
Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thrombotic Complications in MPN Patients
3. Thrombotic Risk Factors in MPN Patients
4. Role of Neutrophils in MPN Thrombosis
5. Neutrophil Extracellular Traps (NETs) Formation
6. Role of NETs in Thrombotic Pathogenesis
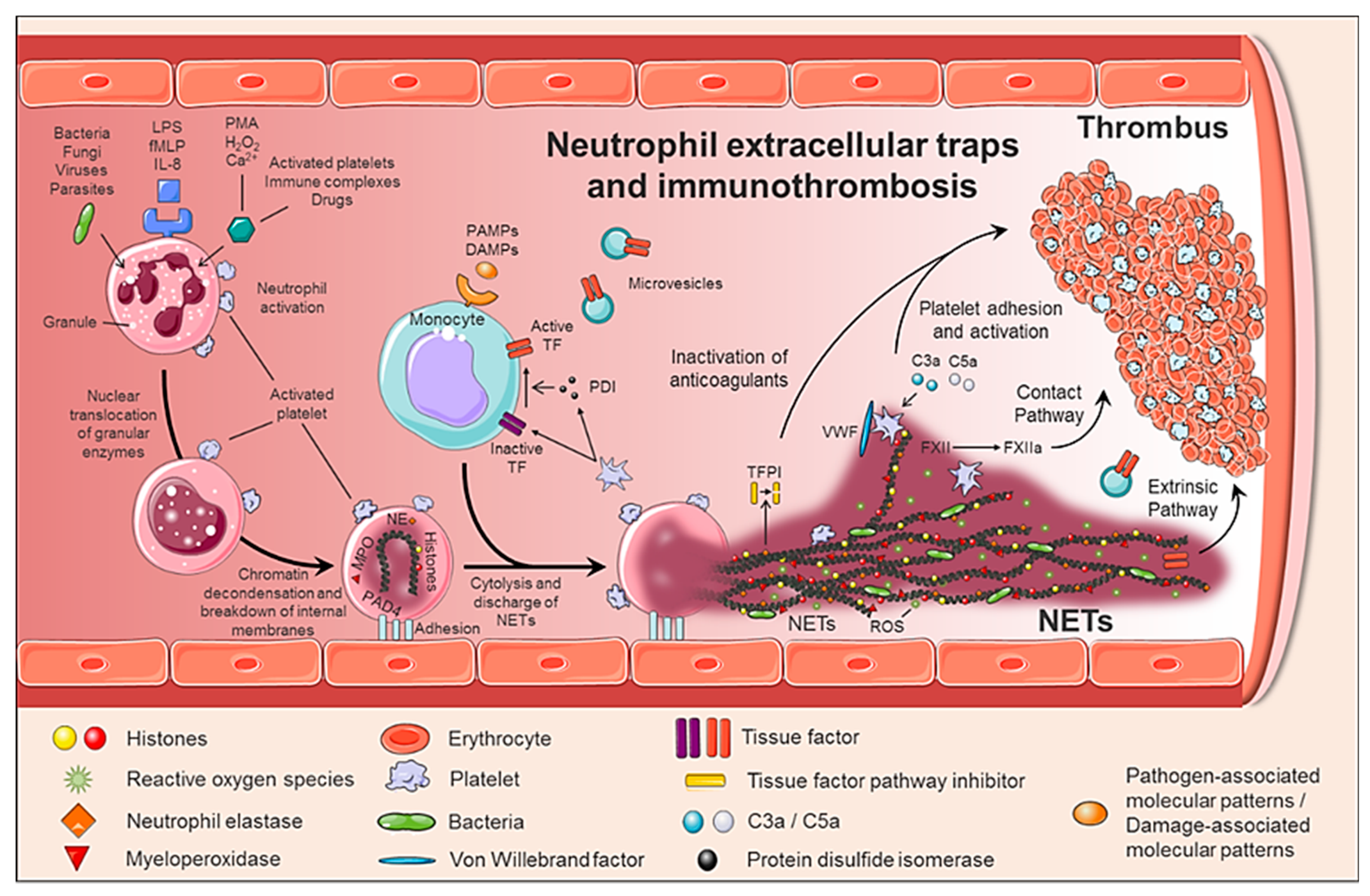
- Any cell death is a potential source of free DNA in plasma, so this is a necessary but not specific finding of NETosis. Although not specific to NETosis, the presence of negative charges (DNA) causes an activation of FXII, a plasma serine protease, initiating the intrinsic pathway of coagulation. This promotes the chain activation of a series of coagulation proteins which in turn results in the formation of fibrin and ultimately the thrombus [47];
- Histones are the most abundant proteins in NETs. They are positively charged and are responsible for packaging the genetic material. It has been shown that histones 3 and 4 (H3 and H4, respectively) are able to activate platelets, favoring their aggregation and contributing to the generation of thrombin [48]. This ability of H3 and H4 to activate platelets seems to be, at least partially, dependent on the signaling pathway of TLR2 and TLR4 receptors, through the transcription factor NF-κB [48]. Alternatively, histones also contribute to thrombin activation by reducing thrombomodulin-dependent protein C activation [49];
- The granule proteases (elastase and Cathepsin G) are enzymes derived from neutrophils and the most abundant proteins in NET after histones. Elastase is located in the acidophilic granules and its function is to eliminate tissue degradation products of pathogens. In the context of thrombogenesis, it causes the degradation and inactivation of two important natural anticoagulants: tissue factor pathway inhibitor (TFPI) and antithrombin (AT) [50,51]. TFPI is the main inhibitor of the TF pathway or extrinsic pathway of coagulation whereas AT blocks thrombin formation, a key step for thrombus formation. In addition, elastase promotes platelet adhesion by facilitating exposure to von Willebrand factor (vWF). Cathepsin G hydrolyzes proteins and also helps block the activity of TFPI and enhance thrombosis by activating protease receptor 4 (PAR4) signaling pathway on platelets. Thus, it was observed that mice deficient in elastase and cathepsin G have defects in TF activation, in fibrin formation, and in thrombus stabilization [52];
- TF, through activation of the extrinsic pathway of coagulation and platelets, promotes thrombus formation. TF has been identified in NETs and it has been documented that this factor comes not only from monocytes that migrate to the inflamed area, but also from neutrophils. This finding was observed in neutrophils isolated from patients with sepsis. The autophagy has been pointed out as the mechanism by which the neutrophil captures the TF that is released during NETosis. In this sense, the TF carried by NETs is capable of stimulating thrombin generation and platelet activation in ex vivo experiments [53,54].
7. NETs in Vascular Pathology
8. Role of NETs in Myeloproliferative Neoplasms
9. New Therapeutic Opportunities to Prevent Thrombosis in MPN
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| MPN | Myeloproliferative neoplasm |
| PV | Polycythemia vera |
| ET | Essential thrombocythemia |
| MF | Myelofibrosis |
| HSC | Hematopoietic stem cell |
| BM | Bone marrow |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription |
| ECLAP | European collaboration on low-dose aspirin in polycythemia vera |
| IWG-MRT | International Working Group for MPN Research and Treatment |
| CYTO-PV | Cytoreductive therapy in PV |
| AVWS | Acquired von Willebrand syndrome |
| ELN | European Leukemia Net |
| WBC | White blood cell |
| WHO | World Health Organization |
| IPSET | International prognostic score for thrombosis in ET |
| Mac-1 | Macrophage-1 antigen |
| PSGL-1 | P-selectin glycoprotein ligand 1 |
| TREM1 | Triggering receptor expressed on myeloid cells 1 |
| LAP | Latency-associated peptide |
| CCL5 | C-C motif chemokine 5 |
| PF4 | Platelet factor 4 |
| GPIbα | PlateletglycoproteinIb alpha chain |
| CALR | Calreticulin |
| ROS | Reactive oxygen species |
| TF | Tissue factor |
| NETs | Neutrophil extracellular traps |
| DNA | Deoxyribonucleic acid |
| MMP9 | Matrix metalloproteinase 9 |
| PAD4 | Peptidyl arginine deaminase 4 |
| LPS | Lipopolysaccharide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| IL-8 | Interleukin 8 |
| TNFα | Tumor necrosis factor alpha |
| FXII | Factor XII |
| TLR2 | Toll-like receptor 2 |
| TLR4 | Toll-like receptor 4 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| TFPI | Tissue factor pathway inhibitor |
| vWF | von Willebrand factor |
| AT | Antithrombin |
| PAR4 | Protease activated receptor 4 |
| DVT | Deep vein thrombosis |
| ApoE | Apolipoprotein E |
| citH3 | Citrullinated histone H3 |
| PMA | Phorbol 12-myristate 13-acetate |
| MPO | Myeloperoxidase |
| WT | Wild type |
| tPA | Tissue plasminogen activator |
| FDA | Food and Drug Administration |
| EMA | European Medicines Agency |
| NAC | N-acetylcysteine |
| VCAM1 | Vascular cell adhesión molecule 1 |
| ICAM1 | Intercellular adhesion molecule 1 |
| fMLP | N-Formylmethionyl-leucyl-phenylalanine |
| H2O2 | Hydrogen peroxide |
| Ca2+ | Calcium ion |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| CatG | Cathepsin G |
| PDI | Protein disulfide isomerase |
References
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoi, K.; Cross, N.C.P. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M. Thrombosis in Myeloproliferative Neoplasms. Semin. Thromb. Hemost. 2014, 40, 348–358. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Sáez Perdomo, M.N.S.; Perera, M.D.M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Rambaldi, A.; Finazzi, G. Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: Is leukocytosis a causative factor? Blood 2009, 114, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.-J.; Forsyth, C.; Harrison, C.N.; Green, A.R. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and Neoplastic Risk in a Large Cohort of Patients With Polycythemia Vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Gruppo Italiano Studio Policitemia Polycythemia Vera: The Natural History of 1213 Patients Followed for 20 Years. Ann. Intern. Med. 1995, 123, 656–664. [CrossRef] [PubMed]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Ofosu, F.; Cortelazzo, S.; Delaini, F.; Marziali, S.; Barbui, T. Hemostatic System Activation in Patients with Lupus Anticoagulant and Essential Thrombocythemia. Semin. Thromb. Hemost. 1994, 20, 324–327. [Google Scholar] [CrossRef]
- Dentali, F.; Ageno, W.; Rumi, E.; Casetti, I.; Poli, D.; Scoditti, U.; Maffioli, M.; di Minno, M.N.D.; Caramazza, D.; Pietra, D.; et al. Cerebral venous thrombosis and myeloproliferative neoplasms: Results from two large databases. Thromb. Res. 2014, 134, 41–43. [Google Scholar] [CrossRef]
- Passamonti, S.M.; Biguzzi, E.; Cazzola, M.; Franchi, F.; Gianniello, F.; Bucciarelli, P.; Pietra, D.; Mannucci, P.M.; Martinelli, I. The JAK2 V617F mutation in patients with cerebral venous thrombosis. J. Thromb. Haemost. 2012, 10, 998–1003. [Google Scholar] [CrossRef]
- Kaifie, A.; Kirschner, M.; Wolf, D.; Maintz, C.; Hänel, M.; Gattermann, N.; Gökkurt, E.; Platzbecker, U.; Hollburg, W.; Göthert, J.R.; et al. Bleeding, thrombosis, and anticoagulation in myeloproliferative neoplasms (MPN): Analysis from the German SAL-MPN-registry. J. Hematol. Oncol. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Geyer, H.L.; Mesa, R.A. Therapy for myeloproliferative neoplasms: When, which agent, and how? Hematology 2014, 2014, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization–essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef] [Green Version]
- Gangat, N.; Wolanskyj, A.P.; Schwager, S.M.; Hanson, C.A.; Tefferi, A. Leukocytosis at diagnosis and the risk of subsequent thrombosis in patients with low-risk essential thrombocythemia and polycythemia vera. Cancer 2009, 115, 5740–5745. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Gangat, N.; Wolanskyj, A. The interaction between leukocytosis and other risk factors for thrombosis in essential thrombocythemia. Blood 2007, 109, 4105. [Google Scholar] [CrossRef]
- Tefferi, A. Leukocytosis as a risk factor for thrombosis in myeloproliferative neoplasms-biologically plausible but clinically uncertain. Am. J. Hematol. 2010, 85, 93–94. [Google Scholar] [CrossRef]
- Landolfi, R.; Di Gennaro, L.; Barbui, T.; De Stefano, V.; Finazzi, G.; Marfisi, R.; Tognoni, G.; Marchioli, R. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007, 109, 2446–2452. [Google Scholar] [CrossRef] [Green Version]
- Ronner, L.; Podoltsev, N.; Gotlib, J.; Heaney, M.L.; Kuykendall, A.T.; O’Connell, C.; Shammo, J.; Fleischman, A.G.; Scherber, R.M.; Mesa, R.; et al. Persistent leukocytosis in polycythemia vera is associated with disease evolution but not thrombosis. Blood 2020, 135, 1696–1703. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Larran, A.; Pereira, A.; Guglielmelli, P.; Hernandez-Boluda, J.C.; Arellano-Rodrigo, E.; Ferrer-Marin, F.; Samah, A.; Griesshammer, M.; Kerguelen, A.; Andreasson, B.; et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica 2016, 101, 926–931. [Google Scholar] [CrossRef]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates β1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule 1. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falanga, A.; Marchetti, M.; Barbui, T.; Smith, C.W. Pathogenesis of Thrombosis in Essential Thrombocythemia and Polycythemia Vera: The Role of Neutrophils. Semin. Hematol. 2005, 42, 239–247. [Google Scholar] [CrossRef] [PubMed]
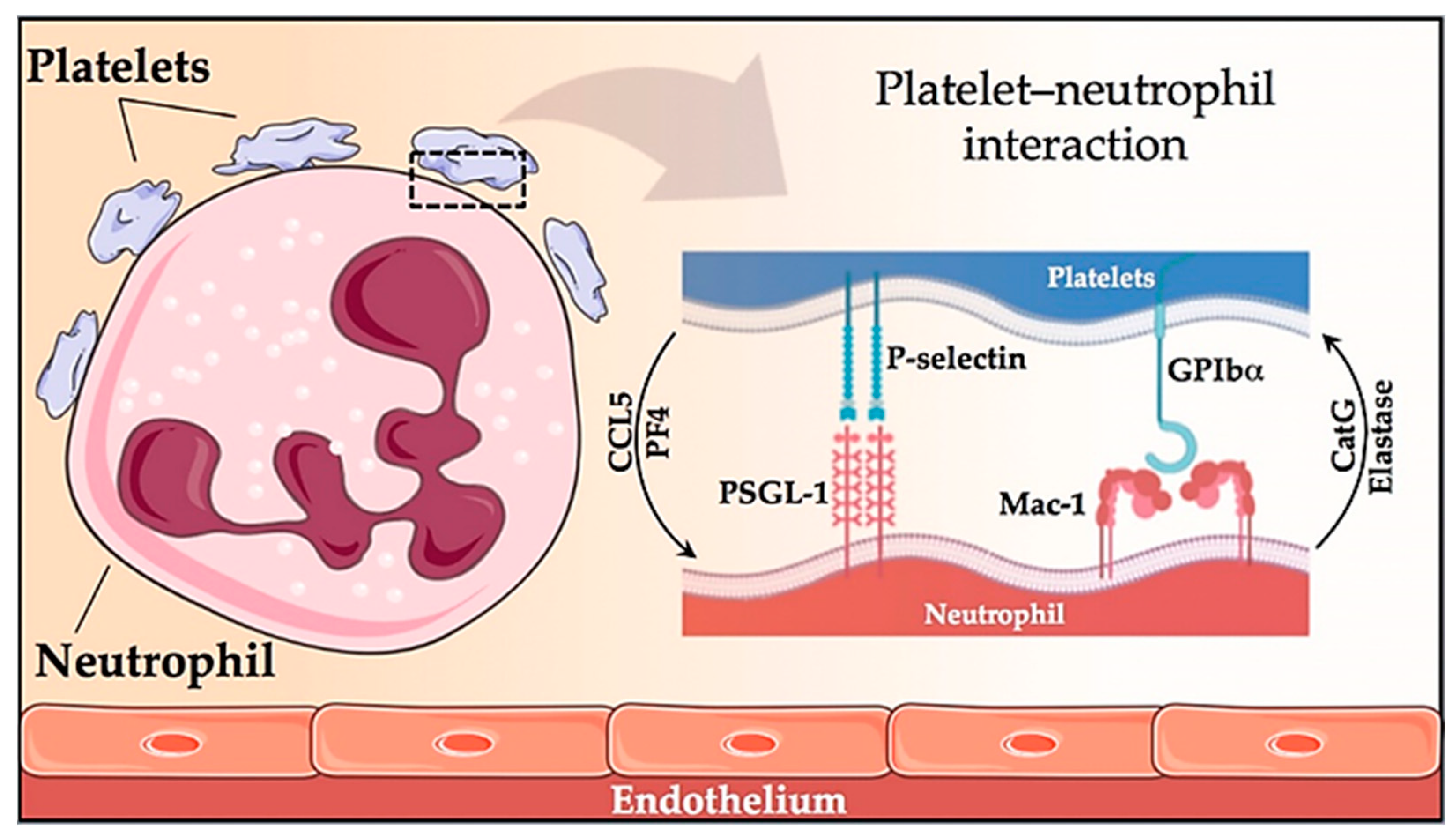
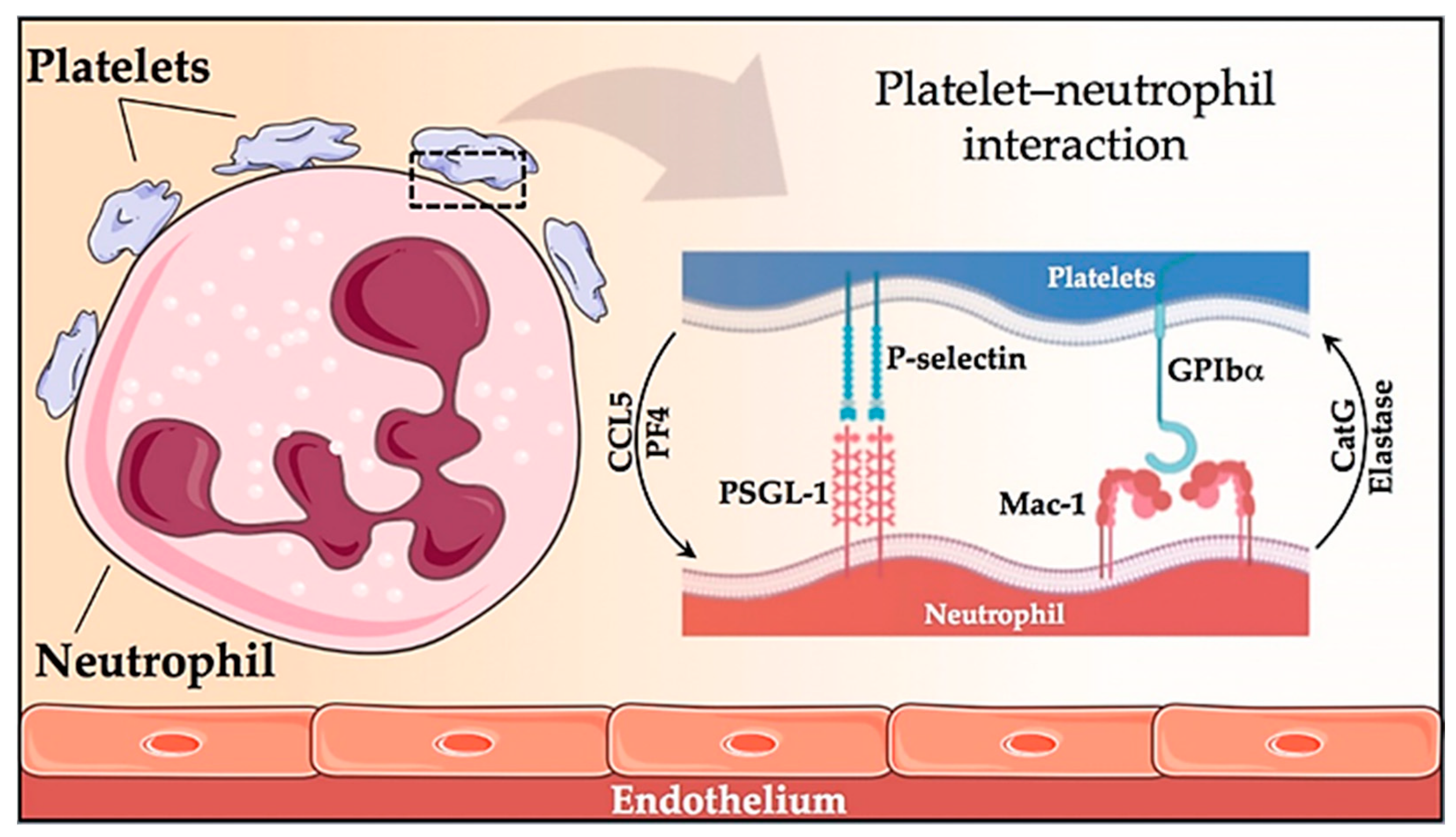
- Lisman, T. Platelet–neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Arellano-Rodrigo, E.; Alvarez-Larrán, A.; Reverter, J.C.; Villamor, N.; Colomer, D.; Cervantes, F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 2006, 91, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa, J.M.; Ferrer-Marín, F.; Lozano, M.L.; Moreno, M.J.; Martinez, C.; Anton, A.I.; Rivera, J.; Vicente, V. Impaired leucocyte activation is underlining the lower thrombotic risk of essential thrombocythaemia patients with CALR mutations as compared with those with the JAK2 mutation. Br. J. Haematol. 2016, 172, 813–815. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Desai, J.; Mulay, S.R.; Nakazawa, D.; Anders, H.-J. Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell. Mol. Life Sci. 2016, 73, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.-D. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (ETs) in Humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawrotniak, M.; Rapala-Kozik, M. Neutrophil extracellular traps (NETs)—Formation and implications. Acta Biochim. Pol. 2013, 60, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrera, C.; Fadeel, B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef] [Green Version]
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Steppich, B.A.; Seitz, I.; Busch, G.; Stein, A.; Ott, I. Modulation of tissue factor and tissue factor pathway inhibitor-1 by neutrophil proteases. Thromb. Haemost. 2008, 100, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Jordan, R.E.; Nelson, R.M.; Kilpatrick, J.; Newgren, J.O.; Esmon, P.C.; Fournel, M.A. Inactivation of human antithrombin by neutrophil elastase. Kinetics of the heparin-dependent reaction. J. Biol. Chem. 1989, 264, 10493–10500. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Kambas, K.; Mitroulis, I.; Ritis, K. The emerging role of neutrophils in thrombosis—the journey of TF through NETs. Front. Immunol. 2012, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy Mediates the Delivery of Thrombogenic Tissue Factor to Neutrophil Extracellular Traps in Human Sepsis. PLoS ONE 2012, 7, e45427. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.N. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases? World J. Cardiol. 2015, 7, 829. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- van Montfoort, M.L.; Stephan, F.; Lauw, M.N.; Hutten, B.A.; Van Mierlo, G.J.; Solati, S.; Middeldorp, S.; Meijers, J.C.M.; Zeerleder, S. Circulating Nucleosomes and Neutrophil Activation as Risk Factors for Deep Vein Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated With Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M.; Swirski, F.K. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 2015, 349, 237–238. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Hashiba, M.; Huq, A.; Tomino, A.; Hirakawa, A.; Hattori, T.; Miyabe, H.; Tsuda, M.; Takeyama, N. Neutrophil extracellular traps in patients with sepsis. J. Surg. Res. 2015, 194, 248–254. [Google Scholar] [CrossRef]
- Ferrer-Marín, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernández-Rivas, J.M.; Hernández-Boluda, J.C.; Fernandez-Rodriguez, C.; Luño, E.; García Hernandez, C.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 34, 2648–2659. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Cedervall, J. NETosis in Cancer—Platelet–Neutrophil Crosstalk Promotes Tumor-Associated Pathology. Front. Immunol. 2016, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [Green Version]
- Mauracher, L.-M.; Posch, F.; Martinod, K.; Grilz, E.; Däullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin Oyarzún, C.P.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Castro Ríos, M.A.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738. [Google Scholar] [CrossRef] [Green Version]
- Guy, A.; Favre, S.; Labrouche-Colomer, S.; Deloison, L.; Gourdou-Latyszenok, V.; Renault, M.-A.; Riviere, E.; James, C. High circulating levels of MPO-DNA are associated with thrombosis in patients with MPN. Leukemia 2019, 33, 2544–2548. [Google Scholar] [CrossRef]
- Kasprzycka, W.; Homa-Mlak, I.; Mlak, R.; Małecka-Massalska, T. Direct and indirect methods of evaluating the NETosis process. J. Pre-Clin. Clin. Res. 2019, 13, 50–56. [Google Scholar] [CrossRef]
- Wolff, L.; Guy, A.; Gourdou-Latyszenokv, V.; Favre, S.; Colomer, S.; Renault, M.A.; Couffinhal, T.; James, C. Neutrophils prothrombotic characteristics during myeloproliferative neoplasms. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 205. [Google Scholar] [CrossRef]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Li, M.; Lin, C.; Deng, H.; Strnad, J.; Bernabei, L.; Vogl, D.T.; Burke, J.J.; Nefedova, Y. A Novel Peptidylarginine Deiminase 4 (PAD4) Inhibitor BMS-P5 Blocks Formation of Neutrophil Extracellular Traps and Delays Progression of Multiple Myeloma. Mol. Cancer Ther. 2020, 19, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-C.; Korinek, M.; Cheng, W.-J.; Hwang, T.-L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 1576. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Mendez Luque, L.F.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer-Marín, F.; Cuenca-Zamora, E.J.; Guijarro-Carrillo, P.J.; Teruel-Montoya, R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031143
Ferrer-Marín F, Cuenca-Zamora EJ, Guijarro-Carrillo PJ, Teruel-Montoya R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2021; 22(3):1143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031143
Chicago/Turabian StyleFerrer-Marín, Francisca, Ernesto José Cuenca-Zamora, Pedro Jesús Guijarro-Carrillo, and Raúl Teruel-Montoya. 2021. "Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms" International Journal of Molecular Sciences 22, no. 3: 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031143