Genomic Adaption and Mutational Patterns in a HaCaT Subline Resistant to Alkylating Agents and Ionizing Radiation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
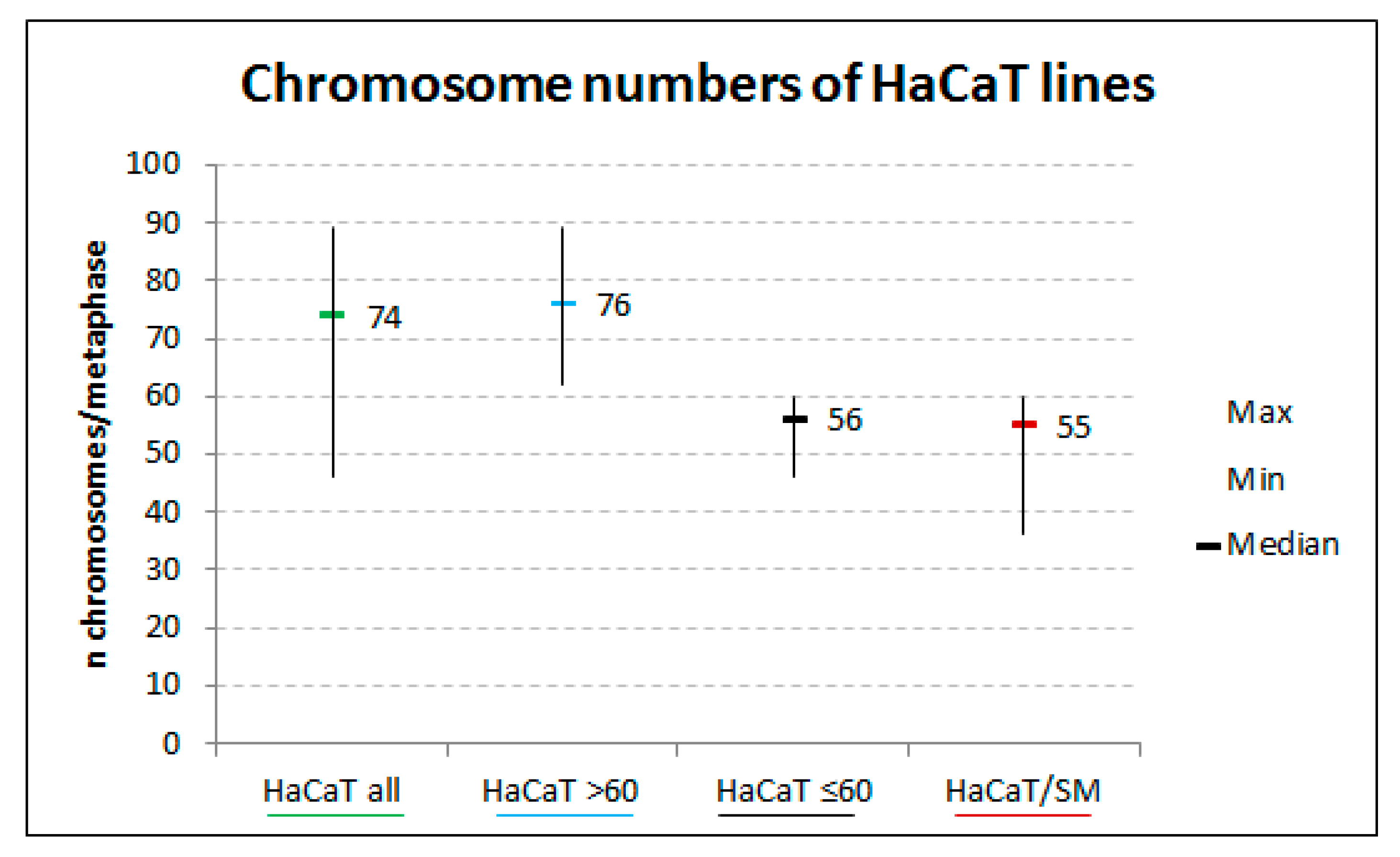
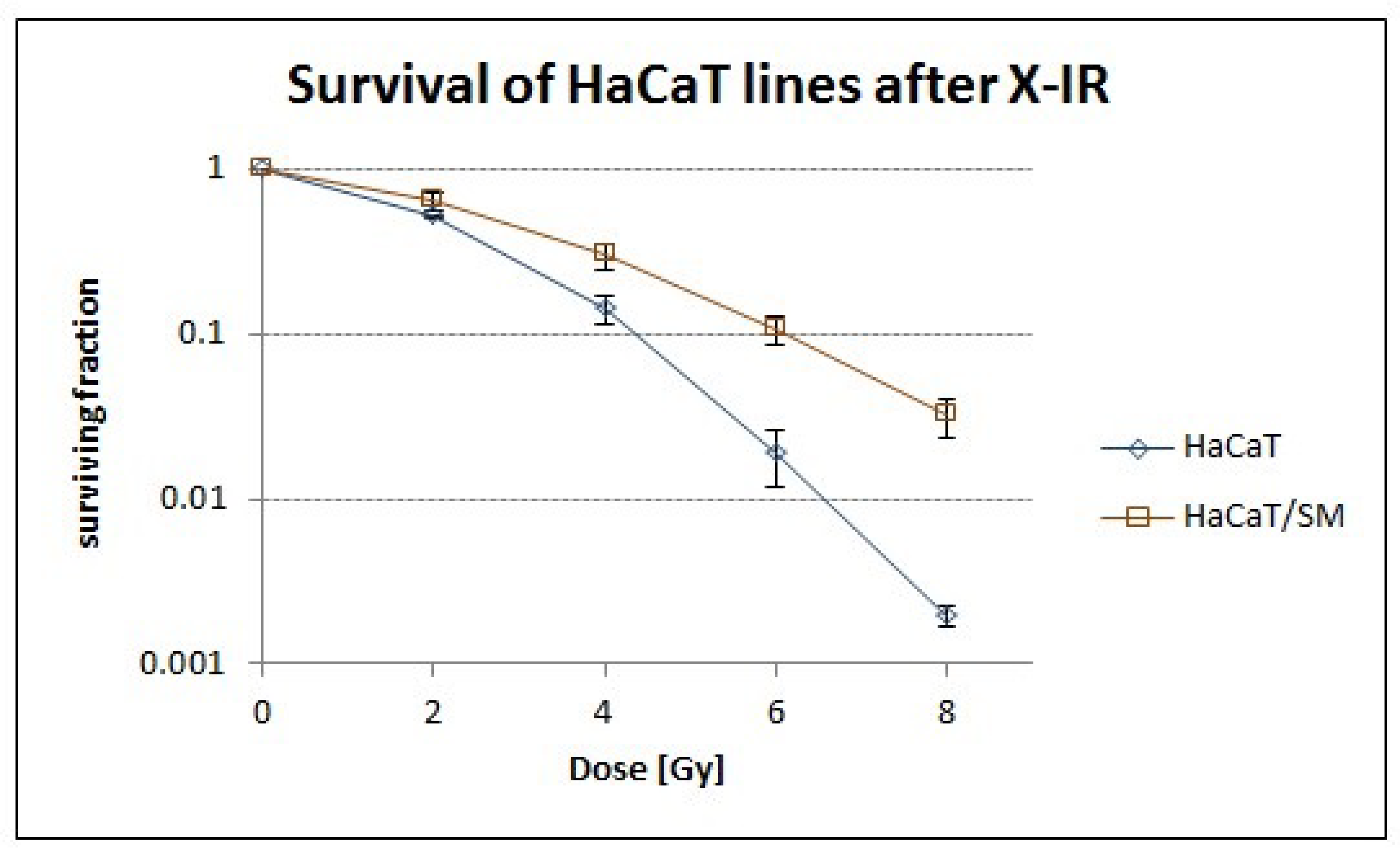
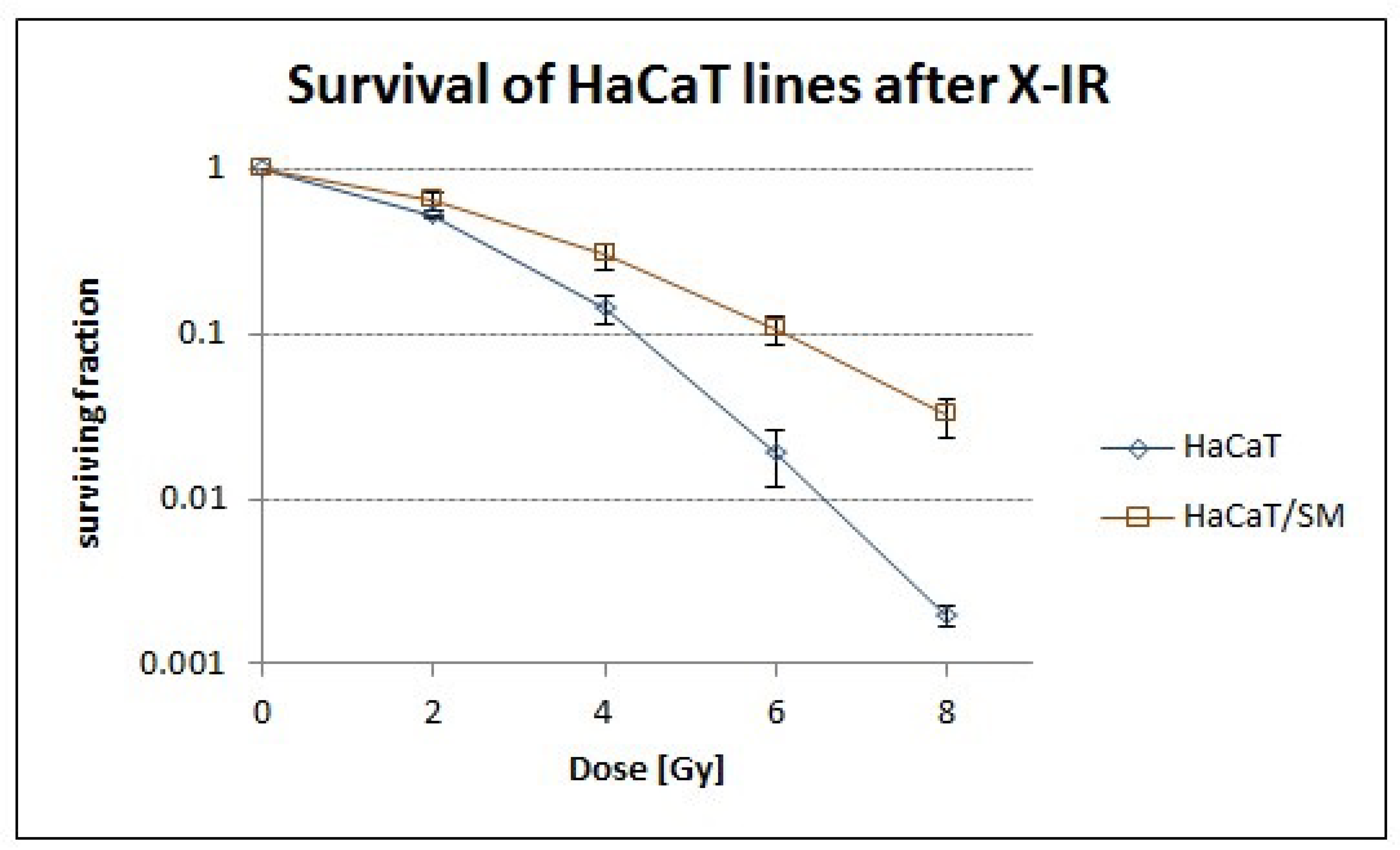
2.1. Sulfur Mustard Exposure Promoted Clonal Expansion of a Hyperdiploid Cell Population with Higher Tolerance to Ionizing Radiation
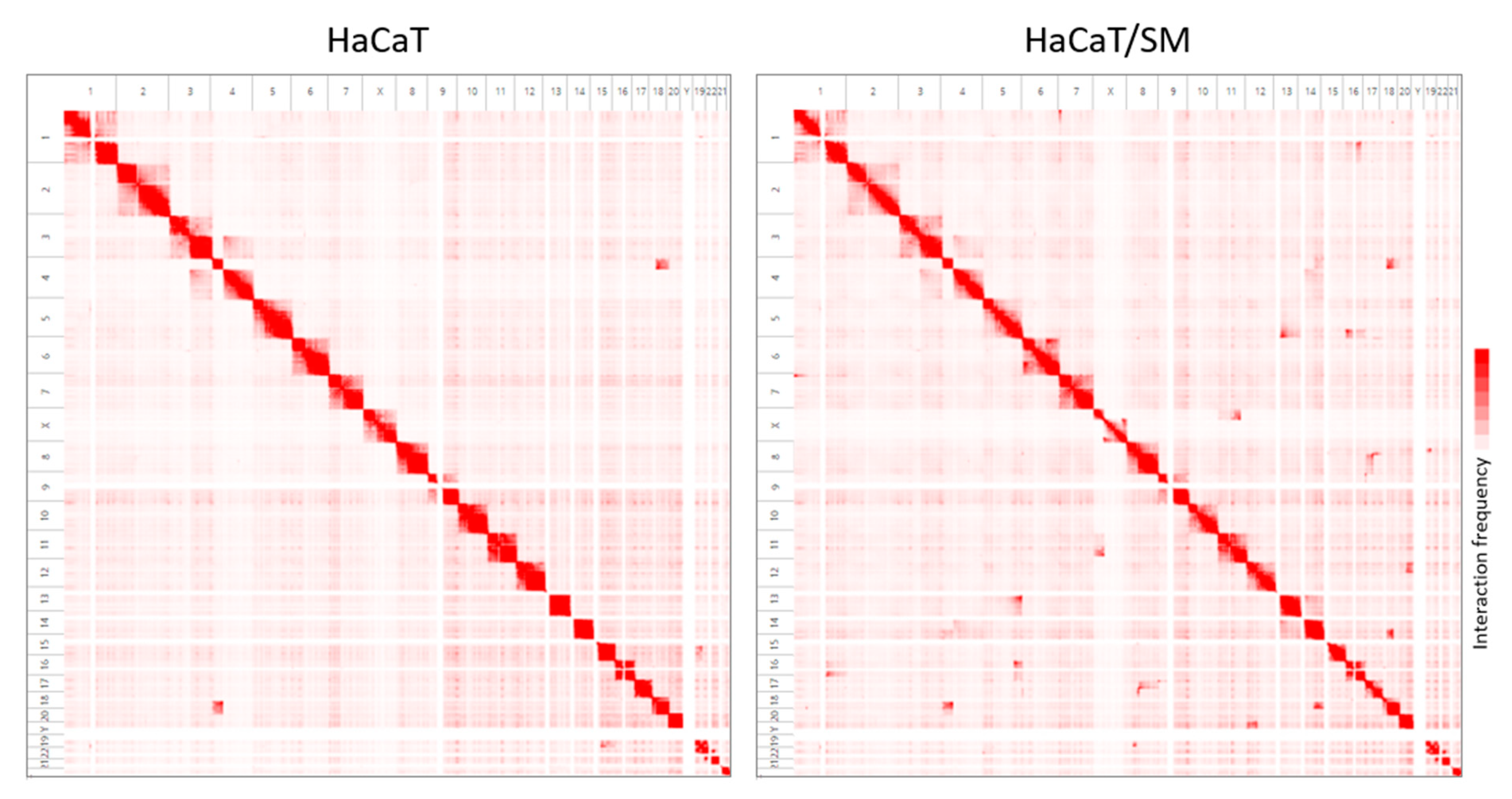
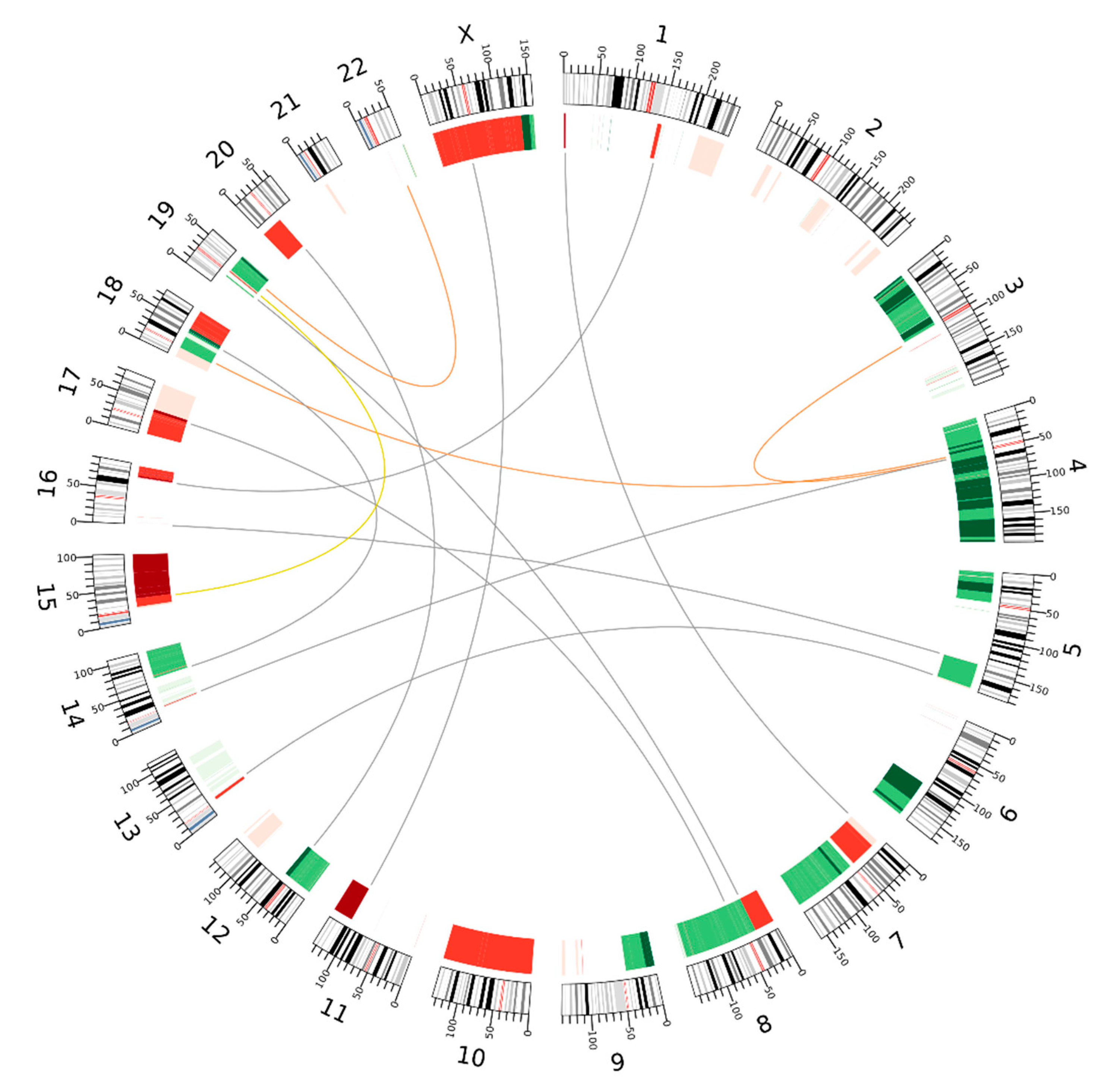
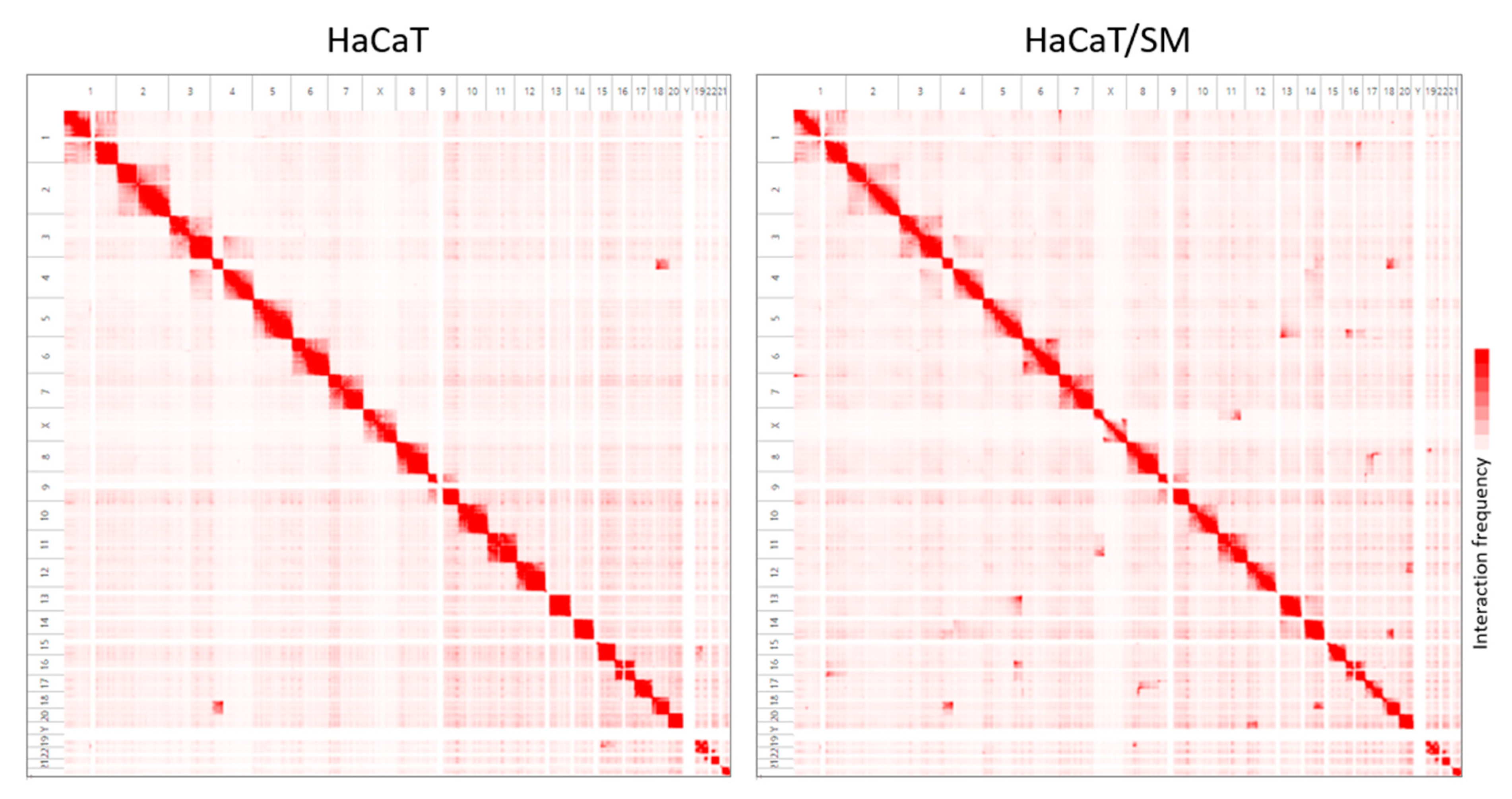
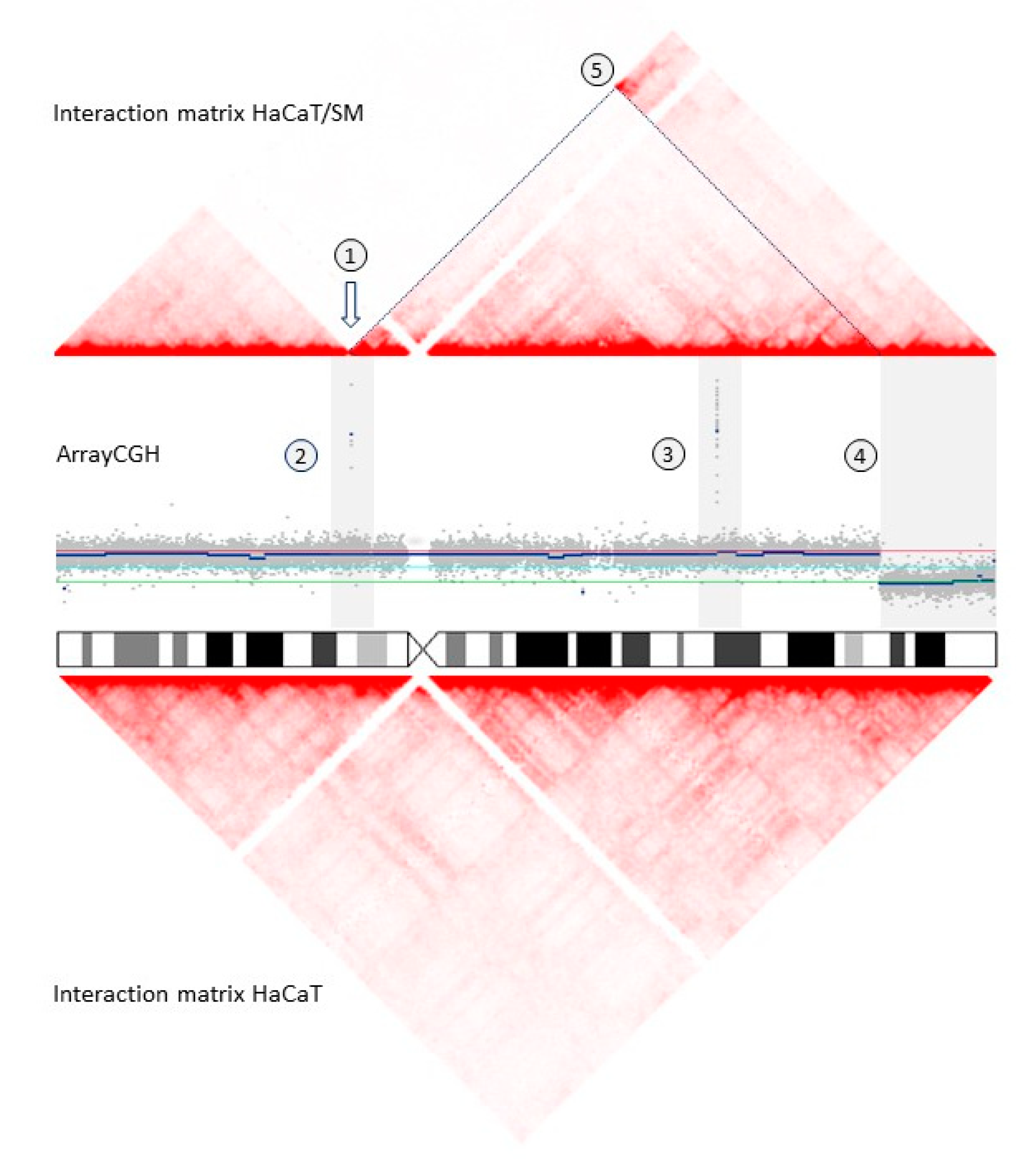
2.2. Shared and Unique Structural Chromosomal Aberrations in HaCaT and HaCaT/SM
2.3. Genomic Characteristics at Sites of DNA Double-Strand Breaks
2.4. Genome-Wide Distribution of Single-Nucleotide Variants
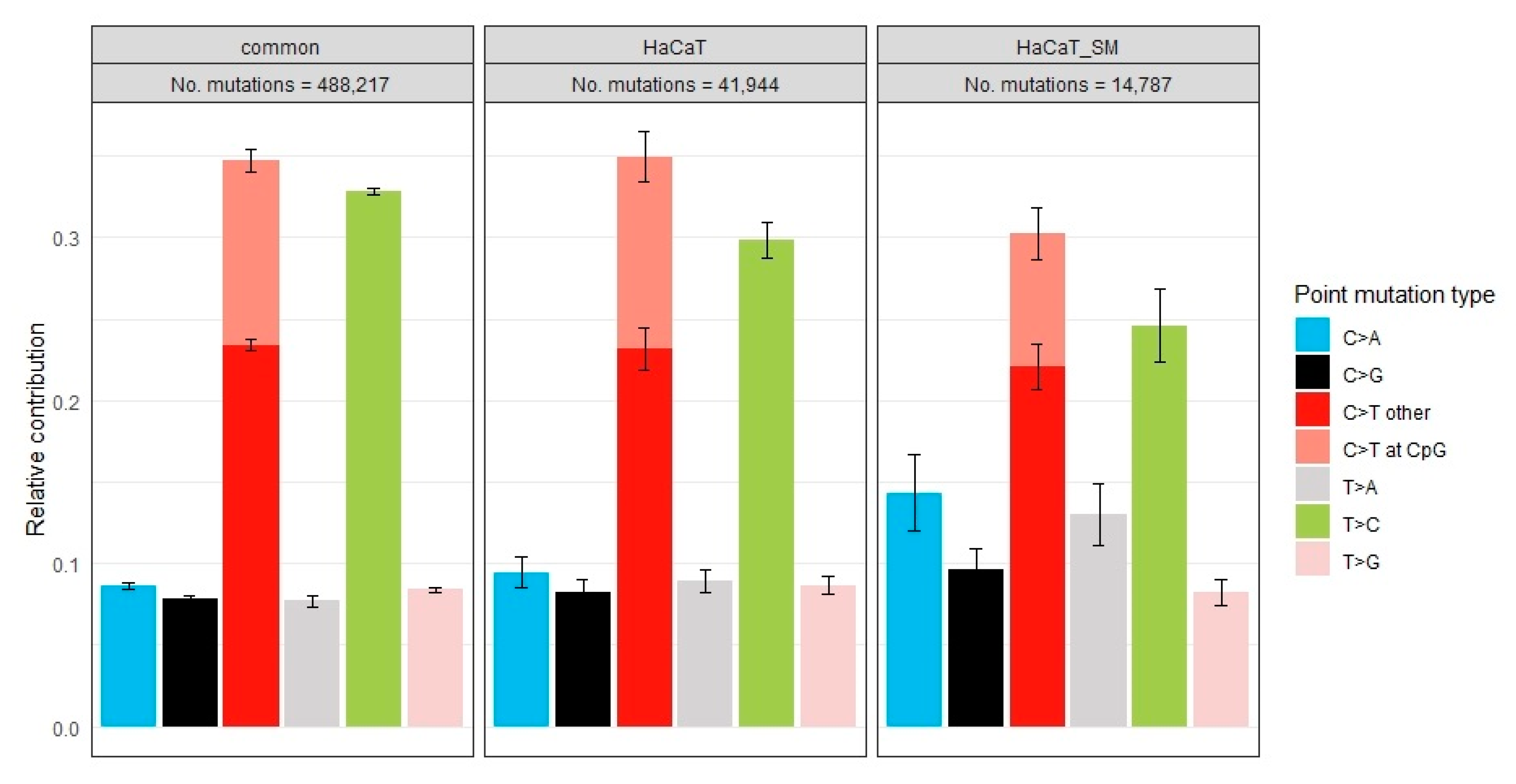
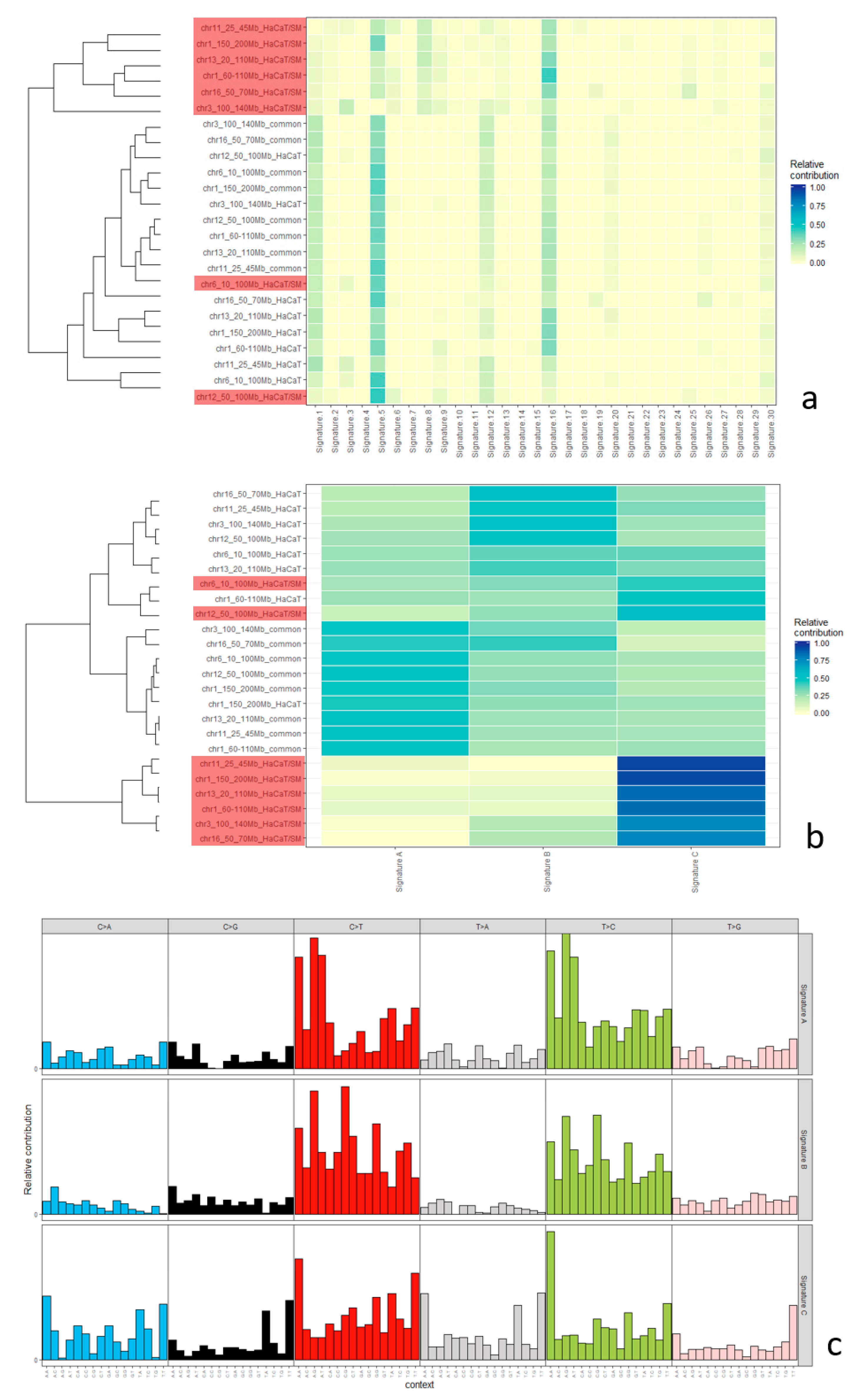
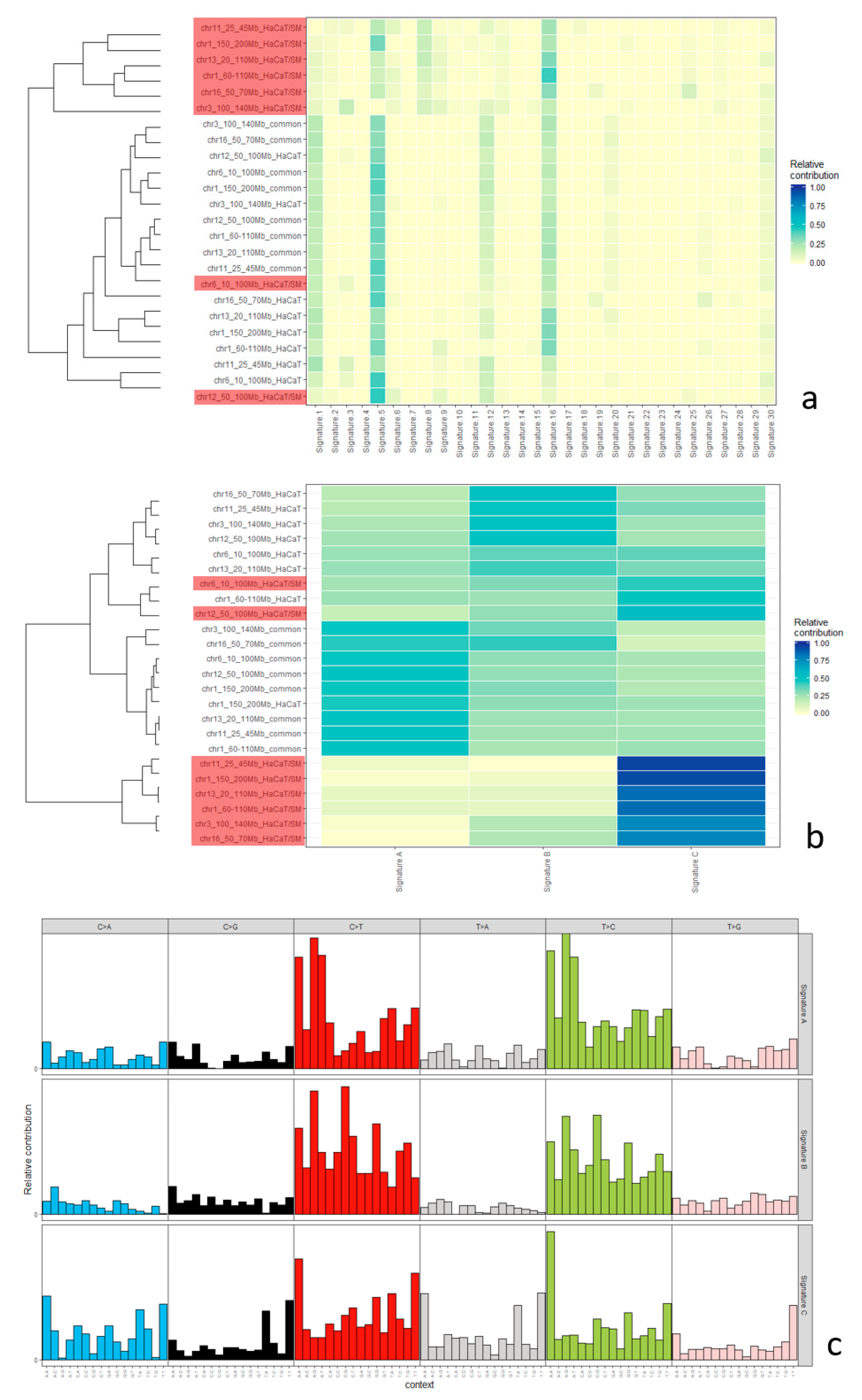
2.5. Mutational Signatures
3. Discussion
3.1. Change in Ploidy and Structural Chromosomal Aberrations
3.2. Distribution of Single-Nucleotide Variants and Mutational Signatures
4. Materials and Methods
4.1. Cultivation and Authentication of HaCaT
4.2. Chromosome Preparation and Quantification of DNA Content
4.3. Whole-Genome Sequencing and Variant Calling
4.4. Analysis of DNA Copy Number Changes
4.5. Genomic Characteristics at Sites of DNA Double-Strand Breaks
4.6. Analysis of Chromosomal Translocations by In Situ Hi-C
4.7. Genomic Distribution of Variants and Mutational Signatures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rall, D.P.; Pechura, C.M. Effects on health of mustard gas. Nature 1993, 366, 398–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szinicz, L. History of chemical and biological warfare agents. Toxicology 2005, 214, 167–181. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Chemical agents and related occupations. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 9–562. [Google Scholar]
- Somani, S.M.; Babu, S.R. Toxicodynamics of sulfur mustard. Int. J. Clin. Pharmacol. Ther. Toxicol. 1989, 27, 419–435. [Google Scholar]
- Dębiak, M.; Panas, A.; Steinritz, D.; Kehe, K.; Bürkle, A. High-throughput analysis of DNA interstrand crosslinks in human peripheral blood mononuclear cells by automated reverse FADU assay. Toxicology 2011, 280, 53–60. [Google Scholar] [CrossRef]
- Roberts, J.J.; Brent, T.P.; Crathorn, A.R. Evidence for the inactivation and repair of the mammalian DNA template after alkylation by mustard gas and half mustard gas. Eur. J. Cancer 1971, 7, 515–524. [Google Scholar] [CrossRef]
- Shahin, S.; Cullinane, C.; Gray, P.J. Mitochondrial and nuclear DNA damage induced by sulphur mustard in keratinocytes. Chem. Biol. Interact. 2001, 138, 231–245. [Google Scholar] [CrossRef]
- Walker, I.G. Intrastrand bifunctional alkylation of DNA in mammalian cells treated with mustard gas. Can. J. Biochem. 1971, 49, 332–336. [Google Scholar] [CrossRef]
- Ludlum, D.B.; Ritchie, P.A.; Hagopian, M.; Niu, T.Q.; Yu, D. Detection of sulfur mustard-induced DNA modifications. Chem. Biol. Interact. 1994, 91, 39–49. [Google Scholar] [CrossRef]
- Fidder, A.; Moes, G.W.; Scheffer, A.G.; van der Schans, G.P.; Baan, R.A.; de Jong, L.P.; Benschop, H.P. Synthesis, characterization, and quantitation of the major adducts formed between sulfur mustard and DNA of calf thymus and human blood. Chem. Res. Toxicol. 1994, 7, 199–204. [Google Scholar] [CrossRef]
- Mood, M.B.; Hefazi, M. The pharmacology, toxicology, and medical treatment of sulphur mustard poisoning. Fundam. Clin. Pharmacol. 2005, 19, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Stetina, R.; Svobodova, H.; Nedecky, B.R.; Jilkova, M.; Sochor, J.; Sobotka, J.; Adam, V.; Kizek, R. Sulfur mustard causes oxidative stress and depletion of antioxidants in muscles, livers, and kidneys of Wistar rats. Drug Chem. Toxicol. 2013, 36, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Layali, I.; Shahriary, A.; Talatappe, N.R.; Tahmasbpour, E.; Rostami, H.; Harchegani, A.B. Sulfur mustard triggers oxidative stress through glutathione depletion and altered expression of glutathione-related enzymes in human airways. Immunopharmacol. Immunotoxicol. 2018, 40, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Harchegani, A.B.; Tahmasbpour, E.; Borna, H.; Imamy, A.; Ghanei, M.; Shahriary, A. Free Radical Production and Oxidative Stress in Lung Tissue of Patients Exposed to Sulfur Mustard: An Overview of Cellular and Molecular Mechanisms. Chem. Res. Toxicol. 2018, 31, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Dube, S.N.; Sugendran, K.; Singh, R.; das Gupta, S.; Somani, S.M. Effect of topically applied sulphur mustard on antioxidant enzymes in blood cells and body tissues of rats. J. Appl. Toxicol. 1996, 16, 245–248. [Google Scholar] [CrossRef]
- Papirmeister, B.; Gross, C.L.; Meier, H.L.; Petrali, J.P.; Johnson, J.B. Molecular basis for mustard-induced vesication. Fundam. Appl. Toxicol. 1985, 5, S134–S149. [Google Scholar] [CrossRef]
- Weiss, A.; Weiss, B. Carcinogenesis due to mustard gas exposure in man, important sign for therapy with alkylating agents. Dtsch. Med. Wochenschr. 1975, 100, 919–923. [Google Scholar] [PubMed]
- Yamada, A.; Hirose, F.; Miyanishi, M. An autopsy case of bronchial carcinoma found in a patient succumbed to occupational mustard gas poisoning. Gan 1953, 44, 216–218. [Google Scholar]
- Tokuoka, S. Early cancer and related changes in the bronchial epithelium of former mustard gas workers. Gan Kagaku Ryoho 1985, 12, 708–713. [Google Scholar]
- Tokuoka, S.; Hayashi, Y.; Inai, K.; Egawa, H.; Aoki, Y.; Akamizu, H.; Eto, R.; Nishida, T.; Ohe, K.; Kobuke, T. Early cancer and related lesions in the bronchial epithelium in former workers of mustard gas factory. Acta Pathol. Jpn. 1986, 36, 533–542. [Google Scholar] [CrossRef]
- Dacre, J.C.; Goldman, M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol. Rev. 1996, 48, 289–326. [Google Scholar] [PubMed]
- World Health Organization. Overall evaluations of carcinogenicity: An updating of IARC Monographs volumes 1 to 42. IARC Monogr. Eval. Carcinog. Risks Hum. Suppl. 1987, 7, 1–440. [Google Scholar]
- de Vita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, L.S.; Wintrobe, M.M.; Dameshek, W.; Goodman, M.J.; Gilman, A.; McLennan, M.T. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Varela, B.D.; Ai, Y.; Liang, D.; Xue, F. Nitrogen Mustards as Anticancer Chemotherapies: Historic Perspective, Current Developments and Future Trends. Curr. Top. Med. Chem. 2019, 19, 691–712. [Google Scholar] [CrossRef]
- Panasci, L.; Xu, Z.-Y.; Bello, V.; Aloyz, R. The role of DNA repair in nitrogen mustard drug resistance. Anticancer Drugs 2002, 13, 211–220. [Google Scholar] [CrossRef]
- Buller, A.L.; Clapper, M.L.; Tew, K.D. Glutathione S-transferases in nitrogen mustard-resistant and -sensitive cell lines. Mol. Pharmacol. 1987, 31, 575–578. [Google Scholar]
- Boffetta, P.; Kaldor, J.M. Secondary malignancies following cancer chemotherapy. Acta Oncol. 1994, 33, 591–598. [Google Scholar] [CrossRef]
- Wolf, M.; Siegert, M.; Rothmiller, S.; Scheithauer, N.; Strobelt, R.; Steinritz, D.; Worek, F.; Thiermann, H.; Schmidt, A. Characterization of sulfur mustard resistant keratinocyte cell line HaCaT/SM. Toxicol. Lett. 2016, 244, 49–55. [Google Scholar] [CrossRef]
- Schmidt, A.; Steinritz, D.; Thiermann, H. Development of the sulfur mustard resistant keratinocyte cell line HaCaT/SM. Toxicol. Lett. 2016, 244, 44–48. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoop, V.M.; Mirancea, N.; Fusenig, N.E. Epidermal organization and differentiation of HaCaT keratinocytes in organotypic coculture with human dermal fibroblasts. J. Investig. Dermatol. 1999, 112, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Popp, S.; Altmeyer, S.; Hülsen, A.; Fasching, C.; Cremer, T.; Fusenig, N.E. Sustained nontumorigenic phenotype correlates with a largely stable chromosome content during long-term culture of the human keratinocyte line HaCaT. Genes Chromosomes Cancer 1997, 19, 201–214. [Google Scholar] [CrossRef]
- Fusenig, N.E.; Boukamp, P. Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol. Carcinog. 1998, 23, 144–158. [Google Scholar] [CrossRef]
- He, Y.Y.; Pi, J.; Huang, J.L.; Diwan, B.A.; Waalkes, M.P.; Chignell, C.F. Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene 2006, 25, 3680–3688. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; He, Y.; Bortner, C.; Huang, J.; Liu, J.; Zhou, T.; Qu, W.; North, S.L.; Kasprzak, K.S.; Diwan, B.A.; et al. Low level, long-term inorganic arsenite exposure causes generalized resistance to apoptosis in cultured human keratinocytes: Potential role in skin co-carcinogenesis. Int. J. Cancer 2005, 116, 20–26. [Google Scholar] [CrossRef]
- Schmidt, A.; Wolf, M.; Rothmiller, S.; Worek, F.; Steinritz, D.; Thiermann, H. Cytostatic resistance profile of the sulfur mustard resistant keratinocyte cell line HaCaT/SM. Toxicol. Lett. 2018, 293, 16–20. [Google Scholar] [CrossRef]
- Harewood, L.; Kishore, K.; Eldridge, M.D.; Wingett, S.; Pearson, D.; Schoenfelder, S.; Collins, V.P.; Fraser, P. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 2017, 18, 125. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [Green Version]
- Barutcu, A.R.; Lajoie, B.R.; McCord, R.P.; Tye, C.E.; Hong, D.; Messier, T.L.; Browne, G.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015, 16, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steininger, A.; Ebert, G.; Becker, B.V.; Assaf, C.; Möbs, M.; Schmidt, C.A.; Grabarczyk, P.; Jensen, L.R.; Przybylski, G.K.; Port, M.; et al. Genome-Wide Analysis of Interchromosomal Interaction Probabilities Reveals Chained Translocations and Overrepresentation of Translocation Breakpoints in Genes in a Cutaneous T-Cell Lymphoma Cell Line. Front. Oncol. 2018, 8, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Turner, D.; Durand, N.C.; Thorvaldsdottir, H.; Mesirov, J.P.; Aiden, E.L. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 2018, 6, 256–258. [Google Scholar] [CrossRef] [Green Version]
- Venkatraman, E.S.; Olshen, A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Peter, W.; Pascheberg, U.; Altmeier, S.; Fasching, C.; Stanbridge, E.J.; Fusenig, N.E. Step-wise progression in human skin carcinogenesis in vitro involves mutational inactivation of p53, rasH oncogene activation and additional chromosome loss. Oncogene 1995, 11, 961–969. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Favorov, A.; Mularoni, L.; Cope, L.M.; Medvedeva, Y.; Mironov, A.A.; Makeev, V.J.; Wheelan, S.J. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012, 8, e1002529. [Google Scholar] [CrossRef] [Green Version]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Zainal, S.N.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Dale, A.-L.B.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Zainal, S.N.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Lim, S.; Ganem, N.J. Tetraploidy and tumor development. Oncotarget 2014, 5, 10959–10960. [Google Scholar] [CrossRef]
- Schoenfelder, K.P.; Fox, D.T. The expanding implications of polyploidy. J. Cell Biol. 2015, 209, 485–491. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.A.; Horton, S.; Saldivar, J.C.; Miuma, S.; Stampfer, M.R.; Heerema, N.A.; Huebner, K. Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer 2013, 52, 1017–1029. [Google Scholar] [CrossRef] [Green Version]
- Schrock, M.S.; Batar, B.; Lee, J.; Druck, T.; Ferguson, B.; Cho, J.H.; Akakpo, K.; Hagrass, H.; Heerema, N.A.; Xia, F.; et al. Wwox-Brca1 interaction: Role in DNA repair pathway choice. Oncogene 2017, 36, 2215–2227. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Wang, Y.; Huebner, K. A Fhit-ing role in the DNA damage checkpoint response. Cell Cycle 2007, 6, 1044–1048. [Google Scholar] [CrossRef]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M.; et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Tudek, B. Imidazole ring-opened DNA purines and their biological significance. J. Biochem. Mol. Biol. 2003, 36, 12–19. [Google Scholar] [PubMed] [Green Version]
- Gehrke, T.H.; Lischke, U.; Gasteiger, K.L.; Schneider, S.; Arnold, S.; Müller, H.C.; Stephenson, D.S.; Zipse, H.; Carell, T. Unexpected non-Hoogsteen-based mutagenicity mechanism of FaPy-DNA lesions. Nat. Chem. Biol. 2013, 9, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836. [Google Scholar] [CrossRef] [Green Version]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Apurinic sites as mutagenic intermediates. Cell 1985, 40, 483–484. [Google Scholar] [CrossRef]
- Gruppi, F.; Hejazi, L.; Christov, P.P.; Krishnamachari, S.; Turesky, R.J.; Rizzo, C.J. Characterization of nitrogen mustard formamidopyrimidine adduct formation of bis(2-chloroethyl)ethylamine with calf thymus DNA and a human mammary cancer cell line. Chem. Res. Toxicol. 2015, 28, 1850–1860. [Google Scholar] [CrossRef] [Green Version]
- Minko, I.G.; Christov, P.P.; Li, L.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Processing of N(5)-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair (Amst.) 2019, 73, 49–54. [Google Scholar] [CrossRef]
- Boysen, G.; Pachkowski, B.F.; Nakamura, J.; Swenberg, J.A. The formation and biological significance of N7-guanine adducts. Mutat. Res. 2009, 678, 76–94. [Google Scholar] [CrossRef] [Green Version]
- Sagher, D.; Strauss, B. Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: Uniqueness of adenine nucleotides. Biochemistry 1983, 22, 4518–4526. [Google Scholar] [CrossRef]
- Schaaper, R.M.; Kunkel, T.A.; Loeb, L.A. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc. Natl. Acad. Sci. USA 1983, 80, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Translesional synthesis on DNA templates containing a single abasic site. A mechanistic study of the “A rule”. J. Biol. Chem. 1997, 272, 13916–13922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, S.; Blatter, N.; Kranaster, R.; Schnur, A.; Diederichs, K.; Welte, W.; Marx, A. Replication through an abasic DNA lesion: Structural basis for adenine selectivity. EMBO J. 2010, 29, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Scherthan, H.; Abend, M.; Muller, K.; Beinke, C.; Braselmann, H.; Zitzelsberger, H.; Kohn, F.M.; Pillekamp, H.; Schiener, R.; Das, O.; et al. Radiation-induced late effects in two affected individuals of the Lilo radiation accident. Radiat. Res. 2007, 167, 615–623. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Poell, J.B.; Mendeville, M.; Sie, D.; Brink, A.; Brakenhoff, R.H.; Ylstra, B. ACE: Absolute copy number estimation from low-coverage whole-genome sequencing data. Bioinformatics 2019, 35, 2847–2849. [Google Scholar] [CrossRef]
- Scheinin, I.; Sie, D.; Bengtsson, H.; van de Wiel, M.A.; Olshen, A.B.; van Thuijl, H.F.; van Essen, H.F.; Eijk, P.P.; Rustenburg, F.; Meijer, G.A.; et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014, 24, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Tebel, K.; Boldt, V.; Steininger, A.; Port, M.; Ebert, G.; Ullmann, R. GenomeCAT: A versatile tool for the analysis and integrative visualization of DNA copy number variants. BMC Bioinform. 2017, 18, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genom. 2019, 19, 985. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.P.; Comoglio, F.; Zhou, T.; Yang, L.; Paro, R.; Rohs, R. DNAshapeR: An R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics 2016, 32, 1211–1213. [Google Scholar] [CrossRef] [Green Version]
- Cer, R.Z.; Donohue, D.E.; Mudunuri, U.S.; Temiz, N.A.; Loss, M.A.; Starner, N.J.; Halusa, G.N.; Volfovsky, N.; Yi, M.; Luke, B.T.; et al. Non-B DB v2.0: A database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013, 41, D94–D100. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zainal, S.N.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullmann, R.; Becker, B.V.; Rothmiller, S.; Schmidt, A.; Thiermann, H.; Kaatsch, H.L.; Schrock, G.; Müller, J.; Jakobi, J.; Obermair, R.; et al. Genomic Adaption and Mutational Patterns in a HaCaT Subline Resistant to Alkylating Agents and Ionizing Radiation. Int. J. Mol. Sci. 2021, 22, 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031146
Ullmann R, Becker BV, Rothmiller S, Schmidt A, Thiermann H, Kaatsch HL, Schrock G, Müller J, Jakobi J, Obermair R, et al. Genomic Adaption and Mutational Patterns in a HaCaT Subline Resistant to Alkylating Agents and Ionizing Radiation. International Journal of Molecular Sciences. 2021; 22(3):1146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031146
Chicago/Turabian StyleUllmann, Reinhard, Benjamin Valentin Becker, Simone Rothmiller, Annette Schmidt, Horst Thiermann, Hanns Leonhard Kaatsch, Gerrit Schrock, Jessica Müller, Julia Jakobi, Richard Obermair, and et al. 2021. "Genomic Adaption and Mutational Patterns in a HaCaT Subline Resistant to Alkylating Agents and Ionizing Radiation" International Journal of Molecular Sciences 22, no. 3: 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031146