
The Biochemical Pathways of Nicotinamide-Derived Pyridones

 , , and
, , and 
Abstract
:
1. Introduction
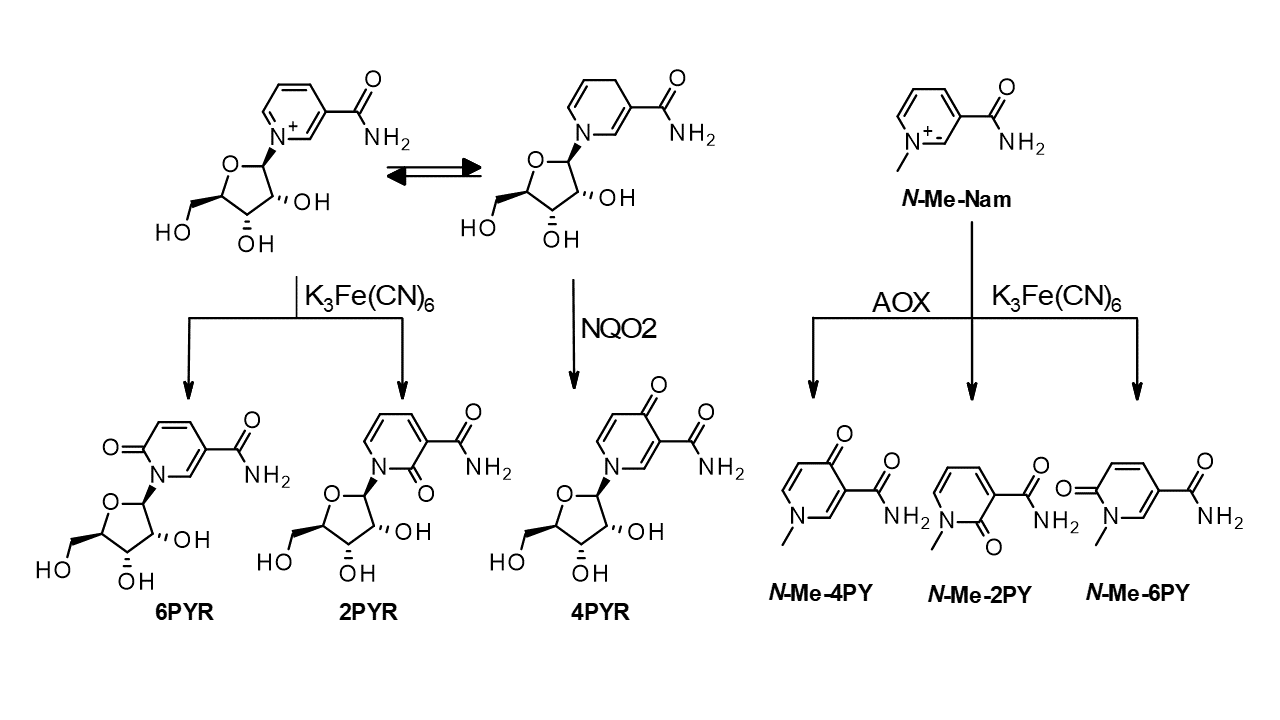
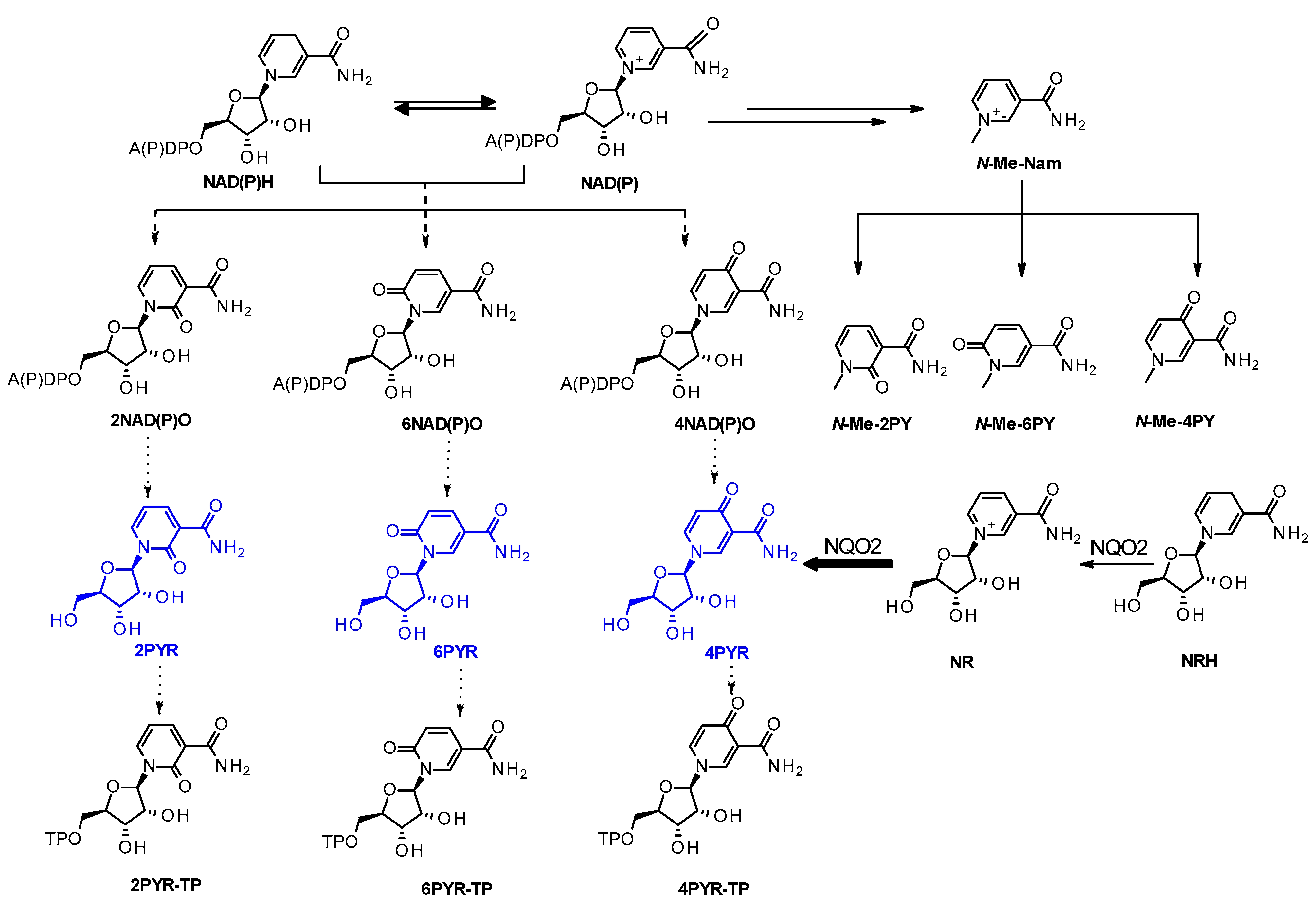
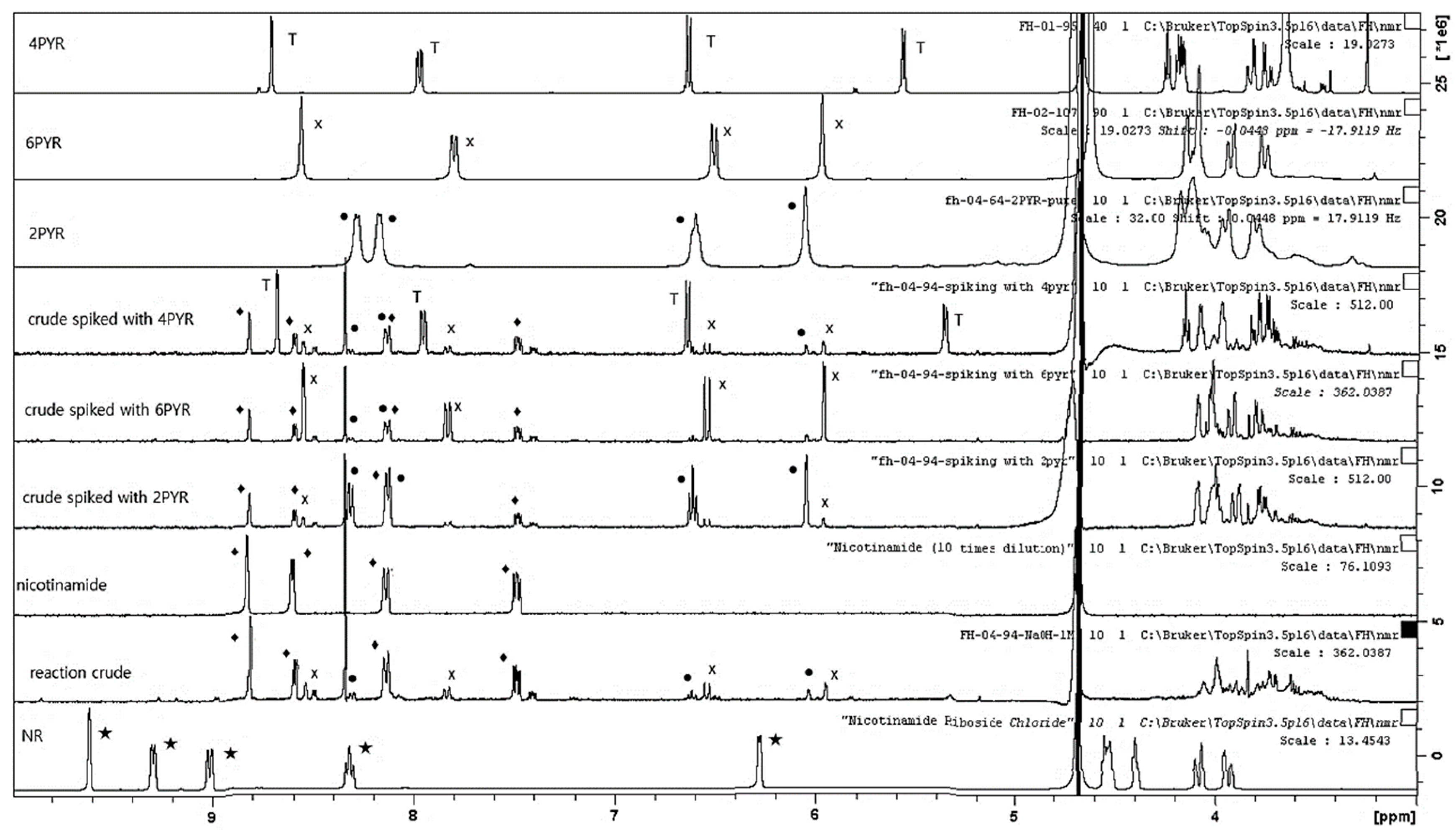
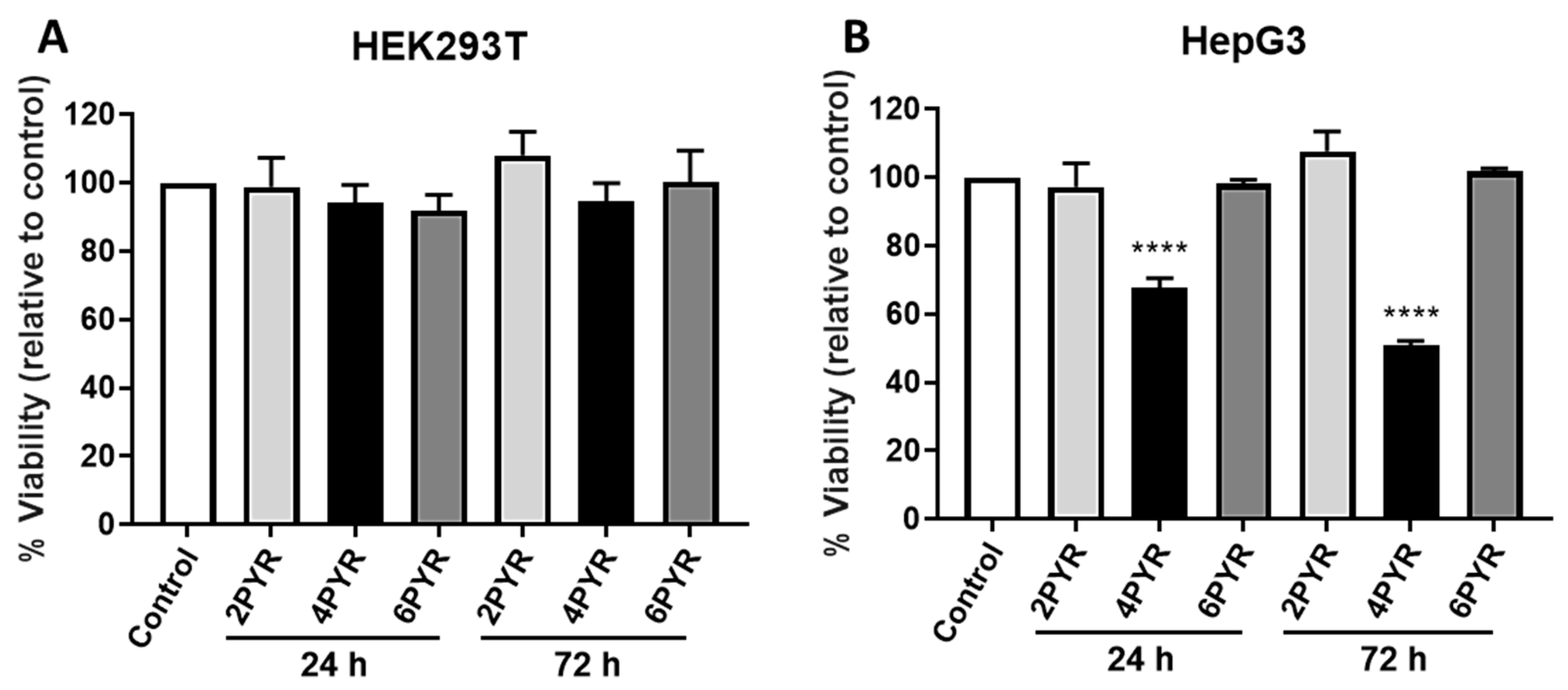
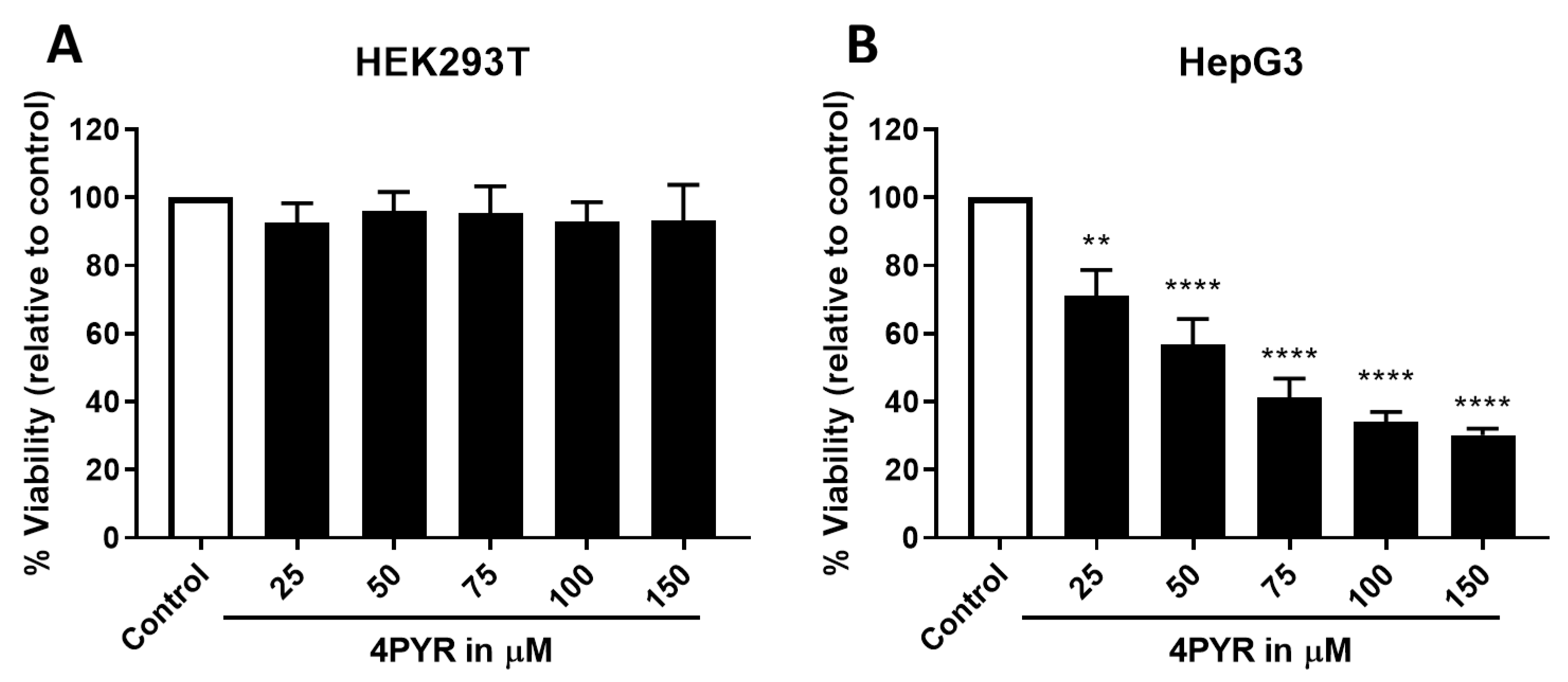
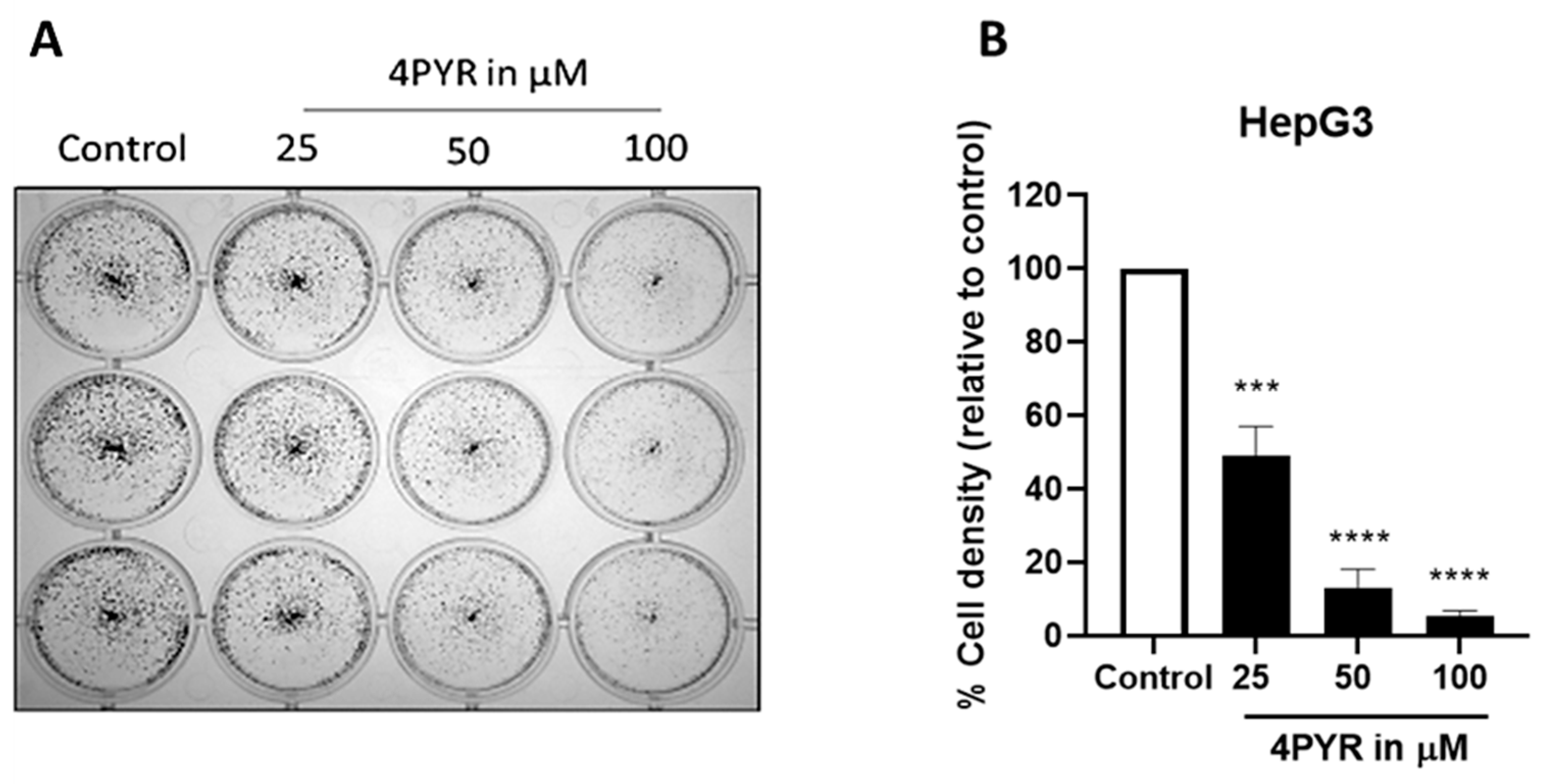
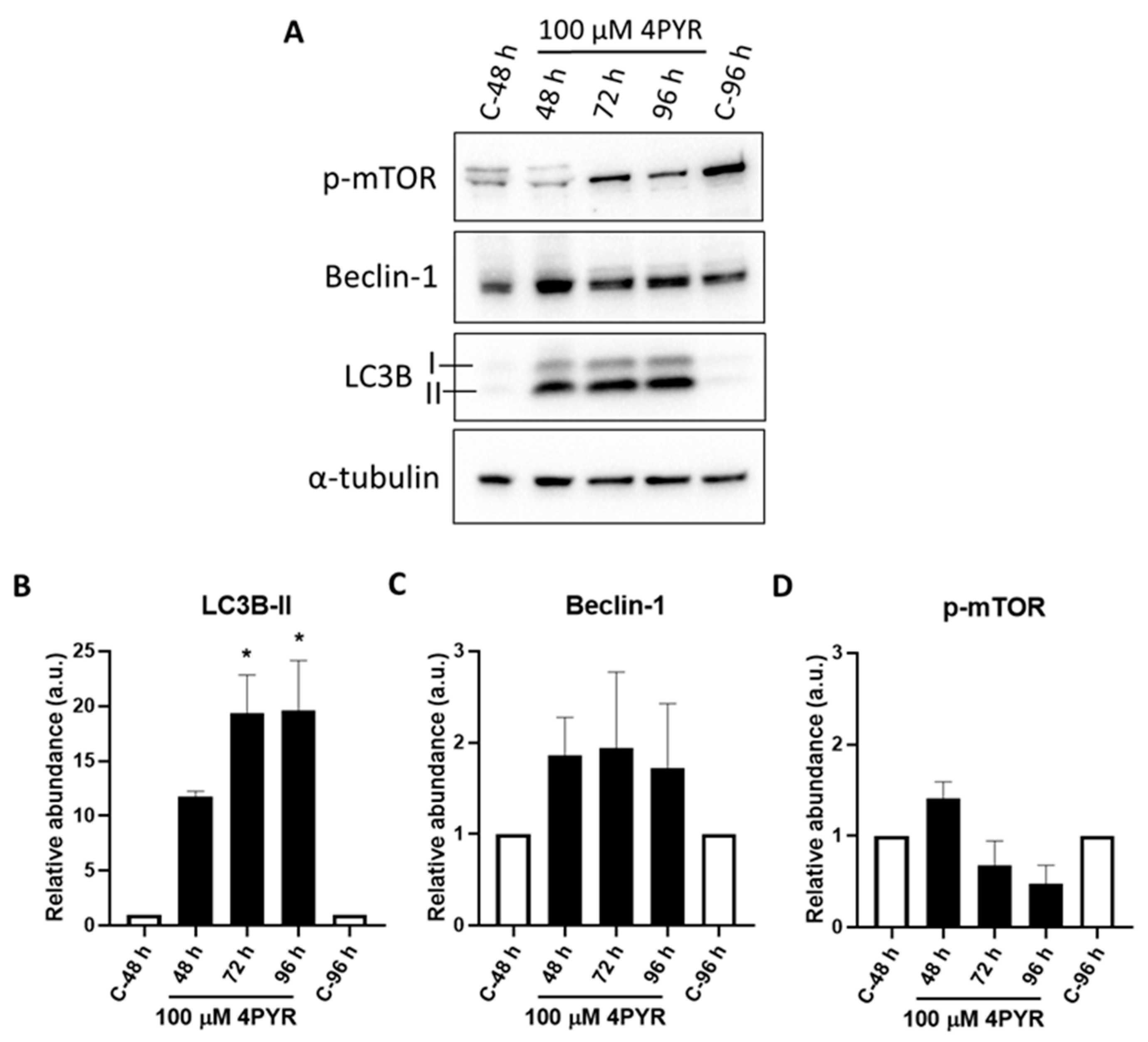
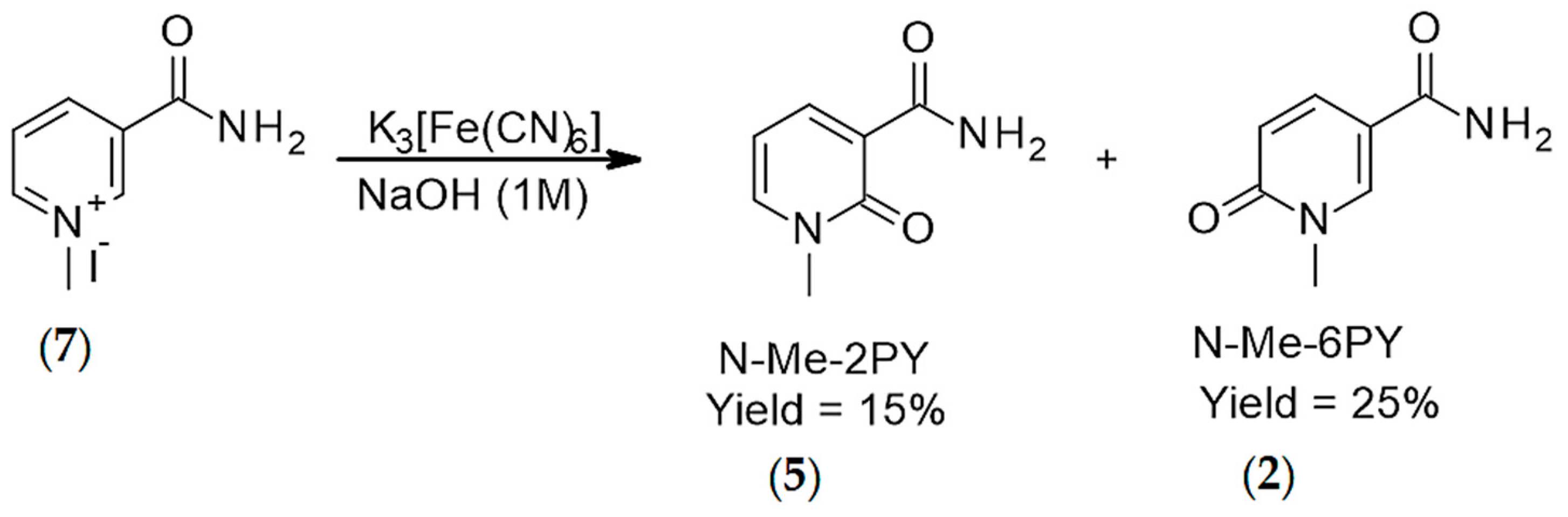
2. Results
3. Discussion
4. Methods
4.1. Syntheses
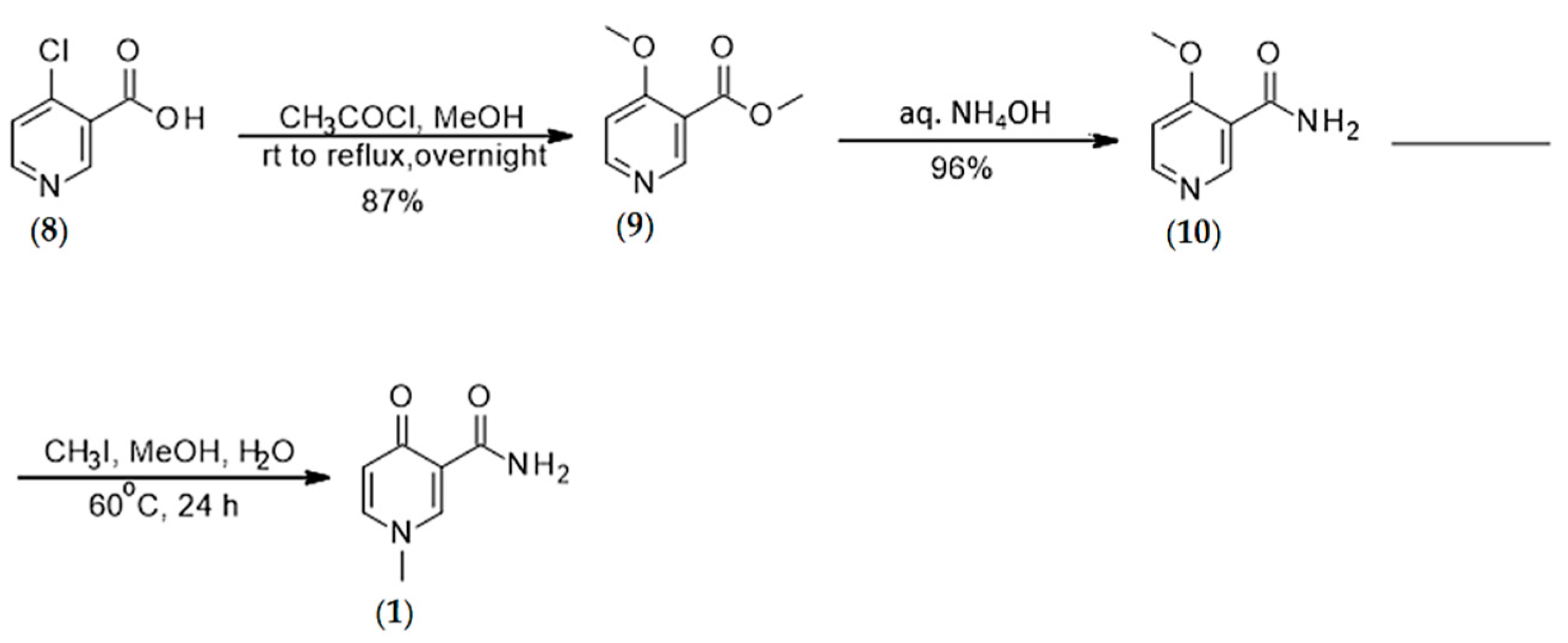
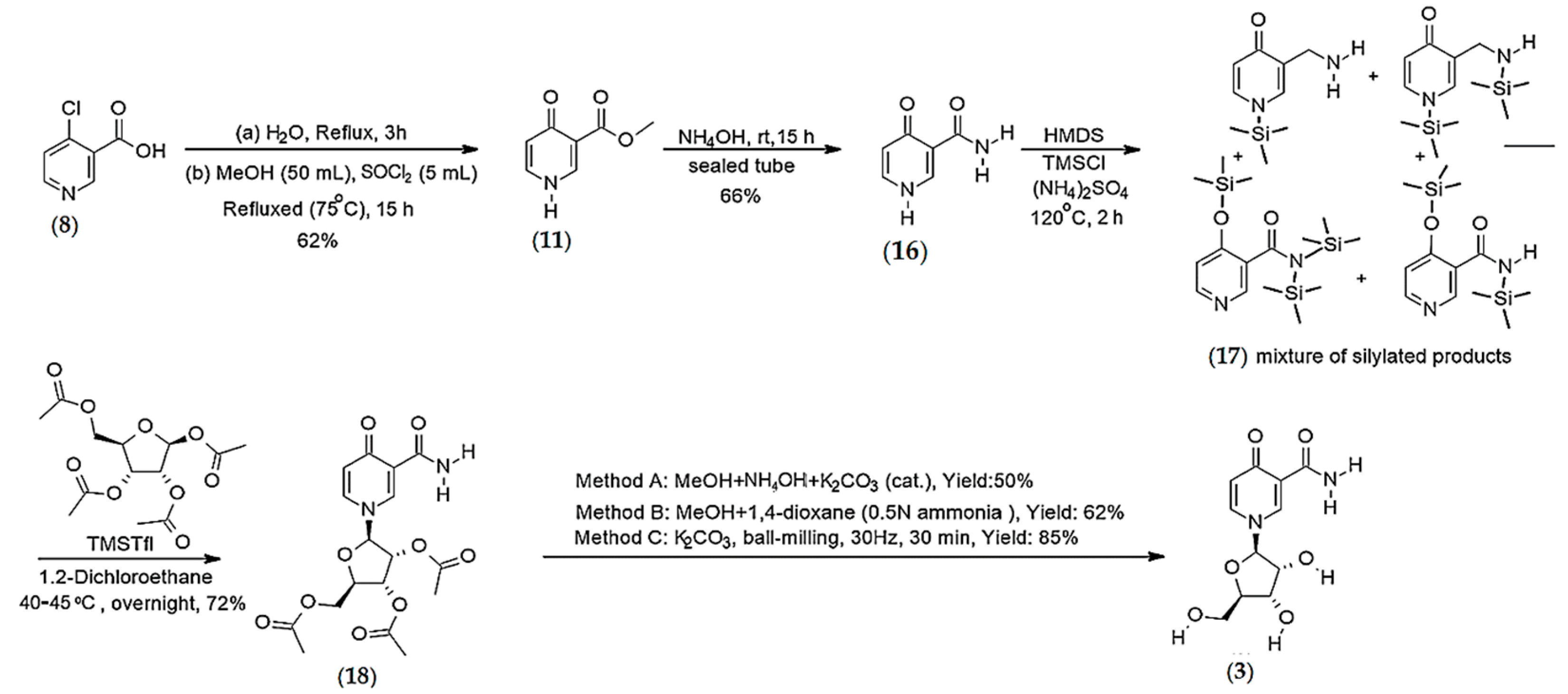
4.1.1. Synthesis of N-methyl-4-pyridone 3-carboxamide (1)
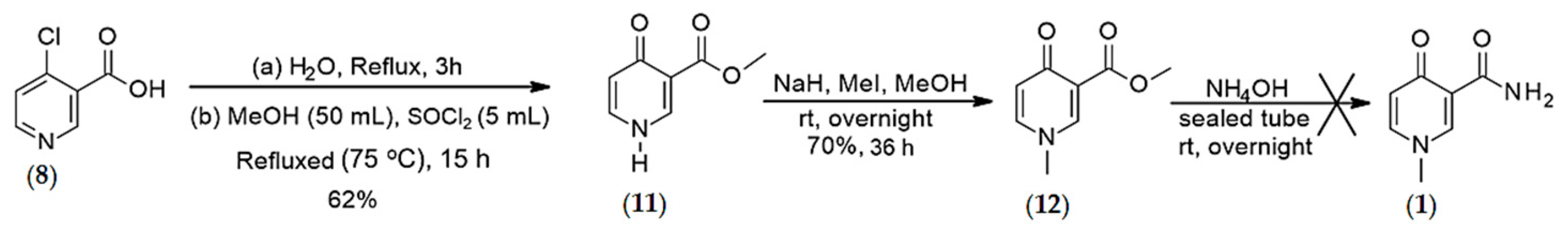
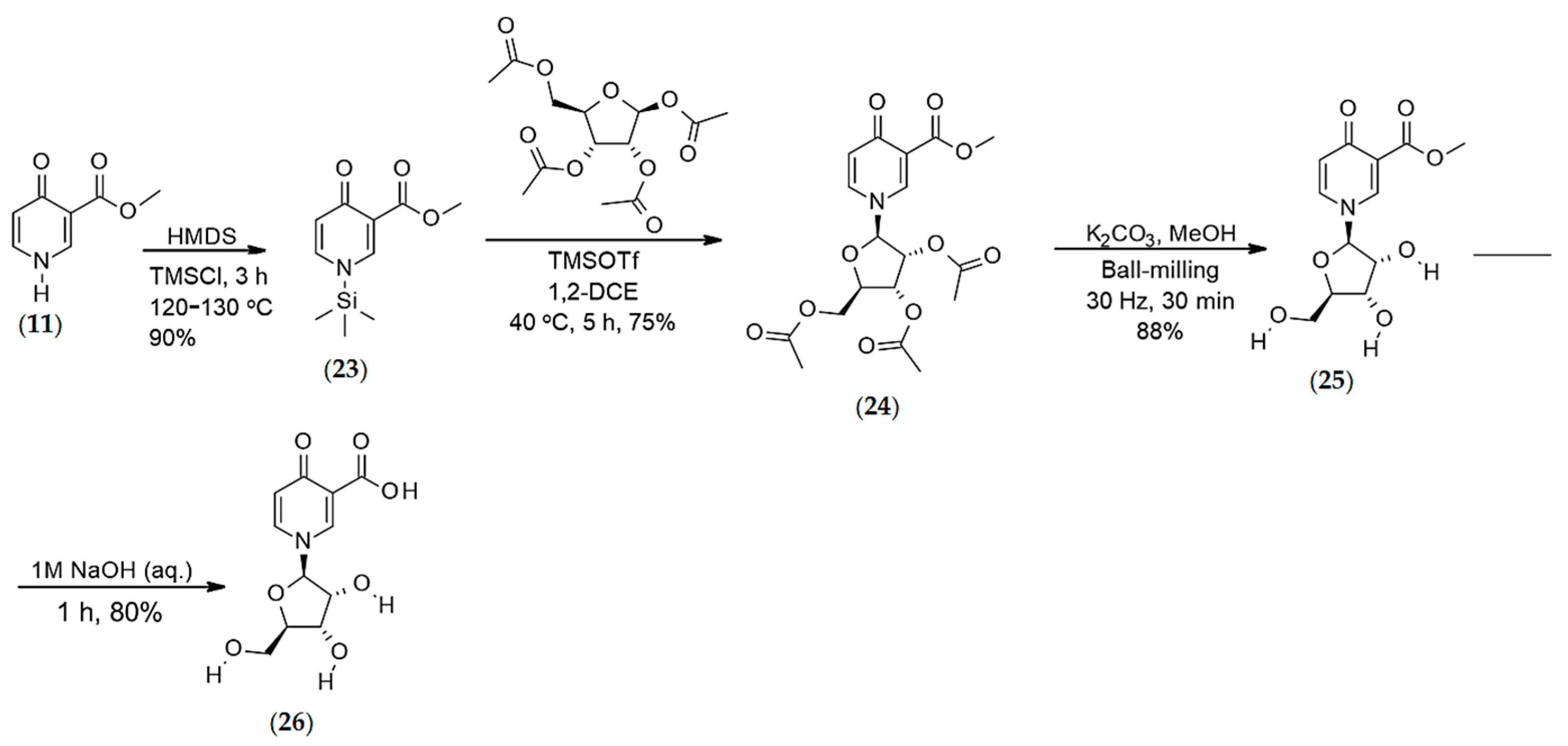
4.1.2. Synthesis of O-methyl, 1-N-methyl-4-oxo-pyridine-3-carboxylate (12)
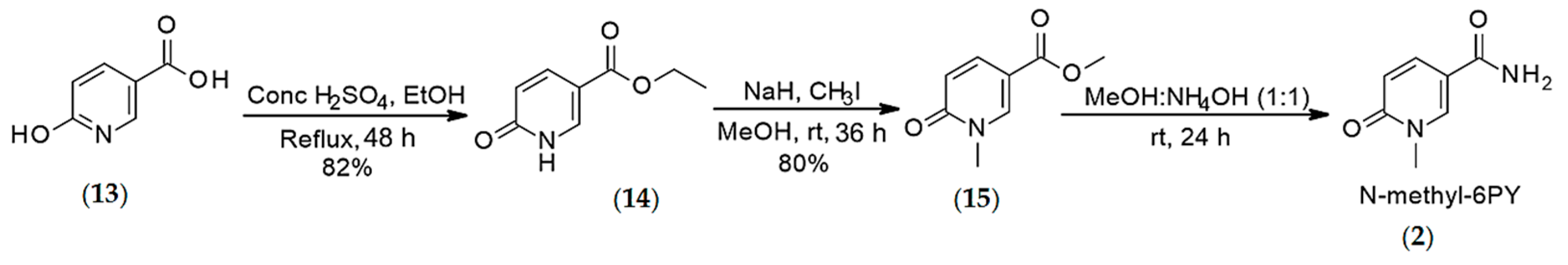
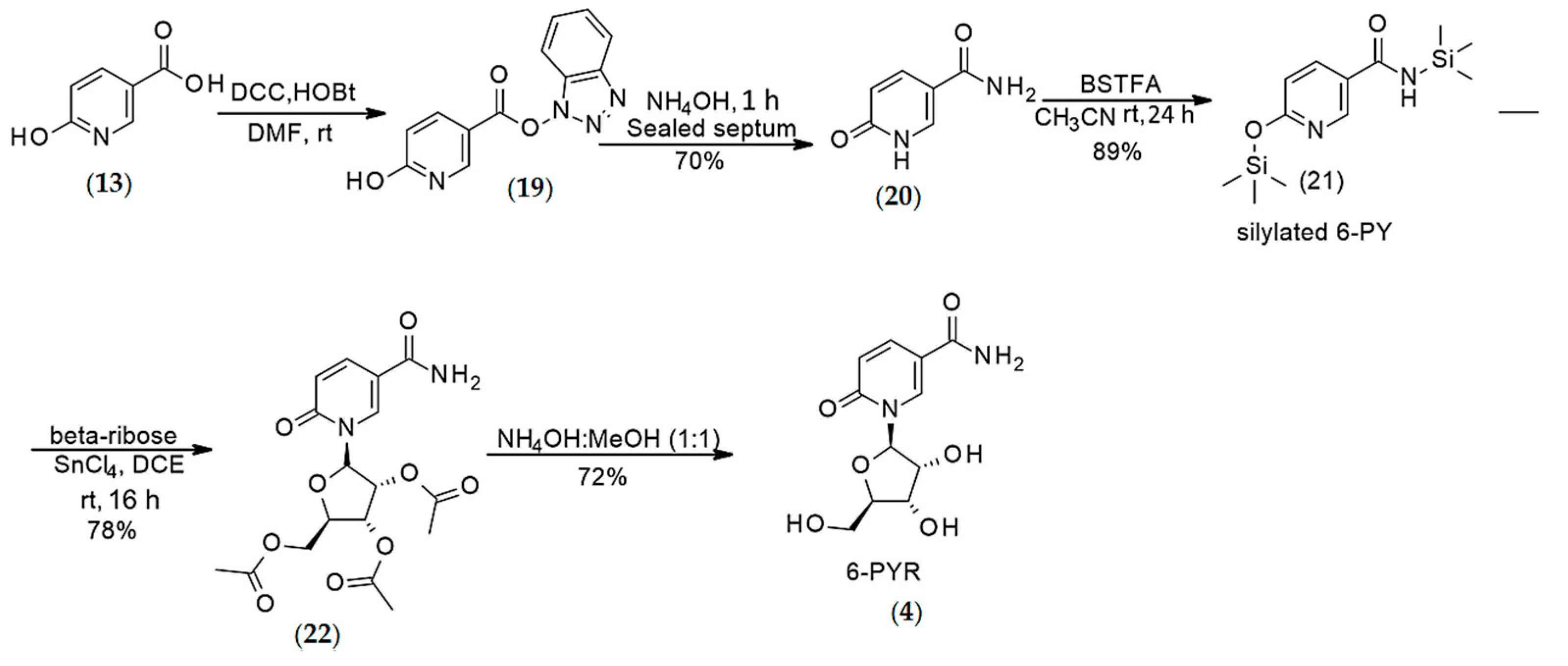
4.1.3. Synthesis of N-methyl-6-pyridone 3-carboxamide (2)
4.1.4. Synthesis of N-methyl-6-pyridone 3-carboxamide (2)
4.1.5. Synthesis of the Ribosylated Pyridone Carboxamides
Synthesis of 4-pyridone-3-carboxamide riboside (3)
Synthesis of 6-pyridone 3-carboxamide riboside (4)
Synthesis of O-methyl-4-pyridone-3-carboxylate-1-β-d-ribofuranoside (26)
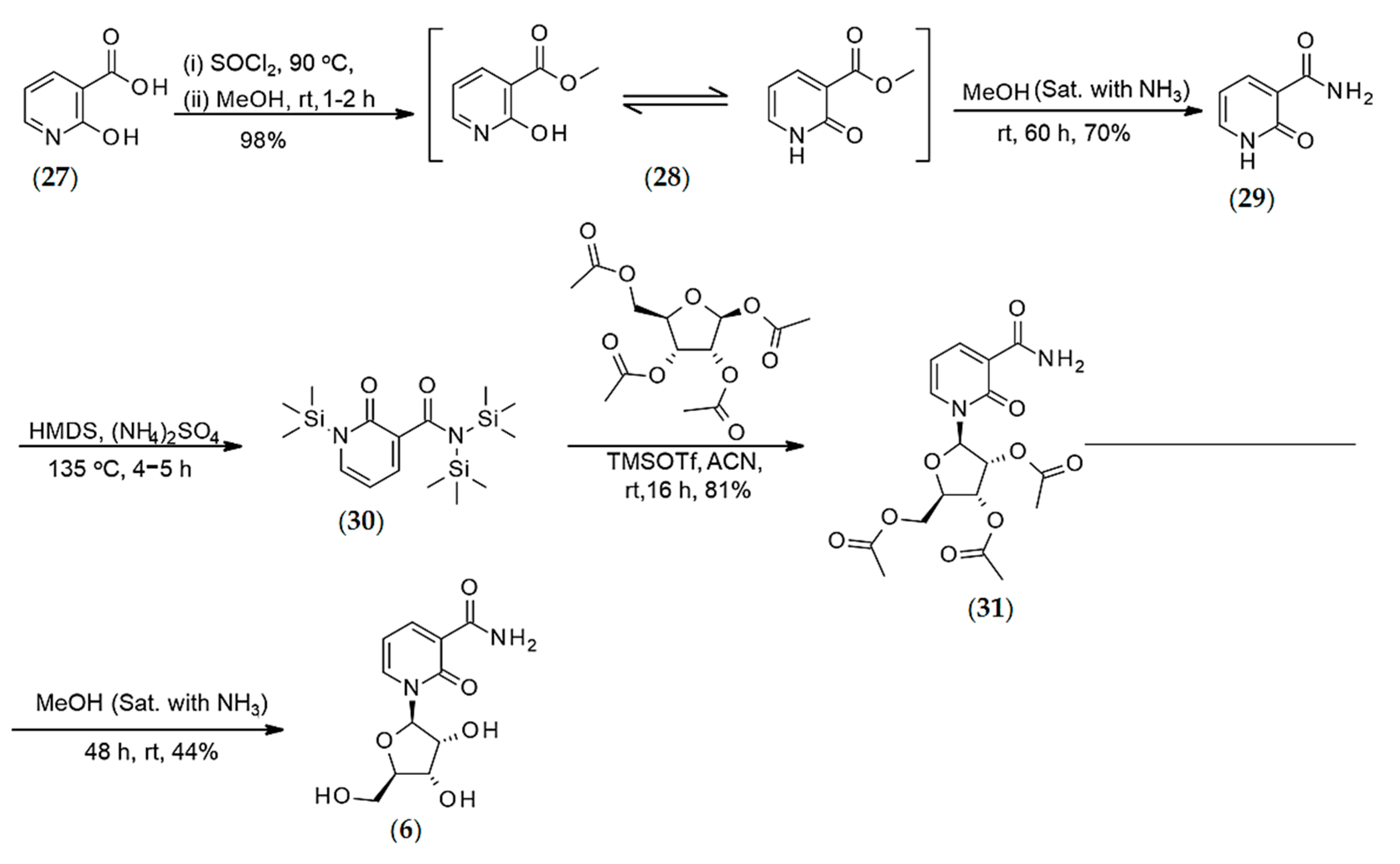
Synthesis of 2-pyridone-3-carboxamide Riboside (6)
Synthesis of 2PYR and 6PYR via Fenton Chemistry
4.2. Enzymatic Conversions
4.2.1. Synthesis of 4PYR via NQO2 Catalyzed Hyper-Oxidation of the Reduced Form of Nicotinamide Riboside (NR), Abbreviated NRH
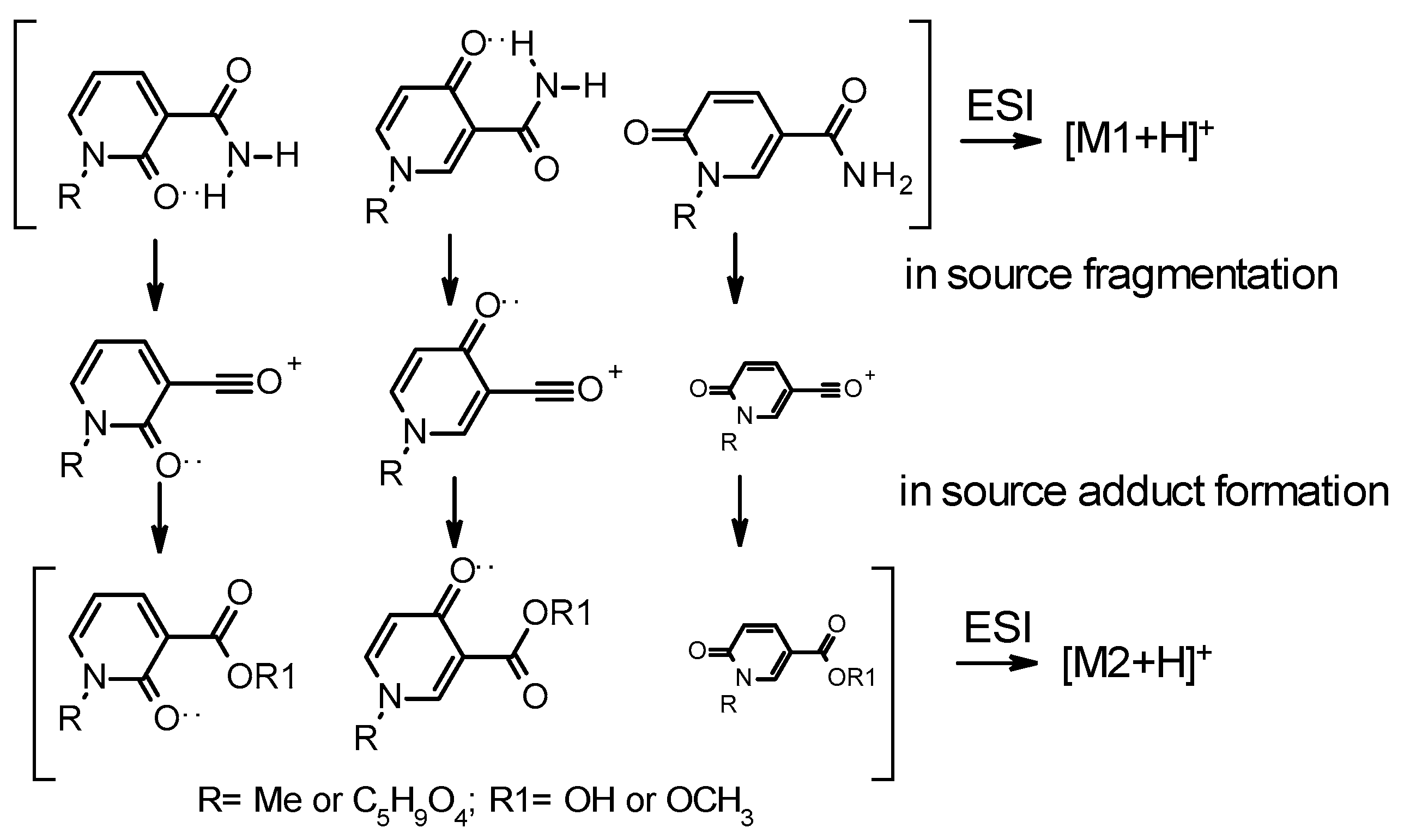
4.2.2. LC-MS/MS Analyses
4.3. Cell-Based Assays
4.3.1. Cell Culture
4.3.2. Cytotoxicity Studies
4.3.3. Clonogenic Assays
4.3.4. Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M. The chemistry of the vitamin B3 metabolome. Biochem. Soc. Trans. 2018, 47, 131–147. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, M.R.; Chellappa, K.; Baur, J.A. Age-related NAD(+) decline. Exp. Gerontol. 2020, 134, 110888. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2PY)—Major Metabolite of Nicotinamide: An Update on an Old Uremic Toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominska, E.M.; Rutkowski, P.; Smolenski, R.T.; Szutowicz, A.; Rutkowski, B.; Swierczynski, J. The age-related increase in N-methyl-2-pyridone-5-carboxamide (NAD catabolite) in human plasma. Mol. Cell. Biochem. 2004, 267, 25–30. [Google Scholar] [CrossRef]
- Rutkowski, B.; Rutkowski, P.; Słomińska, E.; Swierczyński, J. Distribution of Purine Nucleotides in Uremic Fluids and Tissues. J. Ren. Nutr. 2010, 20, 7–10. [Google Scholar] [CrossRef]
- Synesiou, E.; Fairbanks, L.D.; Simmonds, H.A.; Slominska, E.M.; Smolenski, R.T.; Carrey, E.A. 4-Pyridone-3-carboxamide-1-beta-D-ribonucleoside triphosphate (4PyTP), a novel NAD metabolite accumulating in erythrocytes of uremic children: A biomarker for a toxic NAD analogue in other tissues? Toxins 2011, 3, 520–537. [Google Scholar] [CrossRef] [Green Version]
- Carrey, E.A.; Synesiou, E.; Simmonds, H.A.; Fairbanks, L.D. The Novel Nucleotide 4KNTP, in High Concentrations in Erythrocytes of Renal Failure Children: A Comparison with Accumulation of Other Putative Precursors in the Plasma. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1051–1054. [Google Scholar] [CrossRef]
- Rutkowski, B.; Rutkowski, P.; Słomińska, E.; Smolenski, R.T.; Świerczyński, J. Cellular Toxicity of Nicotinamide Metabolites. J. Ren. Nutr. 2012, 22, 95–97. [Google Scholar] [CrossRef]
- Delaney, J.; Hodson, M.P.; Thakkar, H.; Connor, S.C.; Sweatman, B.C.; Kenny, S.P.; McGill, P.J.; Holder, J.C.; Hutton, K.A.; Haselden, J.N.; et al. Tryptophan-NAD+ pathway metabolites as putative biomarkers and predictors of peroxisome proliferation. Arch. Toxicol. 2004, 79, 208–223. [Google Scholar] [CrossRef]
- Deen, C.P.J.; van der Veen, A.; Gomes-Neto, A.W.; Geleijnse, J.M.; Berg, K.J.B.-V.D.; Heiner-Fokkema, M.R.; Kema, I.P.; Bakker, S.J.L. Urinary Excretion of N1-methyl-2-pyridone-5-carboxamide and N1-methylnicotinamide in Renal Transplant Recipients and Donors. J. Clin. Med. 2020, 9, 437. [Google Scholar] [CrossRef] [Green Version]
- Deen, C.P.J.; Veen, A.V.; Gomes-Neto, A.W.; Geleijnse, J.M.; Berg, K.; Heiner-Fokkema, M.R.; Kema, I.P.; Bakker, S.J.L. Urinary Excretion of N(1)-Methylnicotinamide and N(1)-Methyl-2-Pyridone-5-Carboxamide and Mortality in Kidney Transplant Recipients. Nutrients 2020, 12, 2059. [Google Scholar] [CrossRef] [PubMed]
- Gooding, J.; Cao, L.; Ahmed, F.; Mwiza, J.M.; Fernander, M.; Whitaker, C.; Acuff, Z.; McRitchie, S.; Sumner, S.; Ongeri, E.M. LC-MS-based metabolomics analysis to identify meprin-beta-associated changes in kidney tissue from mice with STZ-induced type 1 diabetes and diabetic kidney injury. Am. J. Physiol. Renal Physiol. 2019, 317, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Kawada, T.; Iwai, K. Microdetermination of N1-methyl-2-pyridone-5-carboxamide, a major metabolite of nicotinic acid and nicotinamide, in urine by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1987, 417, 173–177. [Google Scholar] [CrossRef]
- Shibata, K.; Matsuo, H. Levels of NAD, NADP and their related compounds in rat blood. Teikoku Gakuen Kiyo 1989, 15, 9–12. [Google Scholar]
- Slominska, E.M.; Adamski, P.; Lipiński, M.; Swierczynski, J.; Smolenski, R.T. Liquid Chromatographic/Mass Spectrometric Procedure for Measurement of NAD Catabolites in Human and Rat Plasma and Urine. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1245–1249. [Google Scholar] [CrossRef]
- Slominska, E.M.; Yuen, A.; Osman, L.; Gebicki, J.; Yacoub, M.H.; Smolenski, R.T. Cytoprotective Effects of Nicotinamide Derivatives in Endothelial Cells. Nucleosides Nucleotides Nucleic Acids 2008, 27, 863–866. [Google Scholar] [CrossRef]
- Slominska, E.M.; Orlewska, C.; Yuen, A.; Osman, L.; Romaszko, P.; Sokolowska, E.; Foks, H.; Simmonds, H.A.; Yacoub, M.H.; Smolenski, R.T. Metabolism of 4-pyridone-3-carboxamide-1-beta-D-ribonucleoside triphosphate and its nucleoside precursor in the erythrocytes. Nucleosides Nucleotides Nucleic Acids 2008, 27, 830–834. [Google Scholar] [CrossRef]
- Garcia-Perez, I.; Posma, J.M.; Serrano-Contreras, J.I.; Boulangé, C.L.; Chan, Q.; Frost, G.; Stamler, J.; Elliott, P.; Lindon, J.C.; Holmes, E.; et al. Identifying unknown metabolites using NMR-based metabolic profiling techniques. Nat. Protoc. 2020, 15, 2538–2567. [Google Scholar] [CrossRef]
- Horitsu, K. Specific basal bioconversion (biochemical conversion) related to nicotinamide methylation (metabolic process) found in the hepatocytes of rat and mouse regarding Ehrlich ascites tumor host. Kenkyu Kiyo—Tokyo Kasei Daigaku 2 Shizen Kagaku 1998, 38, 1–6. [Google Scholar]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of evo-lutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loring, H.S.; Thompson, P.R. Kinetic Mechanism of Nicotinamide N-Methyltransferase. Biochemistry 2018, 57, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Mierzejewska, P.; Gawlik-Jakubczak, T.; Jablonska, P.; Czajkowski, M.; Kutryb-Zajac, B.; Smolenski, R.T.; Matuszewski, M.; Slominska, E.M. Nicotinamide metabolism alterations in bladder cancer: Preliminary studies. Nucleosides Nucleotides Nucleic Acids 2018, 37, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, P.; Mierzejewska, P.; Kutryb-Zajac, B.; Rzyman, W.; Dziadziuszko, R.; Polanska, J.; Sitkiewicz, M.; Smolenski, R.T.; Slominska, E.M. Increased plasma concentration of 4-pyridone-3-carboxamide-1-ss-D-ribonucleoside (4PYR) in lung cancer. Pre-liminary studies. Nucleosides Nucleotides Nucleic Acids 2019, 38, 781–787. [Google Scholar] [CrossRef]
- Slominska, E.M.; Carrey, E.A.; Foks, H.; Orlewska, C.; Wieczerzak, E.; Sowinski, P.; Yacoub, M.H.; Marinaki, A.M.; Simmonds, H.A.; Smolenski, R.T. A novel nucleotide found in human erythrocytes, 4-pyridone-3-carboxamide-1-beta-D-ribonucleoside triphosphate. J. Biol. Chem. 2006, 281, 32057–32064. [Google Scholar] [CrossRef] [Green Version]
- Pelikant-Malecka, I.; Sielicka, A.; Kaniewska, E.; Smolenski, R.T.; Slominska, E.M. 4-Pyridone-3-carboxamide-1beta-D-ribonucleoside metabolism in endothelial cells and its impact on cellular energetic balance. Nucleosides Nucleotides Nucleic Acids 2014, 33, 338–341. [Google Scholar] [CrossRef]
- Pelikant-Malecka, I.; Kaniewska-Bednarczuk, E.; Szrok, S.; Sielicka, A.; Sledzinski, M.; Orlewska, C.; Smolenski, R.T.; Slominska, E.M. Metabolic pathway of 4-pyridone-3-carboxamide-1beta-d-ribonucleoside and its effects on cellular energetics. Int. J. Biochem. Cell. Biol. 2017, 88, 31–43. [Google Scholar] [CrossRef]
- Pelikant-Malecka, I.; Smolenski, R.T.; Slominska, E.M. Metabolism of 4-pyridone-3-carboxamide-1beta-d-ribonucleoside (4PYR) in primary murine brain microvascular endothelial cells (mBMECs). Nucleosides Nucleotides Nucleic Acids 2018, 37, 639–644. [Google Scholar] [CrossRef]
- Lang, R.; Wahl, A.; Skurk, T.; Yagar, E.F.; Schmiech, L.; Eggers, R.; Hauner, H.; Hofmann, T. Development of a Hydrophilic Liquid Interaction Chromatography−High-Performance Liquid Chromatography−Tandem Mass Spectrometry Based Stable Isotope Dilution Analysis and Pharmacokinetic Studies on Bioactive Pyridines in Human Plasma and Urine after Coffee Consumption. Anal. Chem. 2010, 82, 1486–1497. [Google Scholar] [CrossRef]
- Landelle, G.; Schmitt, E.; Panossian, A.; Vors, J.-P.; Pazenok, S.; Jeschke, P.; Gutbrod, O.; Leroux, F.R. Tri- and difluoro-methoxylated N-based heterocycles—Synthesis and insecticidal activity of novel F3CO—And F2HCO—Analogues of Imidacloprid and Thiacloprid. J. Fluor. Chem. 2017, 203, 155–165. [Google Scholar] [CrossRef]
- Frister, H.; Kemper, K.; Boos, K.-S.; Schlimme, E. Darstellung des Coenzymmetaboliten 1,6-Dihydro-6-oxo-1-(β-D-ribofuranosyl)-3-pyridincarbonsäureamid. Eur. J. Org. Chem. 1985, 1985, 510–516. [Google Scholar] [CrossRef]
- Hanna, N.B.; Joshi, R.V.; Larson, S.B.; Robins, R.K.; Revankar, G.R. Synthesis of certain 1-β-D-ribofuranosyl-1,2-dihydro-2-oxopyridines structurally related to nicotinamide ribonucleoside. J. Heterocycl. Chem. 1989, 26, 1835–1843. [Google Scholar] [CrossRef]
- Holman, W.I.M.; Wiegand, C. The chemical conversion of nicotinic acid and nicotinamide to derivatives of N-methyl-2-pyridone by methylation and oxidation. Biochem. J. 1948, 43, 423–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, C.; Holman, W.I.M. Synthesis of Derivatives of N-Methyl-2-Pyridone from Nicotinic Acid and Nicotinamide. Nat. Cell. Biol. 1948, 162, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Peretz, H.; Watson, D.G.; Blackburn, G.; Zhang, T.; Lagziel, A.; Shtauber-Naamati, M.; Morad, T.; Keren-Tardai, E.; Greenshpun, V.; Usher, S.; et al. Urine metabolomics reveals novel physiologic functions of human aldehyde oxidase and provides biomarkers for typing xanthinuria. Metabolomics 2011, 8, 951–959. [Google Scholar] [CrossRef]
- Kitamura, S.; Nitta, K.; Tayama, Y.; Tanoue, C.; Sugihara, K.; Inoue, T.; Horie, T.; Ohta, S. Aldehyde Oxidase-Catalyzed Metabolism of N1-Methylnicotinamide in Vivo and in Vitro in Chimeric Mice with Humanized Liver. Drug Metab. Dispos. 2008, 36, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Kitamura, S.; Tatsumi, K.; Asahara, T.; Dohi, K. Differences in aldehyde oxidase activity in cytosolic prepara-tions of human and monkey liver. Biochem. Mol. Biol. Int. 1997, 41, 1153–1160. [Google Scholar]
- Hanukoglu, I. Conservation of the Enzyme—Coenzyme Interfaces in FAD and NADP Binding Adrenodoxin Reductase—A Ubiquitous Enzyme. J. Mol. Evol. 2017, 85, 205–218. [Google Scholar] [CrossRef]
- de Rosa, M.; Pennati, A.; Pandini, V.; Monzani, E.; Zanetti, G.; Aliverti, A. Enzymatic oxidation of NADP+ to its 4-oxo de-rivative is a side-reaction displayed only by the adrenodoxin reductase type of ferredoxin-NADP+ reductases. FEBS J. 2007, 274, 3998–4007. [Google Scholar] [CrossRef]
- Bossi, R.T.; Aliverti, A.; Raimondi, D.; Fischer, F.; Zanetti, G.; Ferrari, D.; Tahallah, N.; Maier, C.S.; Heck, A.J.R.; Rizzi, M.; et al. A covalent modification of NADP+ revealed by the atomic resolution structure of FprA, a Mycobacterium tuberculosis oxidoreductase. Biochemistry 2002, 41, 8807–8818. [Google Scholar] [CrossRef]
- Huntley, C.M.; Cotterill, A.S.; Maillard, J.-Y.; Balzarini, J.; Simons, C. Synthesis and biological evaluation of pyridine-2-one nucleosides. Nucleosides Nucleotides Nucleic Acids 2001, 20, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.T.; Eberlin, M.N.; Simionato, A.V.C. Targeted metabolomics: Liquid chromatography coupled to mass spectrometry method development and validation for the identification and quantitation of modified nucleosides as putative cancer biomarkers. Talanta 2020, 210, 120640. [Google Scholar] [CrossRef] [PubMed]
- Willmann, L.; Erbes, T.; Krieger, S.; Trafkowski, J.; Rodamer, M.; Kammerer, B. Metabolome analysis via comprehensive two-dimensional liquid chromatography: Identification of modified nucleosides from RNA metabolism. Anal. Bioanal. Chem. 2015, 407, 3555–3566. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Koenigs, L.L.; Thompson, S.J.; Peter, R.M.; Rettie, A.E.; Trager, W.F.; Nelson, S.D. Oxidation of Acetaminophen to Its Toxic Quinone Imine and Nontoxic Catechol Metabolites by Baculovirus-Expressed and Purified Human Cytochromes P450 2E1 and 2A6. Chem. Res. Toxicol. 1998, 11, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Braver-Sewradj, S.P.D.; Braver, M.W.D.; Toorneman, R.M.; van Leeuwen, S.; Zhang, Y.; Dekker, S.J.; Vermeulen, N.P.E.; Commandeur, J.N.M.; Vos, J.C. Reduction and Scavenging of Chemically Reactive Drug Metabolites by NAD(P)H:Quinone Oxidoreductase 1 and NRH:Quinone Oxidoreductase 2 and Variability in Hepatic Concentrations. Chem. Res. Toxicol. 2018, 31, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Slominska, E.M.; Borkowski, T.; Rybakowska, I.; Abramowicz-Glinka, M.; Orlewska, C.; Smolenski, R.T. In Vitro and Cellular Effects of 4-pyridone-3-carboxamide riboside on Enzymes of Nucleotide Metabolism. Nucleosides Nucleotides Nucleic Acids 2014, 33, 353–357. [Google Scholar] [CrossRef]
- Sonavane, M.; Hayat, F.; Makarov, M.; Migaud, M.E.; Gassman, N. Dihydronicotinamide riboside promotes cell-specific cytotoxicity by tipping the balance between metabolic regulation and oxidative stress. PLoS ONE 2020, 15, e0242174. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, C.; Sano, M.; Fukuwatari, T.; Shibata, K. Time-Dependent Effects of L-Tryptophan Administration on Urinary Excretion of L-Tryptophan Metabolites. J. Nutr. Sci. Vitaminol. 2014, 60, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Pelantová, H.; Bugáňová, M.; Holubová, M.; Sediva, B.; Zemenová, J.; Sýkora, D.; Kavalkova, P.; Haluzík, M.; Železná, B.; Maletínská, L.; et al. Urinary metabolomic profiling in mice with diet-induced obesity and type 2 diabetes mellitus after treatment with metformin, vildagliptin and their combination. Mol. Cell. Endocrinol. 2016, 431, 88–100. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.S.; Sordillo, J.; Lasky-Su, J.; Dahlin, A.; Perng, W.; Rifas-Shiman, S.L.; Weiss, S.T.; Gold, D.R.; Litonjua, A.A.; Hivert, M.-F.; et al. Plasma metabolite profiles in children with current asthma. Clin. Exp. Allergy 2018, 48, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Joo, J.; Park, B.; Park, S.J.; Lee, W.J.; Han, S.S.; Kim, T.H.; Hong, E.K.; Woo, S.M.; Yoo, B.C. Reduced levels of N’-methyl-2-pyridone-5-carboxamide and lysophosphatidylcholine 16:0 in the serum of patients with intrahepatic cholangiocarci-noma, and the correlation with recurrence-free survival. Oncotarget 2017, 8, 112598–112609. [Google Scholar] [CrossRef] [Green Version]
- Lenglet, A.; Liabeuf, S.; el Esper, N.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.-S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The NICOREN study. Nephrol. Dial. Transplant. 2016, 32, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Tsalik, E.L.; Willig, L.K.; Rice, B.J.; van Velkinburgh, J.C.; Mohney, R.P.; McDunn, J.E.; Dinwiddie, D.L.; Miller, N.A.; Mayer, E.S.; Glickman, S.W.; et al. Renal systems biology of patients with systemic inflammatory response syndrome. Kidney Int. 2015, 88, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Fukuwatari, T.; Suzuki, C. Pharmacological doses of nicotinic acid and nicotinamide are independently metab-olized in rats. J. Nutr. Sci. Vitaminol. 2014, 60, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Gillmor, H.A.; Bolton, C.H.; Hopton, M.; Moore, W.P.T.; Perrett, D.; Bingley, P.J.; Gale, E.A. Measurement of nicotinamide and N-methyl-2-pyridone-5-carboxamide in plasma by high performance liquid chromatography. Biomed. Chromatogr. 1999, 13, 360–362. [Google Scholar] [CrossRef]
- Peron, G.; Zengin, G.; Sut, S. Supplementation with resveratrol as Polygonum cuspidatum Sieb. et Zucc. extract induces changes in the excretion of urinary markers associated to aging in rats. Fitoterapia 2018, 129, 154–161. [Google Scholar] [CrossRef]
- Zhou, H.; Li, L.; Wu, C.; Kurtán, T.; Mándi, A.; Liu, Y.; Gu, Q.; Zhu, T.; Guo, P.; Li, D. Penipyridones A–F, Pyridone Alkaloids from Penicillium funiculosum. J. Nat. Prod. 2016, 79, 1783–1790. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Philip, R.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Maeta, A.; Sano, M.; Fukuwatari, T.; Shibata, K. Simultaneous measurement of nicotinamide and its catabolites, nicotinamideN-oxide, N1-methyl-2-pyridone-5-carboxamide, andN1-methyl-4-pyridone-3-carboxamide, in mice urine. Biosci. Biotechnol. Biochem. 2014, 78, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Steckel, A.; Schlosser, G. An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation. Molecules 2019, 24, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trammell, S.A.; Brenner, C. Targeted, LCMS-based Metabolomics for Quantitative Measurement of NAD(+) Metabolites. Comput. Struct. Biotechnol. J. 2013, 4, e201301012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, J.I.; Gömpel, K.; Bakuradze, T.; Eisenbrand, G.; Richling, E. Urinary Excretion of Niacin Metabolites in Humans After Coffee Consumption. Mol. Nutr. Food Res. 2018, 62, e1700735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelikant-Malecka, I.; Sielicka, A.; Kaniewska, E.; Smolenski, R.T.; Slominska, E.M. Influence of 4-pyridone-3-carboxamide-1Beta-D-ribonucleoside (4PYR) on activities of extracellular enzymes in endothelial human cells. Nucleosides Nucleotides Nucleic Acids 2016, 35, 732–736. [Google Scholar] [CrossRef]
- Shibata, K.; Fukuwatari, T. Pyridone compounds, catabolites of NAD are new uremic toxins. Bitamin 2007, 81, 571–574. [Google Scholar]
- Yagi, K.; Ohishi, N. Hydroxylation of Riboflavin 7- and 8-Methyl Groups in Mammals; De Gruyter: Berlin, Germany, 1984; pp. 819–832. [Google Scholar]
- Houee-Levin, C.; Bobrowski, K.; Horakova, L.; Karademir, B.; Schoeneich, C.; Davies, M.J.; Spickett, C.M. Exploring oxidative modifications of tyrosine: An update on mechanisms of formation, advances in analysis and biological consequences. Free Radic. Res. 2015, 49, 347–373. [Google Scholar]
- Halliwell, B. The Chemistry of Free Radicals. Toxicol. Ind. Health 1993, 9, 1–21. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (Solvent) | Chemical Shift (Multiplicity, J Values in Hz) | |||||
|---|---|---|---|---|---|---|
| NH | H2 | H3 | H4 | H5 | H6 | |
| 2PY (DMSO-d6) | 12.3 (s), 9.05 (s), 7.51 (s) | 7.66 (d, J = 4.32 Hz) | 6.41 (t, J = 6.66 Hz) | 8.29 (d, J = 5.28 Hz) | ||
| 2PY (DMSO-d6) [25] | 11.80 (s), 9.02 (s), 7.57 (s) | 7.69 (q, 1H) | 6.44 (t) | 8.31 (q) | ||
| 4PY (CD3OD) | 8.54 (s) | 6.56 (d, J = 6.53 Hz) | 7.79 (d, J = 5.56 Hz) | |||
| 4PY (CD3OD) [25] | 8.56 (d, J = 1.7 Hz) | 6.58 (d, J = 7.2 Hz) | 7.80 (dd, J = 7.2, 1.7 Hz) | |||
| 6PY (DMSO-d6) | 11.92(s), 7.72 (s) 7.18 (s) | 8.00 (s) | 7.85 (d, J = 9.6 Hz) | 6.32 (d, J = 9.56 Hz) | ||
| 6PY (DMSO-d6) [31] | 11.84 (s), 7.71 (s), 7.18 (s) | 7.99 (d, J = 2.6 Hz) | 7.82–7.87 (dd, J = 2.6 & 9.5 Hz) | 6.32 (d, J = 9.5 Hz) | ||
| Compound (Solvent) | Chemical Shift (Multiplicity, J Values in Hz) | ||||||
|---|---|---|---|---|---|---|---|
| NH | H2 | H3 | H4 | H5 | H6 | CH3 | |
| N-Me-2PY (DMSO-d6) | 9.06 (s) 7.54 (s) | 8.01 (dd, J = 2.16 & 2.16 Hz) | 6.44 (t, J = 6.88 Hz) | 8.27 (dd, J = 2.20; 2.16 Hz) | 3.08 (s) | ||
| N-Me-2PY * (DMSO-d6) [29] | 9.09 (brs, 1H, H-Nb), 7.57 (brs, 1H, H-Na) | 8.31 (dd, J = 7.21, 2.21 Hz) | 6.47 (dd, J = 7.10, 7.10 Hz) | 8.04 (dd, J = 6.55, 2.21 Hz) | 3.56 (s) | ||
| N-Me-4PY (DMSO-d6) | 9.36 (s), 7.54 (s) | 8.54 (s) | 6.53 (d, J = 7.4 Hz) | 7.87 (d, J = 5.08 Hz) | 3.82 (s) | ||
| N-Me-4PY * (DMSO-d6)2 [29] | 9.56 (brs), 7.41 (brs) | 7.74 (dd, J = 2.44, 7.49 Hz) | 6.38 (d, J = 7.49 Hz) | 8.44 (d, J = 2.44 Hz) | 3.75 (s) | ||
| N- Me-6PY (D2O) | 8.38 (s) | 7.95 (d, 1H, J = 6.69 Hz) | 6.53 (d, J = 9.44 Hz) | 3.62 (s) | |||
| N- Me-6PY * (DMSO-d6) [29] | 7.23 (brs), 7.69 (brs) | 6.38 (d, J = 9.49 Hz) | 7.85 (dd, J = 9.49, 2.54 Hz) | 8.36 (d, J = 2.54 Hz) | 3.46 (s) | ||
| Compound (Solvent) | Chemical Shift (Multiplicity, J Values in Hz) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NH | H2 | H3 | H4 | H5 | H6 | H1’ | H2’ | H3’ | H4’ | H5’ | CH3 | |
| 2PYR (D2O) | 8.46 (dd, J = 5.4, 7.04 Hz, 2H, H4 & H6), | 6.14 (s), | 4.12 (bs, 3H, C2’,3’,4’H) | 3.97 (d, J = 12.9 Hz), 3.80 (d, J = 11.56 Hz) | ||||||||
| 6.55 (t, J = 6.96 Hz) | ||||||||||||
| 2PYR (DMSO-d6) [32] | 7.63 and 8.96 (2 brs, CONH2) | 8.32 (m) | 6.55 (t) | 8.42 (m) | 6.08 (d, J = 1.17 Hz) | 5.55 (d, 1H (C2’OH) | 5.10 (d, 1H, C3’OH) | 5.23 (t, 1H, C5’OH), 3.64 (m) | ||||
| 3.79–3.97 (m, 3H) | ||||||||||||
| 4PYR (D2O) | 8.67 (d, J = 2.24 Hz) | 6.65 (d, J = 7.6 Hz) | 7.94 (dd, J = 2.24 Hz, 2.24 Hz) | 5.55 (d, J = 5.5 Hz) | 4.23 (t, J = 5.28 Hz) | 4.19–4.13 (m) | 3.81 and 3.73 (ABX, 2H, JAB = 12.7 Hz, JAX = 3.04 Hz, JBX = 4.0 Hz) | |||||
| 4PYR (D2O) [25] | 8.66 (d, J = 2.4 Hz) | 6.58 (d, J = 7.8) | 7.93 (dd, J = 7.8, 2.4 Hz | 5.53 (d, J = 5.9) | 4.22 (t, J = 5.4) | 4.16 (dd, J = 5.1, 3.5) | 4.14 (ddd, J = 4.0, 3.5, 3.0 Hz) | 3.72 (dd, J = 12.6, 3.0 Hz), 3.79 (dd, J = 12.6, 3.0 Hz) | ||||
| 6PYR (D2O) | 8.60 (s) | 7.84 (d, J = 9.2 Hz) | 6.54 (d, J = 9.44 Hz) | 6.01 (s) | 4.19–4.13 (m) | 3.97 (d, J = 12.92 Hz), 3.80 (d, J = 12.88 Hz) | ||||||
| 6PYR (DMSO-d6) [31] | 7.54(1 H, Amid-NH), 7.30 (s) | 8.52 (d, J = 1.9 Hz) | 7.81–7.86 (dd, J = 1.9 Hz, 9.5 Hz) | 6.40 (d, J = 9.5 Hz) | 6.01 (d, J = 3.5 Hz) | 5.45 (d, J = 4.6 Hz,1H, C2’ OH) | 5.07 (d, J = 4.9 Hz, 1 H, C3’ OH) | 5.17 (t, J = 4.5 Hz, C5’OH, 3.57–3.76 (mb) | ||||
| 3.92–4.08 (m, 3H, C2’,3’,4’H) | ||||||||||||
| 4PYR ACID (D2O) | 8.21 (d, J = 2.4 Hz) | 6.52 (d, J = 7.6 Hz) | 7.89 (dd, J = 7.6, 2.4 Hz) | 5.51 (d, J = 5.6 Hz) | 4.26 (dd, J = 5.6 and 5.3 Hz) | 4.20 (dd, J = 5.3 and 3.6 Hz) | 4.15 (dd, J = 8.3, 3.6 Hz) | 3.82 (dd, J = 12.8, 4.5 Hz), 3.74 (dd, J = 12.8, 4.5 Hz) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayat, F.; Sonavane, M.; Makarov, M.V.; Trammell, S.A.J.; McPherson, P.; Gassman, N.R.; Migaud, M.E. The Biochemical Pathways of Nicotinamide-Derived Pyridones. Int. J. Mol. Sci. 2021, 22, 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031145
Hayat F, Sonavane M, Makarov MV, Trammell SAJ, McPherson P, Gassman NR, Migaud ME. The Biochemical Pathways of Nicotinamide-Derived Pyridones. International Journal of Molecular Sciences. 2021; 22(3):1145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031145
Chicago/Turabian StyleHayat, Faisal, Manoj Sonavane, Mikhail V. Makarov, Samuel A. J. Trammell, Pamela McPherson, Natalie R. Gassman, and Marie E. Migaud. 2021. "The Biochemical Pathways of Nicotinamide-Derived Pyridones" International Journal of Molecular Sciences 22, no. 3: 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031145