, M.; McDonnell, L.A. Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.-C.; Iacob, R.E.; Sankaranarayanan, S.; Yang, L.; Ahlijanian, M.; Tao, L.; Tymiak, A.A.; Chen, G. Probing Conformational Dynamics of Tau Protein by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 29, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, F. Highlights on the capacities of “Gel-based” proteomics. Proteome Sci. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Zong, C.; Young, G.W.; Wang, Y.; Lu, H.; Deng, N.; Drews, O.; Ping, P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008, 8, 5025–5037. [Google Scholar] [CrossRef] [Green Version]
- Nizhnikov, A.A.; Alexandrov, A.I.; Ryzhova, T.A.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D.; Galkin, A.P. Proteomic Screening for Amyloid Proteins. PLoS ONE 2014, 9, e116003. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, B.; Shtrasburg, S.; Pras, M. Micropurification techniques in the analysis of amyloid proteins. J. Clin. Pathol. 2003, 56, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Esparza, T.J.; Wildburger, N.C.; Jiang, H.; Gangolli, M.; Cairns, N.J.; Bateman, R.J.; Brody, D.L. Soluble Amyloid-beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep. 2016, 6, 38187. [Google Scholar] [CrossRef]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] Prion Aggregates Are Formed by Small Sup35 Polymers Fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [Green Version]
- Schmittschmitt, J.P.; Scholtz, J.M. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci. 2009, 12, 2374–2378. [Google Scholar] [CrossRef] [Green Version]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef] [Green Version]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–1841. [Google Scholar] [CrossRef]
- Kang, S.-C.; Li, R.; Wang, C.; Pan, T.; Liu, T.; Rubenstein, R.; Barnard, G.; Wong, B.-S.; Sy, M.-S. Guanidine hydrochloride extraction and detection of prion proteins in mouse and hamster prion diseases by ELISA. J. Pathol. 2003, 199, 534–541. [Google Scholar] [CrossRef]
- Kim, J.R.; Muresan, A.; Lee, K.Y.C.; Murphy, R.M. Urea modulation of β-amyloid fibril growth: Experimental studies and kinetic models. Protein Sci. 2008, 13, 2888–2898. [Google Scholar] [CrossRef] [Green Version]
- Vernaglia, B.A.; Huang, J.; Clark, E.D. Guanidine Hydrochloride Can Induce Amyloid Fibril Formation from Hen Egg-White Lysozyme. Biomacromolecules 2004, 5, 1362–1370. [Google Scholar] [CrossRef]
- Maler, J.M.; Klafki, H.-W.; Paul, S.; Spitzer, P.; Groemer, T.W.; Henkel, A.W.; Esselmann, H.; Lewczuk, P.; Kornhuber, J.; Wiltfang, J. Urea-based two-dimensional electrophoresis of beta-amyloid peptides in human plasma: Evidence for novel Aβ species. Proteomics 2007, 7, 3815–3820. [Google Scholar] [CrossRef]
- Kawooya, J.K.; Emmons, T.L.; A Gonzalez-DeWhitt, P.; Camp, M.C.; D’Andrea, S.C. Electrophoretic mobility of Alzheimer’s amyloid-β peptides in urea–sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Anal. Biochem. 2003, 323, 103–113. [Google Scholar] [CrossRef]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Schmidt, B.; Klafki, H.-W.; Maler, M.; Dyrks, T.; et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef]
- Katorcha, E.; Baskakov, I.V. Analysis of Covalent Modifications of Amyloidogenic Proteins Using Two-Dimensional Electrophoresis: Prion Protein and Its Sialylation. Toxic. Assess. 2018, 1779, 241–255. [Google Scholar] [CrossRef]
- Fiorini, M.; Bongianni, M.; Monaco, S.; Zanusso, G. Biochemical Characterization of Prions. Prog. Mol. Biol. Transl. Sci. 2017, 150, 389–407. [Google Scholar] [CrossRef]
- Shen, P.; Dang, J.; Wang, Z.; Zhang, W.; Yuan, J.; Lang, Y.; Ding, M.; Mitchell, M.; Kong, Q.; Feng, J.; et al. Characterization of Anchorless Human PrP With Q227X Stop Mutation Linked to Gerstmann-Sträussler-Scheinker Syndrome In Vivo and In Vitro. Mol. Neurobiol. 2021, 58, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Shelanski, M.L. Microheterogeneity of Micro tubule-Associated ? Proteins Is Due to Differences in Phosphorylation. J. Neurochem. 1986, 47, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, A.; Nishizawa, Y.; Matsuura, I.; Hayashi, F.; Usukura, J.; Bondarenko, V.A. Microtubule-associated protein tau in bovine retinal photoreceptor rod outer segments: Comparison with brain tau. Biochim. Biophys. Acta Bioenerg. 2013, 1832, 1549–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeant, N.; Fernández-Gómez, F.J.; Obriot, H.; Eddarkaoui, S.; Buée-Scherrer, V.; Buée, L. Two-Dimensional Electrophoresis Protocols to Analyze the Microtubule-Associated Tau Proteins from Several Biological Sources. Adv. Struct. Saf. Stud. 2016, 1523, 251–261. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; A Garcia, B.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, N.L.; Costello, C.A.; Begley, T.P.; McLafferty, F.W. Thiaminase I (42 kDa) heterogeneity, sequence refinement, and active site location from high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 1995, 6, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Mortz, E.; O’Connor, P.B.; Roepstorff, P.; Kelleher, N.L.; Wood, T.D.; McLafferty, F.W.; Mann, M. Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases. Proc. Natl. Acad. Sci. USA 1996, 93, 8264–8267. [Google Scholar] [CrossRef] [Green Version]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Brown, J.N.; Tolić, N.; Meng, D.; Liu, X.; Zhang, H.; Zhao, R.; Moore, R.J.; Pevzner, P.; Smith, R.D.; et al. Quantitative analysis of human salivary gland-derived intact proteome using top-down mass spectrometry. Proteom. 2014, 14, 1211–1222. [Google Scholar] [CrossRef]
- Brodie, N.I.; Huguet, R.; Zhang, T.; Viner, R.; Zabrouskov, V.; Pan, J.; Borchers, C.H. Top-down hydrogen–deuterium ex-change analysis of protein structures using ultraviolet photodissociation. Anal. Chem. 2018, 90, 3079–3082. [Google Scholar] [CrossRef]
- Davis, R.G.; Park, H.M.; Kim, K.; Greer, J.B.; Fellers, R.T.; LeDuc, R.D.; Yau, P.M. Top-Down Proteomics Enables Com-parative Analysis of Brain Proteoforms Between Mouse Strains. Anal. Chem. 2018, 90, 3802–3810. [Google Scholar] [CrossRef] [PubMed]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Nshanian, M.; Lantz, C.; Wongkongkathep, P.; Schrader, T.; Klärner, F.-G.; Blümke, A.; Despres, C.; Ehrmann, M.; Smet-Nocca, C.; Bitan, G.; et al. Native Top-Down Mass Spectrometry and Ion Mobility Spectrometry of the Interaction of Tau Protein with a Molecular Tweezer Assembly Modulator. J. Am. Soc. Mass Spectrom. 2018, 30, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Cardiff, R.D. Methods of Immunohistochemistry and Immunofluorescence: Converting Invisible to Visible. Toxic. Assess. 2016, 1458, 1–12. [Google Scholar] [CrossRef]
- Ucal, Y.; Durer, Z.A.; Atak, H.; Kadioglu, E.; Sahin, B.; Coskun, A.; Baykal, A.T.; Ozpinar, A. Clinical applications of MALDI imaging technologies in cancer and neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 795–816. [Google Scholar] [CrossRef]
- Winter, M.; Tholey, A.; Kristen, A.; Röcken, C. MALDI Mass Spectrometry Imaging: A Novel Tool for the Identification and Classification of Amyloidosis. Proteomics 2017, 17, 1700236. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.M.; Norris-Caneda, K.; Drake, R.R. In Situ Imaging of Tryptic Peptides by MALDI Imaging Mass Spectrometry Using Fresh-Frozen or Formalin-Fixed, Paraffin-Embedded Tissue. Curr. Protoc. Protein Sci. 2018, 94, e65. [Google Scholar] [CrossRef]
- Ikegawa, M.; Nirasawa, T.; Kakuda, N.; Miyasaka, T.; Kuzuhara, Y.; Murayama, S.; Ihara, Y. Visualization of Amyloid β Deposits in the Human Brain with Matrix-assisted Laser Desorption/Ionization Imaging Mass Spectrometry. J. Vis. Exp. 2019, 145, e57645. [Google Scholar] [CrossRef] [Green Version]
- Michno, W.; Wehrli, P.M.; Blennow, K.; Zetterberg, H.; Hanrieder, J. Molecular imaging mass spectrometry for probing protein dynamics in neurodegenerative disease pathology. J. Neurochem. 2019, 151, 488–506. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.J.; Spraggins, J.M.; Caprioli, R.M. Protein identification strategies in MALDI imaging mass spectrometry: A brief review. Curr. Opin. Chem. Biol. 2019, 48, 64–72. [Google Scholar] [CrossRef]
- Carlred, L.; Michno, W.; Kaya, I.; Sjövall, P.; Syvänen, S.; Hanrieder, J. Probing amyloid-β pathology in transgenic Alzheimer’s disease (tgArcSwe) mice using MALDI imaging mass spectrometry. J. Neurochem. 2016, 138, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakuda, N.; Miyasaka, T.; Iwasaki, N.; Nirasawa, T.; Wada-Kakuda, S.; Takahashi-Fujigasaki, J.; Murayama, S.; Ihara, Y.; Ikegawa, M. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribat, Y.; Talmat-Amar, Y.; Paucard, A.; Lesport, P.; Bonneaud, N.; Bauer, C.; Bec, N.; Parmentier, M.-L.; Benigno, L.; Larroque, C.; et al. Systemic delivery of P42 peptide: A new weapon to fight Huntington’s disease. Acta Neuropathol. Commun. 2014, 2, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, M.; de Marais, N.J.; Faull, R.L.; Grey, A.C.; Curtis, M.A. Subventricular zone lipidomic architecture loss in Huntington’s disease. J. Neurochem. 2018, 146, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Eraña, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.R.; Surewicz, W.K.; Sigurdson, C.J. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci. Rep. 2017, 7, srep43295. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Smirnovas, V.; Baron, G.S.; Offerdahl, D.K.; Raymond, G.J.; Caughey, B.; Surewicz, W.K. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Rempel, D.L.; Zhang, J.; Sharma, A.K.; Mirica, L.M.; Gross, M.L. Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Moulick, R.; Das, R.; Udgaonkar, J.B. Partially Unfolded Forms of the Prion Protein Populated under Misfolding-promoting Conditions. J. Biol. Chem. 2015, 290, 25227–25240. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, F.; Xiao, X.; Kim, C.; Bohon, J.; Kiselar, J.; Safar, J.G.; Ma, J.; Surewicz, W.K. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J. Biol. Chem. 2018, 293, 18494–18503. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The mechanism of formation of amyloids: The organization of proteins into globular, partially folded or intrinsically disordered functional conformations is a tightly regulated process. However, mutations, aberrant cleavage or cellular stress can cause the formation of altered monomeric species. This event, known as monomer activation, destabilizes the native structure and forms the thermodynamically favorable β-sheets rich structure. The combination of these altered structures, or primary nucleation, leads to the formation of various multimers, protofibrils (2.5 to 3 nm in diameter), and fibrils (6 to 10 nm). The intertwining of protofibrils and fibrils leads to the formation of highly stable mature amyloid fibrils (60–120 nm). The primary event of nucleation and fibril formation is relatively slow and is referred to as the lag phase of growth. As more native proteins mimic the structure of misfolded seeds, the growth of amyloid fibrils reaches an exponential phase leading to rapid accumulation of aggregates [7,8].
Figure 1.
The mechanism of formation of amyloids: The organization of proteins into globular, partially folded or intrinsically disordered functional conformations is a tightly regulated process. However, mutations, aberrant cleavage or cellular stress can cause the formation of altered monomeric species. This event, known as monomer activation, destabilizes the native structure and forms the thermodynamically favorable β-sheets rich structure. The combination of these altered structures, or primary nucleation, leads to the formation of various multimers, protofibrils (2.5 to 3 nm in diameter), and fibrils (6 to 10 nm). The intertwining of protofibrils and fibrils leads to the formation of highly stable mature amyloid fibrils (60–120 nm). The primary event of nucleation and fibril formation is relatively slow and is referred to as the lag phase of growth. As more native proteins mimic the structure of misfolded seeds, the growth of amyloid fibrils reaches an exponential phase leading to rapid accumulation of aggregates [7,8].

Figure 2.
Involvement of known prion-like proteins in multiple neurodegenerative disorders. The figure depicts the overlapping pathological profile of PrP (green circle), α-Synuclein (red circle), Aβ (blue circle), Tau (yellow circle), and TDP-43 (black circle). Each of the stated disorders have further clinical variants (as shown in the case of AD), thereby complicating the role of prion-like proteins in bringing about the observed pathology. PDD—Parkinson’s disease with dementia; DS—Down’s syndrome; FTD-T—frontotemporal dementia with tau pathology; fAD—familial AD; sAD—sporadic AD; rpAD—rapidly-progressive AD; PCA-AD—posterior cortical atrophy–AD; PPA-AD—primary progressive aphasia with AD.
Figure 2.
Involvement of known prion-like proteins in multiple neurodegenerative disorders. The figure depicts the overlapping pathological profile of PrP (green circle), α-Synuclein (red circle), Aβ (blue circle), Tau (yellow circle), and TDP-43 (black circle). Each of the stated disorders have further clinical variants (as shown in the case of AD), thereby complicating the role of prion-like proteins in bringing about the observed pathology. PDD—Parkinson’s disease with dementia; DS—Down’s syndrome; FTD-T—frontotemporal dementia with tau pathology; fAD—familial AD; sAD—sporadic AD; rpAD—rapidly-progressive AD; PCA-AD—posterior cortical atrophy–AD; PPA-AD—primary progressive aphasia with AD.

Figure 3.
Virtual 2D map for AD-associated prion-like proteins. Differences in electrophoretic mobility of Aβ proteoforms based on variations in post-translational cleavage.
Figure 3.
Virtual 2D map for AD-associated prion-like proteins. Differences in electrophoretic mobility of Aβ proteoforms based on variations in post-translational cleavage.

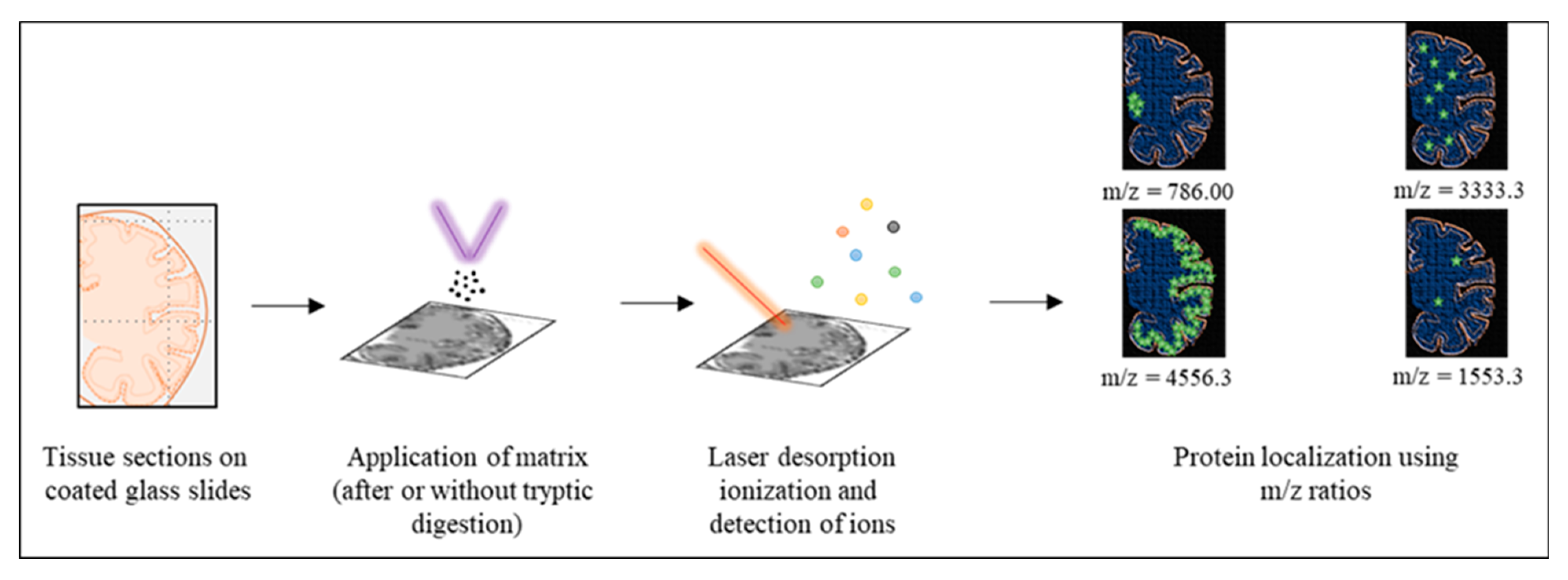
Figure 4.
Summarized methodology matrix-assisted laser desorption ionization-imaging mass spectrometry (MALDI-IMS) for Aβ proteoforms. A section of the tissue is deposited onto conductive slides (step 1) and coated uniformly with the appropriate matrix solution (step 2). Matrix and sample are desorbed and ionized upon the application of ultraviolet laser (red line; step 3). These gaseous ions (colored dots) are then analyzed. Targeted proteins can be localized by selecting the m/z value corresponding to their ions as shown for the m/z ratio of 786, 3333.3, 4556.3 and 1553.3 with green stars in the figure. The relative number of stars depicts the hypothetical signal intensity for the selected m/z ratio.
Figure 4.
Summarized methodology matrix-assisted laser desorption ionization-imaging mass spectrometry (MALDI-IMS) for Aβ proteoforms. A section of the tissue is deposited onto conductive slides (step 1) and coated uniformly with the appropriate matrix solution (step 2). Matrix and sample are desorbed and ionized upon the application of ultraviolet laser (red line; step 3). These gaseous ions (colored dots) are then analyzed. Targeted proteins can be localized by selecting the m/z value corresponding to their ions as shown for the m/z ratio of 786, 3333.3, 4556.3 and 1553.3 with green stars in the figure. The relative number of stars depicts the hypothetical signal intensity for the selected m/z ratio.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Age at onset, affected brain regions, and common symptoms of major neurodegenerative proteinopathies. Age at onset represents a range, rather than mean, due to multiple clinical variants of each of these disorders. * sporadic CJD.
Table 1.
Age at onset, affected brain regions, and common symptoms of major neurodegenerative proteinopathies. Age at onset represents a range, rather than mean, due to multiple clinical variants of each of these disorders. * sporadic CJD.
| Proteinopathy | Age at Onset (Years) | Primary Region | Common Symptoms |
|---|---|---|---|
| AD | 40–65 (early and late-onset variants) | Hippocampus and entorhinal cortex. | Memory and language impairment and visuospatial deficits. [16,17] |
| PD | 40–50 | Substantia nigra (midbrain). | Rigidity, resting tremor and bradykinesia. [18] |
| sCJD * | 44–70 (depends on subtype) | Cerebral cortex and cerebellum. | Cognitive impairment and myoclonus. [19] |
| DLB | 50–80 | Midbrain and neocortex. | Parkinsonian syndrome, autonomic and sleep fluctuations and hallucinations. [20] |
| HD | 20–49 | Caudate nucleus and putamen (basal ganglia). | Choreiform movements, emotional and behavioral alterations, bradykinesia. [21] |
| ALS | 45–55 | Motor neurons. | Focal muscle wasting, spasticity and flexor spasms. [22,23] |
Table 2.
A summary of the structure and variants of major amyloidogenic proteins associated with neurodegenerative proteinopathies.
Table 2.
A summary of the structure and variants of major amyloidogenic proteins associated with neurodegenerative proteinopathies.
| Amyloids | Precursor Protein | Associated Diseases | Proteoforms or Other Variants |
|---|---|---|---|
| Aβ | Amyloid beta A4 protein: Intrinsically disordered protein with 770 residues | AD, Cerebral amyloid angiopathy (CAA) [24,25]. | 26 differentially truncated and post translationally modified proteoforms [26] |
| α-Synuclein | Alpha Synuclein: Intrinsically disordered protein with 140 residues | PD and DLB [27] | 11 differentially truncated and post translationally modified proteoforms [28] |
| PrPSc | Major prion protein: Intrinsically disordered protein with 253 amino acids | CJD, Fatal Familial Insomnia (FFI), Gerstmann-Straussler-Scheinker disease (GSS), Huntington disease-like type 1 (HDL1), Kuru and Spongiform encephalopathy [29] | 2 Proteoforms based on Proteinase-K resistance Genetic variants (codon 129 polymorphism). [30] |
| ASOD | Superoxide dismutase: Intrinsically disordered protein with 154 amino acids | ALS—TDP-43 amyloids also involved. [31,32] | Genetic variants. No proteoforms reported yet. [33] |
| ATau | Microtubule-associated protein tau: Intrinsically disordered protein with 758 amino acids | Frontotemporal dementia (FTD), AD, Progressive Supranuclear Palsy (PSP), Corticobasal degeneration (CBD), Pick’s disease, Argyrophilic grain disease, Dementia with Lewy bodies and Parkinsonism linked to chromosome 17. [34] | Six isoforms. Differentially post translationally modified proteoforms. [35] |
| ATTR | Transthyretin: Mostly β-sheet with 147 amino acids | Familial Amyloid polyneuropathy, Leptomeningeal amyloidosis. [36] | Differentially oxidized proteoforms. [37] |
| AHtt | Huntington: Intrinsically disordered protein with 3142 residues | Huntington disease. [38] | Differentially post translationally modified proteoforms. [39] |
Table 3.
A summary of the capabilities and considerations for the proteomic tools discussed in this review. The previously studied amyloids, using each of these techniques, have also been stated.
Table 3.
A summary of the capabilities and considerations for the proteomic tools discussed in this review. The previously studied amyloids, using each of these techniques, have also been stated.
| Technique | Utility for Amyloids | Samples | Previously Targetted Amyloids |
|---|---|---|---|
| 2D-GE | Resolves minor biochemical variations among proteoforms by targeting isoelectric points and molecular weights. | Solubilized proteins with native charges preferably monomeric species (multimeric species may provide misleading results if more than one proteoform is involved). Buffers selected must not induce aggregation under experimental conditions. | PrP, Aβ, Tau |
| Top-Down MS | Identifies proteoforms and their post-translational modifications in their native forms. | Undigested proteins in their native conformations. Buffers that prevent aggregation but do not affect the spectrum of target. In case of MALDI, matices have to be tested for their capability to ionize the target. | Aβ, tau, α-Synuclein |
| MALDI IMS | Locates proteins via in situ identification of proteoforms. | Paraffin-embedded or frozen tissue sections. Matices have to be tested for their capability to ionize the target. | Aβ, tau |
| HDX-MS | Depicts 3D structures of proteins. | Undenatured, undigested proteins in their native conformations. Experimental conditions have to be carefully controlled to prevent uneven deutrium labelling among replicates. | PrP, Aβ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Noor, A.; Zafar, S.; Zerr, I. Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. Int. J. Mol. Sci. 2021, 22, 1085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031085
AMA Style
Noor A, Zafar S, Zerr I. Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. International Journal of Molecular Sciences. 2021; 22(3):1085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031085
Chicago/Turabian StyleNoor, Aneeqa, Saima Zafar, and Inga Zerr. 2021. "Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges" International Journal of Molecular Sciences 22, no. 3: 1085. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031085
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.