Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy

Abstract
:1. Introduction
2. Etiology of Neovascular AMD (nvAMD)
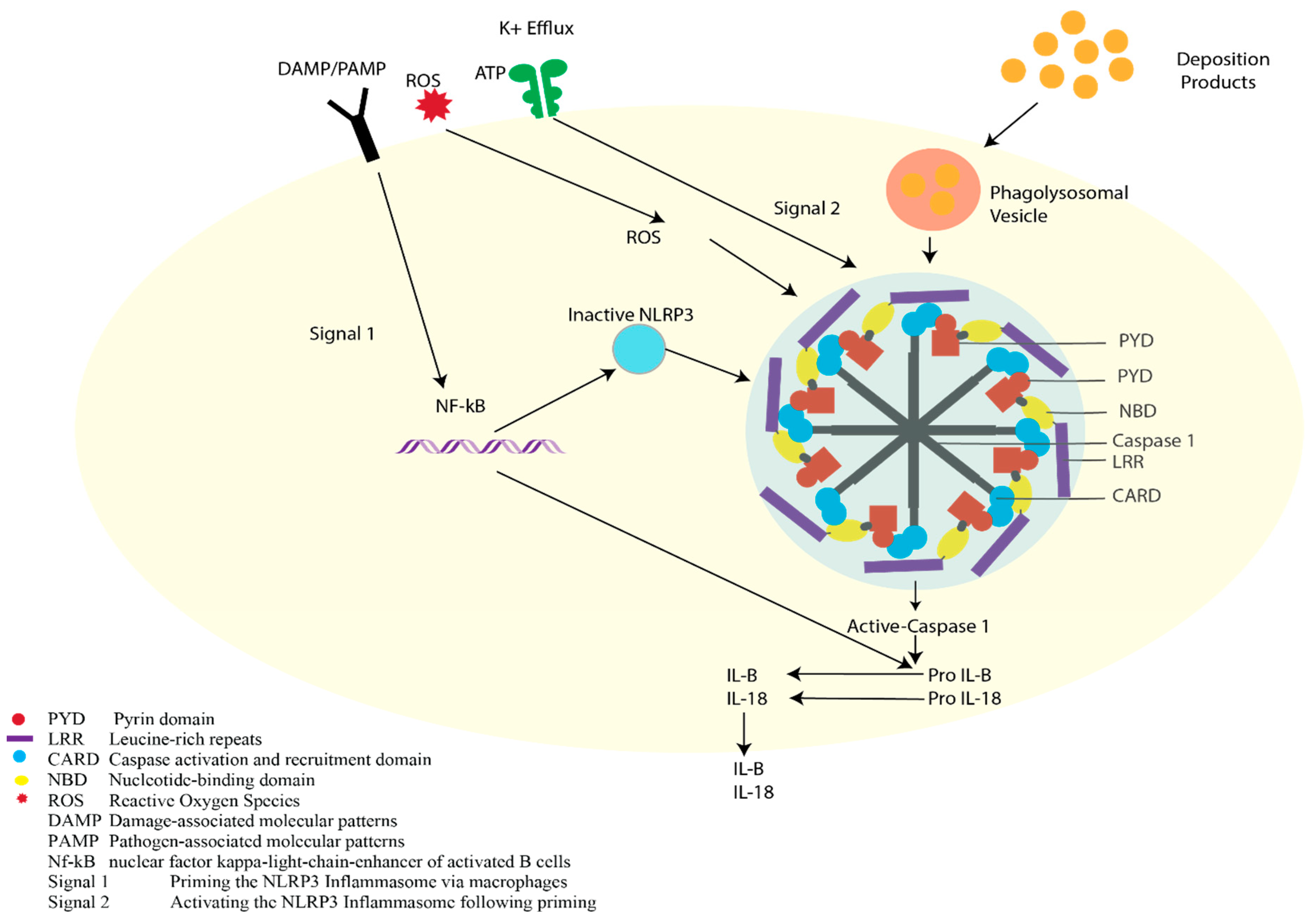
3. Pathophysiology
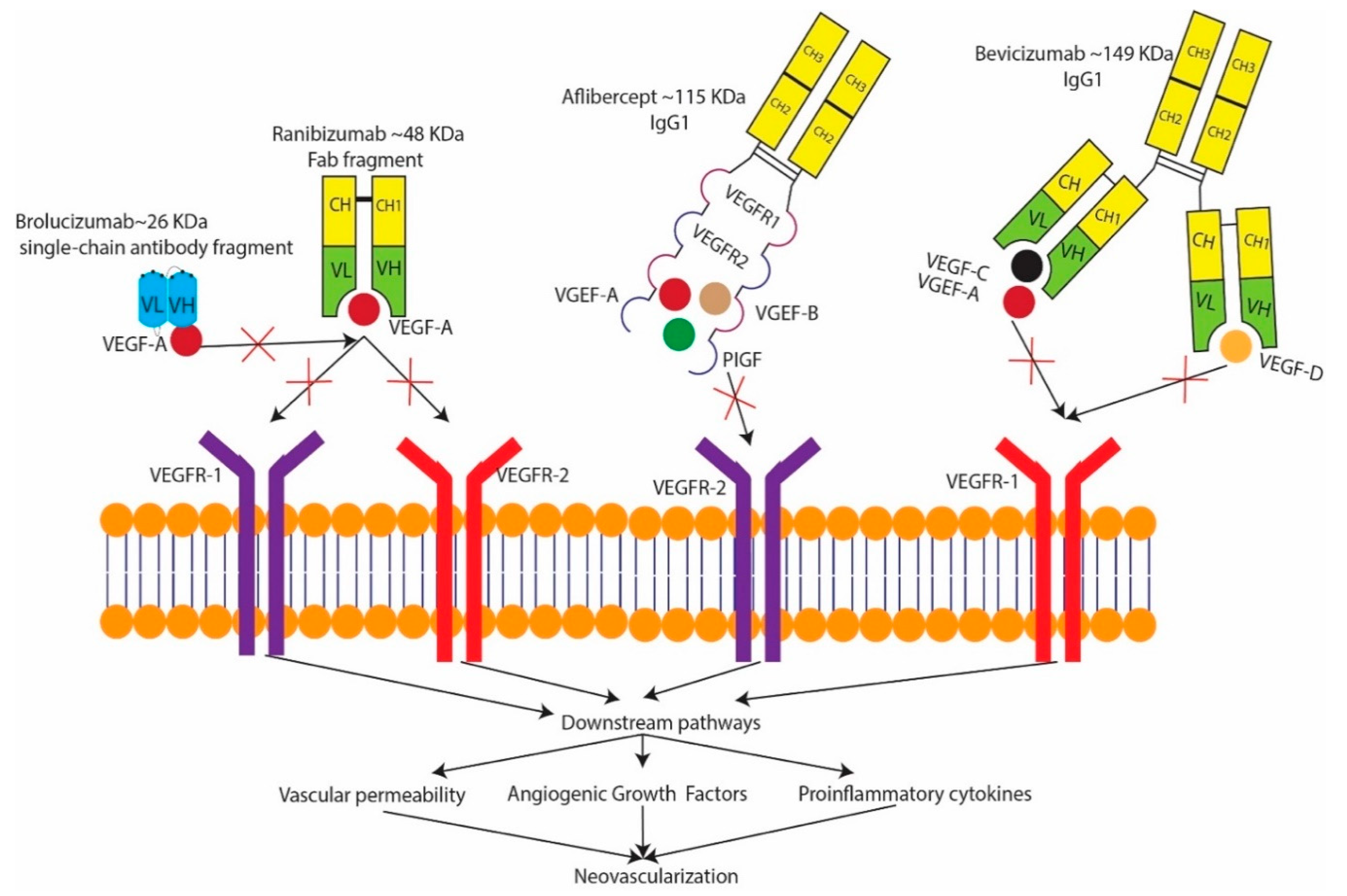
4. Treatment
Future Advancements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMD | Age-related macular degeneration |
| nvAMD | Neovascular age-related macular degeneration |
| CNV | Choroidal neovascularization |
| BrM | Bruch’s Membrane |
| GA | Geographic atrophy |
| OS | Outer photoreceptor segments |
| RPE | Retinal pigment epithelium |
| MHC | Major histocompatibility complex |
| IL | Interleukin |
| A2E | N-Retinylidene-N-Retinylethanolamine |
| PUFA | Polyunsaturated fatty acids |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
| PIGF | Placental growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| Th2 | Helper T-cells type 1 |
| Th2 | Helper T-cells type 2 |
| Tie2 | Tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains 2 |
| Ang | Angiopoietin |
| PDT | Photodynamic therapy |
| SNRVM | Sub-retinal neovascular membrane |
| Anti-VEGF | Anti-vascular endothelial growth factor |
| AAV | Aden-associated virus |
| Rock | Rho-associated protein kinase |
| OR | Odds ratio |
| HR | Hazard ratio |
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Chapman, N.A.; Jacobs, R.J.; Braakhuis, A.J. Role of diet and food intake in age-related macular degeneration: A systematic review. Clin. Exp. Ophthalmol. 2019, 47, 106–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokotas, H.; Grigoriadou, M.; Petersen, M.B. Age-related macular degeneration: Genetic and clinical findings. Clin. Chem. Lab. Med. 2011, 49, 601–616. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar]
- Davis, M.D.; Gangnon, R.E.; Lee, L.-Y.; Hubbard, L.D.; Klein, B.E.K.; Klein, R.; Ferris, F.L.; Bressler, S.B.; Milton, R.C.; Age-Related Eye Disease Study, G. The Age-Related Eye Disease Study severity scale for age-related macular degeneration: AREDS Report No. 17. Arch. Ophthalmol. 2005, 123, 1484–1498. [Google Scholar] [PubMed] [Green Version]
- Coleman, H.R.; Chan, C.-C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- The Age-Related Eye Disease Study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: The Age-Related Eye Disease Study Report Number 6. Am. J. Ophthalmol. 2001, 132, 668–681. [CrossRef]
- Hyman, L.; Neborsky, R. Risk factors for age-related macular degeneration: An update. Curr. Opin. Ophthalmol. 2002, 13, 171–175. [Google Scholar] [CrossRef]
- Ristau, T.; Ersoy, L.; Hahn, M.; den Hollander, A.I.; Kirchhof, B.; Liakopoulos, S.; Fauser, S. Nongenetic Risk Factors for Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5228–5232. [Google Scholar] [CrossRef] [Green Version]
- Velilla, S.; García-Medina, J.J.; García-Layana, A.; Dolz-Marco, R.; Pons-Vázquez, S.; Pinazo-Durán, M.D.; Gómez-Ulla, F.; Arévalo, J.F.; Díaz-Llopis, M.; Gallego-Pinazo, R. Smoking and age-related macular degeneration: Review and update. J. Ophthalmol. 2013, 2013, 895147. [Google Scholar] [CrossRef] [PubMed]
- Hyman, L.; Schachat, A.P.; He, Q.; Leske, M.C. Hypertension, cardiovascular disease, and age-related macular degeneration. Age-Related Macular Degeneration Risk Factors Study Group. Arch. Ophthalmol. 2000, 118, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnicka, A.R.; Jarrar, Z.; Wormald, R.; Cook, D.G.; Fletcher, A.; Owen, C.G. Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: A meta-analysis. Ophthalmology 2012, 119, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-D.; Presley, J.B.; Chimento, M.F.; Curcio, C.A.; Johnson, M. Age-related changes in human macular Bruch’s membrane as seen by quick-freeze/deep-etch. Exp. Eye Res. 2007, 85, 202–218. [Google Scholar] [CrossRef] [Green Version]
- Buch, H.; Vinding, T.; la Cour, M.; Jensen, G.B.; Prause, J.U.; Nielsen, N.V. Risk factors for age-related maculopathy in a 14-year follow-up study: The Copenhagen City Eye Study. Acta Ophthalmol. Scand. 2005, 83, 409–418. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Vingerling, J.R.; Dielemans, I.; Bots, M.L.; Hofman, A.; Grobbee, D.E.; de Jong, P.T.V.M. Age-related Macular Degeneration Is Associated with Atherosclerosis: The Rotterdam Study. Am. J. Epidemiol. 1995, 142, 404–409. [Google Scholar] [CrossRef]
- Buford, T.W. Hypertension and aging. Ageing Res. Rev. 2016, 26, 96–111. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.S.; Katz, J.; Bressler, N.M.; Rahmani, B.; Tielsch, J.M. Racial differences in the prevalence of age-related macular degeneration: The Baltimore eye survey. Ophthalmology 1999, 106, 1049–1055. [Google Scholar] [CrossRef]
- Bressler, S.B.; Muñoz, B.; Solomon, S.D.; West, S.K. The Salisbury Eye Evaluation (SEE) Study Team. Racial Differences in the Prevalence of Age-Related Macular Degeneration: The Salisbury Eye Evaluation (SEE) Project. Arch. Ophthalmol. 2008, 126, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Clemons, T.E.; Milton, R.C.; Klein, R.; Seddon, J.M.; Ferris, F.L., 3rd. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology 2005, 112, 533–539. [Google Scholar] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Wong, T.Y.; Cotch, M.F.; Liu, K.; Burke, G.; Saad, M.F.; Jacobs, D.R., Jr. Prevalence of age-related macular degeneration in 4 racial/ethnic groups in the multi-ethnic study of atherosclerosis. Ophthalmology 2006, 113, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, B.; Klein, R.; Rodriguez, J.; Snyder, R.; West, S.K. Prevalence of age-related macular degeneration in a population-based sample of Hispanic people in Arizona: Proyecto VER. Arch. Ophthalmol. 2005, 123, 1575–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Linton, K.L. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology 1992, 99, 933–943. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Jensen, S.C.; Mares-Perlman, J.A.; Cruickshanks, K.J.; Palta, M. Age-related maculopathy in a multiracial United States population: The National Health and Nutrition Examination Survey III. Ophthalmology 1999, 106, 1056–1065. [Google Scholar] [CrossRef]
- Varma, R.; Fraser-Bell, S.; Tan, S.; Klein, R.; Azen, S.P. Prevalence of age-related macular degeneration in Latinos: The Los Angeles Latino eye study. Ophthalmology 2004, 111, 1288–1297. [Google Scholar] [CrossRef]
- Oshima, Y.; Ishibashi, T.; Murata, T.; Tahara, Y.; Kiyohara, Y.; Kubota, T. Prevalence of age related maculopathy in a representative Japanese population: The Hisayama study. Br. J. Ophthalmol. 2001, 85, 1153–1157. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, M.A.; Gaudio, A.R.; Miller, S.; Weiner, A. Iris pigmentation and extent of disease in patients with neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2734–2740. [Google Scholar]
- Nicolas, C.M.; Robman, L.D.; Tikellis, G.; Dimitrov, P.N.; Dowrick, A.; Guymer, R.H.; McCarty, C.A. Iris colour, ethnic origin and progression of age-related macular degeneration. Clin. Exp. Ophthalmol. 2003, 31, 465–469. [Google Scholar] [CrossRef]
- Tomany, S.C.; Klein, R.; Klein, B.E. The relationship between iris color, hair color, and skin sun sensitivity and the 10-year incidence of age-related maculopathy: The Beaver Dam Eye Study. Ophthalmology 2003, 110, 1526–1533. [Google Scholar] [CrossRef]
- Deangelis, M.M.; Silveira, A.C.; Carr, E.A.; Kim, I.K. Genetics of age-related macular degeneration: Current concepts, future directions. Semin. Ophthalmol. 2011, 26, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.G.; Ying, G.-S.; Jaffe, G.J.; Toth, C.A.; Daniel, E.; Grunwald, J.; Martin, D.F.; Hagstrom, S.A.; Group, C.R. Single-Nucleotide Polymorphisms Associated With Age-Related Macular Degeneration and Lesion Phenotypes in the Comparison of Age-Related Macular Degeneration Treatments Trials. JAMA Ophthalmol. 2016, 134, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.; Wang, J.J.; Smith, W.; Leeder, S.R. Smoking and the 5-Year Incidence of Age-Related Maculopathy: The Blue Mountains Eye Study. Arch. Ophthalmol. 2002, 120, 1357–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Meuer, S.M.; Swift, M.; Gangnon, R.E. Fifteen-year cumulative incidence of age-related macular degeneration: The Beaver Dam Eye Study. Ophthalmology 2007, 114, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Klein, B.E.K.; Moss, S.E. Relation of Smoking to the Incidence of Age-related Maculopathy: The Beaver Dam Eye Study. Am. J. Epidemiol. 1998, 147, 103–110. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.K.; Tomany, S.C.; Moss, S.E. Ten-Year Incidence of Age-related Maculopathy and Smoking and Drinking: The Beaver Dam Eye Study. Am. J. Epidemiol. 2002, 156, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.A.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef]
- Hara, K. Effects of cigarette smoking on ocular circulation chronic effect on choroidal circulation. Nippon Ganka Gakkai Zasshi 1991, 95, 939–943. [Google Scholar]
- Fields, M.A.; Bowrey, H.E.; Gong, J.; Moreira, E.F.; Cai, H.; Del Priore, L.V. Extracellular matrix nitration alters growth factor release and activates bioactive complement in human retinal pigment epithelial cells. PLoS ONE 2017, 12, e0177763. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, M.; Nagai, N.; Sussan, T.E.; Biswal, S.; Handa, J.T. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE 2008, 3, e3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.; Assink, J.; Klein, R.; Mitchell, P.; Klaver, C.C.W.; Klein, B.E.K.; Hofman, A.; Jensen, S.; Wang, J.J.; de Jong, P.T.V.M. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology 2001, 108, 697–704. [Google Scholar] [CrossRef]
- Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Yates, J.R.W.; Bradley, M.; Moore, A.T.; Bird, A.C. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 2006, 90, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suner, I.J.; Espinosa–Heidmann, D.G.; Pereira–Simon, S.; Piña, Y.; Cousins, S.W. Cigarette Smoke Increases Severity of Experimental Choroidal Neovascularization (CNV): Role of Inflammation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3507. [Google Scholar]
- Lee, J.; Cooke, J.P. Nicotine and pathological angiogenesis. Life Sci. 2012, 91, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.A.; Bressler, S.B.; Munoz, B.; West, S.K. Racial Differences and Other Risk Factors for Incidence and Progression of Age-Related Macular Degeneration: Salisbury Eye Evaluation (SEE) Project. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Akishima, S.; Matsushita, S.; Sato, F.; Hyodo, K.; Imazuru, T.; Enomoto, Y.; Noma, M.; Hiramatsu, Y.; Shigeta, O.; Sakakibara, Y. Cigarette-smoke-induced vasoconstriction of peripheral arteries: Evaluation by synchrotron radiation microangiography. Circ. J. 2007, 71, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Role of lipid peroxidation and oxidative stress in alcohol toxicity. Free Radic. Biol. Med. 1989, 7, 537–539. [Google Scholar] [CrossRef]
- Chong, E.W.; Kreis, A.J.; Wong, T.Y.; Simpson, J.A.; Guymer, R.H. Alcohol consumption and the risk of age-related macular degeneration: A systematic review and meta-analysis. Am. J. Ophthalmol. 2008, 145, 707–715. [Google Scholar] [CrossRef]
- Arnarsson, A.; Sverrisson, T.; Stefánsson, E.; Sigurdsson, H.; Sasaki, H.; Sasaki, K.; Jonasson, F. Risk Factors for Five-Year Incident Age-related Macular Degeneration: The Reykjavik Eye Study. Am. J. Ophthalmol. 2006, 142, 419–428.e411. [Google Scholar] [CrossRef]
- Coleman, A.L.; Seitzman, R.L.; Cummings, S.R.; Yu, F.; Cauley, J.A.; Ensrud, K.E.; Stone, K.L.; Hochberg, M.C.; Pedula, K.L.; Thomas, E.L.; et al. The Association of Smoking and Alcohol Use With Age-related Macular Degeneration in the Oldest Old: The Study of Osteoporotic Fractures. Am. J. Ophthalmol. 2010, 149, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudtson, M.D.; Klein, R.; Klein, B.E.K. Alcohol consumption and the 15-year cumulative incidence of age-related macular degeneration. Am. J. Ophthalmol. 2007, 143, 1026–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.K.M.; Chong, E.W.; Williamson, E.; Aung, K.Z.; Makeyeva, G.A.; Giles, G.G.; English, D.R.; Hopper, J.; Guymer, R.H.; Baird, P.N.; et al. 20/20—Alcohol and Age-related Macular Degeneration: The Melbourne Collaborative Cohort Study. Am. J. Epidemiol. 2012, 176, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Tie, L.J.; Wu, S.S.; Lv, P.L.; Huang, H.W.; Wang, W.Q.; Wang, H.; Ma, L. Overweight, Obesity, and Risk of Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1276–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, K.P.; Klein, B.E.K.; Lee, K.E.; Klein, R. Measures of body shape and adiposity as related to incidence of age-related eye diseases: Observations from the Beaver Dam Eye Study. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2592–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingerling, J.R.; Hofman, A.; Grobbee, D.E.; de Jong, P.T.V.M. Age-Related Macular Degeneration and Smoking: The Rotterdam Study. Arch. Ophthalmol. 1996, 114, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Alberg, A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology 2002, 180, 121–137. [Google Scholar] [CrossRef]
- Rohrer, B.; Frazer-Abel, A.; Leonard, A.; Ratnapriya, R.; Ward, T.; Pietraszkiewicz, A.; O’Quinn, E.; Adams, K.; Swaroop, A.; Wolf, B.J. Association of age-related macular degeneration with complement activation products, smoking, and single nucleotide polymorphisms in South Carolinians of European and African descent. Mol. Vis. 2019, 25, 79–92. [Google Scholar]
- Lee, J.; Cooke, J.P. The role of nicotine in the pathogenesis of atherosclerosis. Atherosclerosis 2011, 215, 281–283. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, H.; Lu, S.C. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001, 15, 1335–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Jones, D.P.; Goldberg, J.; Ziegler, T.R.; Bostick, R.M.; Wilson, P.W.; Manatunga, A.K.; Shallenberger, L.; Jones, L.; Vaccarino, V. Association between adherence to the Mediterranean diet and oxidative stress. Am. J. Clin. Nutr. 2008, 88, 1364–1370. [Google Scholar] [PubMed]
- Cougnard-Grégoire, A.; Merle, B.M.; Korobelnik, J.F.; Rougier, M.B.; Delyfer, M.N.; Le Goff, M.; Samieri, C.; Dartigues, J.F.; Delcourt, C. Olive Oil Consumption and Age-Related Macular Degeneration: The Alienor Study. PLoS ONE 2016, 11, e0160240. [Google Scholar] [CrossRef] [PubMed]
- Merle, B.M.J.; Silver, R.E.; Rosner, B.; Seddon, J.M. Adherence to a Mediterranean diet, genetic susceptibility, and progression to advanced macular degeneration: A prospective cohort study. Am. J. Clin. Nutr. 2015, 102, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-J.; Chang, M.-L.; Zhang, F.F.; Li, T.; Gensler, G.; Schleicher, M.; Taylor, A. The relationship of major American dietary patterns to age-related macular degeneration. Am. J. Ophthalmol. 2014, 158, 118–127.e111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnolo, D.F.; Selmin, O.I. Mediterranean Diet and Prevention of Chronic Diseases. Nutr. Today 2017, 52, 208–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Quezada, I.; Román-Viñas, B.; Serra-Majem, L. The Mediterranean diet and nutritional adequacy: A review. Nutrients 2014, 6, 231–248. [Google Scholar] [CrossRef] [Green Version]
- Serra-Majem, L.; Bes-Rastrollo, M.; Román-Viñas, B.; Pfrimer, K.; Sánchez-Villegas, A.; Martínez-González, M.A. Dietary patterns and nutritional adequacy in a Mediterranean country. Br. J. Nutr. 2009, 101, S21–S28. [Google Scholar] [CrossRef] [Green Version]
- Stevens, G.A.; Singh, G.M.; Lu, Y.; Danaei, G.; Lin, J.K.; Finucane, M.M.; Bahalim, A.N.; McIntire, R.K.; Gutierrez, H.R.; Cowan, M.; et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul. Health Metr. 2012, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Gunes, F.E.; Bekiroglu, N.; Imeryuz, N.; Agirbasli, M. Relation between eating habits and a high body mass index among freshman students: A cross-sectional study. J. Am. Coll. Nutr. 2012, 31, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Aoki, A.; Inoue, M.; Nguyen, E.; Obata, R.; Kadonosono, K.; Shinkai, S.; Hashimoto, H.; Sasaki, S.; Yanagi, Y. Dietary n-3 Fatty Acid, α-Tocopherol, Zinc, vitamin D, vitamin C and β-carotene are Associated with Age-Related Macular Degeneration in Japan. Sci. Rep. 2016, 6, 20723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, R.E.; Woodside, J.V.; McGrath, A.; Young, I.S.; Vioque, J.L.; Chakravarthy, U.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; et al. Mediterranean Diet Score and Its Association with Age-Related Macular Degeneration: The European Eye Study. Ophthalmology 2017, 124, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SanGiovanni, J.P.; Chew, E.Y.; Clemons, T.E.; Davis, M.D.; Ferris, F.L., 3rd; Gensler, G.R.; Kurinij, N.; Lindblad, A.S.; Milton, R.C.; Seddon, J.M.; et al. The relationship of dietary lipid intake and age-related macular degeneration in a case-control study: AREDS Report No. 20. Arch. Ophthalmol. 2007, 125, 671–679. [Google Scholar] [PubMed]
- Okubo, A.; Rosa, R.H., Jr.; Bunce, C.V.; Alexander, R.A.; Fan, J.T.; Bird, A.C.; Luthert, P.J. The relationships of age changes in retinal pigment epithelium and Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 443–449. [Google Scholar]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Zimbrón, L.F.; Zamora-Alvarado, R.; Ochoa-De la Paz, L.; Velez-Montoya, R.; Zenteno, E.; Gulias-Cañizo, R.; Quiroz-Mercado, H.; Gonzalez-Salinas, R. Age-Related Macular Degeneration: New Paradigms for Treatment and Management of AMD. Oxid. Med. Cell. Longev. 2018, 2018, 8374647. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.-O.; Williams, M. (Eds.) Anatomical Distribution of Rods and Cones. In Neuroscience, 2th ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Katz, M.L.; Robison, W.G., Jr. What is lipofuscin? Defining characteristics and differentiation from other autofluorescent lysosomal storage bodies. Arch. Gerontol. Geriatr. 2002, 34, 169–184. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Wu, Y.; Kim, C.Y.; Zhou, J. Phospholipid meets all-trans-retinal: The making of RPE bisretinoids. J. Lipid. Res. 2010, 51, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, C.J.; Rakoczy, P.E.; Constable, I.J. Lipofuscin of the retinal pigment epithelium: A review. Eye (Lond) 1995, 9, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnemann, S.; Leung, L.; Rodriguez-Boulan, E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 3842–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharm. Rep. 2006, 58, 353–363. [Google Scholar]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant Lipid and Protein Components of Drusen. PLoS ONE 2010, 5, e10329. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Jorgenson, E.; Melles, R.B.; Hoffmann, T.J.; Jia, X.; Sakoda, L.C.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Shen, L. Common coding variants in the HLA-DQB1 region confer susceptibility to age-related macular degeneration. Eur. J. Hum. Genet. 2016, 24, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Nita, M.; Strzałka-Mrozik, B.; Grzybowski, A.; Mazurek, U.; Romaniuk, W. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Monit. 2014, 20, 1003–1016. [Google Scholar]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef]
- Kijlstra, A.; La Heij, E.; Hendrikse, F. Immunological factors in the pathogenesis and treatment of age-related macular degeneration. Ocul. Immunol. Inflamm. 2005, 13, 3–11. [Google Scholar] [CrossRef]
- Hoppe, G.; O’Neil, J.; Hoff, H.F.; Sears, J. Accumulation of oxidized lipid-protein complexes alters phagosome maturation in retinal pigment epithelium. Cell. Mol. Life. Sci. 2004, 61, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Iriyama, A.; Yanagi, Y.; Inoue, Y.; Takahashi, H.; Tamaki, Y. Role of A2E in the Pathogenesis of CNV. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4146. [Google Scholar]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Loskutova, E.; Nolan, J.; Howard, A.; Beatty, S. Macular pigment and its contribution to vision. Nutrients 2013, 5, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Drobek-Słowik, M.; Karczewicz, D.; Safranow, K. The potential role of oxidative stress in the pathogenesis of the age-related macular degeneration (AMD). Postepy Hig. Med. Dosw. (Online) 2007, 61, 28–37. [Google Scholar] [PubMed]
- Wiktorowska-Owczarek, A.; Nowak, J.Z. Pathogenesis and prophylaxis of AMD: Focus on oxidative stress and antioxidants. Postepy Hig. Med. Dosw. (Online) 2010, 64, 333–343. [Google Scholar] [PubMed]
- Hammond, B.R., Jr.; Wooten, B.R.; Snodderly, D.M. Cigarette smoking and retinal carotenoids: Implications for age-related macular degeneration. Vis. Res 1996, 36, 3003–3009. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Cho, E.; Willett, W.C.; Sastry, S.M.; Schaumberg, D.A. Intakes of Lutein, Zeaxanthin, and Other Carotenoids and Age-Related Macular Degeneration During 2 Decades of Prospective Follow-up. JAMA Ophthalmol. 2015, 133, 1415–1424. [Google Scholar] [CrossRef]
- Marie, M.; Bigot, K.; Angebault, C.; Barrau, C.; Gondouin, P.; Pagan, D.; Fouquet, S.; Villette, T.; Sahel, J.-A.; Lenaers, G.; et al. Light action spectrum on oxidative stress and mitochondrial damage in A2E-loaded retinal pigment epithelium cells. Cell Death Dis. 2018, 9, 287. [Google Scholar] [CrossRef]
- Dong, A.; Xie, B.; Shen, J.; Yoshida, T.; Yokoi, K.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress promotes ocular neovascularization. J. Cell Physiol. 2009, 219, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Heidmann, D.G.; Suner, I.J.; Catanuto, P.; Hernandez, E.P.; Marin-Castano, M.E.; Cousins, S.W. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Investig. Ophthalmol. Vis. Sci. 2006, 47, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, H.E.; Liu, Z.; Crott, J.W.; Choi, S.-W.; Song, B.C.; Mason, J.B.; Johnson, E.J. A Comparison of Carotenoids, Retinoids, and Tocopherols in the Serum and Buccal Mucosa of Chronic Cigarette Smokers versus Nonsmokers. Cancer Epidemiol. Biomark. Amplif. Amplif. Prev. 2006, 15, 993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimm, E.; Colditz, G. Smoking, Alcohol, and Plasma Levels of Carotenes and Vitamin Ea. Ann. N. Y. Acad. Sci. 1993, 686, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic. Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoh Kam, J.; Lenassi, E.; Jeffery, G. Viewing ageing eyes: Diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS ONE 2010, 5, e13127. [Google Scholar] [CrossRef] [PubMed]
- Fisichella, V.; Giurdanella, G.; Platania, C.B.; Romano, G.L.; Leggio, G.M.; Salomone, S.; Drago, F.; Caraci, F.; Bucolo, C. TGF-β1 prevents rat retinal insult induced by amyloid-β (1-42) oligomers. Eur. J. Pharm. 2016, 787, 72–77. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. The Inflammasomes. PLoS Pathog. 2009, 5, e1000510. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447s–455s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.; Ting, J.P.Y. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J. Immunol. 2009, 183, 7623–7629. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br. J. Pharm. 2014, 171, 5589–5602. [Google Scholar] [CrossRef]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Cho, D.; Kim, T.S. Efficient induction of T helper type 1-mediated immune responses in antigen-primed mice by anti-CD3 single-chain Fv/interleukin-18 fusion DNA. Immunology 2004, 111, 27–34. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.-S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Corridoni, D.; Pizarro, T.T.; Cominelli, F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012, 59, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, O.A.; Finkelstein, A.; Shima, D.T. A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS ONE 2013, 8, e67263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. 2013, 4, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Compan, V.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Martínez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazán, E.; Reimers, D.; et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Platania, C.B.M.; Giurdanella, G.; Di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem. Pharm. 2017, 138, 130–139. [Google Scholar] [CrossRef]
- Kim, Y.; Tarallo, V.; Kerur, N.; Yasuma, T.; Gelfand, B.D.; Bastos-Carvalho, A.; Hirano, Y.; Yasuma, R.; Mizutani, T.; Fowler, B.J.; et al. DICER1/Alu RNA dysmetabolism induces Caspase-8–mediated cell death in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 16082. [Google Scholar]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häsler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic. Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lee, J.K.; Tjhen, R.; Stroud, R.M.; James, T.L. Structural and biochemical insights into the dicing mechanism of mouse Dicer: A conserved lysine is critical for dsRNA cleavage. Proc. Natl. Acad. Sci. USA 2008, 105, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.; Fowler, B.; Cho, W.; Kleinman, M.; Ponicsan, S.; Hauswirth, W.; Chiodo, V.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.B.; Uehara, H.; Kim, Y.; Yasuma, T.; Yasuma, R.; Hirahara, S.; Makin, R.D.; Apicella, I.; Pereira, F.; Nagasaka, Y.; et al. Chronic Dicer1 deficiency promotes atrophic and neovascular outer retinal pathologies in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 2579–2587. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Jang, Y.P.; Kim, S.R.; Sparrow, J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2006, 103, 16182–16187. [Google Scholar] [CrossRef] [Green Version]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Heidmann, D.G.; Suner, I.J.; Hernandez, E.P.; Monroy, D.; Csaky, K.G.; Cousins, S.W. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3586–3592. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, E.; Anand, A.; Ambati, B.K.; van Rooijen, N.; Ambati, J. Macrophage depletion inhibits experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3578–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambati, J.; Anand, A.; Fernandez, S.; Sakurai, E.; Lynn, B.C.; Kuziel, W.A.; Rollins, B.J.; Ambati, B.K. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Yoshimoto, T.; Hayashi, N.; Mizutani, H.; Nakanishi, K. Induction of allergic inflammation by interleukin-18 in experimental animal models. Immunol. Rev. 2004, 202, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zandi, S.; Nakao, S.; Chun, K.-H.; Fiorina, P.; Sun, D.; Arita, R.; Zhao, M.; Kim, E.; Schueller, O.; Campbell, S.; et al. ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep. 2015, 10, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Tatar, O.; Yoeruek, E.; Szurman, P.; Bartz-Schmidt, K.U.; Adam, A.; Shinoda, K.; Eckardt, C.; Boeyden, V.; Claes, C.; Pertile, G.; et al. Effect of Bevacizumab on Inflammation and Proliferation in Human Choroidal Neovascularization. Arch. Ophthalmol. 2008, 126, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Walshe, T.E.; Dole, V.S.; Maharaj, A.S.R.; Patten, I.S.; Wagner, D.D.; D’Amore, P.A. Inhibition of VEGF or TGF-{beta} signaling activates endothelium and increases leukocyte rolling. Arter. Thromb. Vasc. Biol. 2009, 29, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar] [CrossRef]
- Leppänen, V.-M.; Saharinen, P.; Alitalo, K. Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proc. Natl. Acad. Sci. USA 2017, 114, 4376. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Nambu, H.; Umeda, N.; Kachi, S.; Oshima, Y.; Akiyama, H.; Nambu, R.; Campochiaro, P.A. Angiopoietin 1 prevents retinal detachment in an aggressive model of proliferative retinopathy, but has no effect on established neovascularization. J. Cell Physiol. 2005, 204, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Daly, C. The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb. Perspect. Med. 2012, 2, a006550. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.S.; Yip, Y.W.; Bakthavatsalam, M.; Chen, L.J.; Ng, T.K.; Lai, T.Y.; Pang, C.P.; Brelén, M.E. Elevated angiopoietin 2 in aqueous of patients with neovascular age related macular degeneration correlates with disease severity at presentation. Sci. Rep. 2017, 7, 45081. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, D.; Suzuma, K.; Suzuma, I.; Ohashi, H.; Ojima, T.; Kurimoto, M.; Murakami, T.; Kimura, T.; Takagi, H. Vitreous levels of angiopoietin 2 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 2005, 139, 476–481. [Google Scholar] [CrossRef]
- Sahni, J.; Patel, S.S.; Dugel, P.U.; Khanani, A.M.; Jhaveri, C.D.; Wykoff, C.C.; Hershberger, V.S.; Pauly-Evers, M.; Sadikhov, S.; Szczesny, P.; et al. Simultaneous Inhibition of Angiopoietin-2 and Vascular Endothelial Growth Factor-A with Faricimab in Diabetic Macular Edema: BOULEVARD Phase 2 Randomized Trial. Ophthalmology 2019, 126, 1155–1170. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Gerber, H.P.; Ferrara, N. The role of VEGF in normal and neoplastic hematopoiesis. J. Mol. Med. (Berl.) 2003, 81, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care (New Rochelle) 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Coultas, L.; Chawengsaksophak, K.; Rossant, J. Endothelial cells and VEGF in vascular development. Nature 2005, 438, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Pinedo, H.M. The role of vascular endothelial growth factor (VEGF) in tumor angiogenesis and early clinical development of VEGF-receptor kinase inhibitors. Clin. Breast Cancer 2000, 1 (Suppl. 1), S80–S84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Eriksson, U. Novel VEGF family members: VEGF-B, VEGF-C and VEGF-D. Int. J. Biochem. Cell Biol. 2001, 33, 421–426. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Oommen, S.; Gupta, S.K.; Vlahakis, N.E. Vascular endothelial growth factor A (VEGF-A) induces endothelial and cancer cell migration through direct binding to integrin {alpha}9{beta}1: Identification of a specific {alpha}9{beta}1 binding site. J. Biol. Chem. 2011, 286, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan-Benson, E.; Hartmann, J.; Vendrov, A.E.; Budd, S.; Byfield, G.; Parker, A.; Ahmad, F.; Huang, W.; Runge, M.; Burridge, K.; et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef]
- Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Monte, M.D.; Casini, G. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy. Cells 2020, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.-O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, A.P.; De Vries, G.W. Role of PLCgamma and Ca(2+) in VEGF- and FGF-induced choroidal endothelial cell proliferation. Am. J. Physiol. Cell Physiol. 2001, 281, C1448–C1456. [Google Scholar] [CrossRef] [PubMed]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayless, K.J.; Johnson, G.A. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J. Vasc. Res. 2011, 48, 369–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Anderson, C.; Zhang, H.; Li, X.; Inglis, F.; Jayagopal, A.; Wang, S. Repression of Choroidal Neovascularization Through Actin Cytoskeleton Pathways by MicroRNA-24. Mol. Ther. 2014, 22, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulak, J.; Józkowicz, A.; Dembinska-Kiec, A.; Guevara, I.; Zdzienicka, A.; Zmudzinska-Grochot, D.; Florek, I.; Wójtowicz, A.; Szuba, A.; Cooke, J.P. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2000, 20, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, M.; Kakinuma, Y.; Ando, M.; Katare, R.G.; Yamasaki, F.; Doi, Y.; Sato, T. Nitric oxide stimulates vascular endothelial growth factor production in cardiomyocytes involved in angiogenesis. J. Physiol. Sci. 2006, 56, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno-Matsui, K.; Morita, I.; Tombran-Tink, J.; Mrazek, D.; Onodera, M.; Uetama, T.; Hayano, M.; Murota, S.I.; Mochizuki, M. Novel mechanism for age-related macular degeneration: An equilibrium shift between the angiogenesis factors VEGF and PEDF. J. Cell Physiol. 2001, 189, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Adamis, A.P.; Shima, D.T.; Yeo, K.T.; Yeo, T.K.; Brown, L.F.; Berse, B.; D’Amore, P.A.; Folkman, J. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 1993, 193, 631–638. [Google Scholar] [CrossRef]
- Dawson, D.W.; Volpert, O.V.; Gillis, P.; Crawford, S.E.; Xu, H.; Benedict, W.; Bouck, N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar] [CrossRef]
- Cai, J.; Jiang, W.G.; Grant, M.B.; Boulton, M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J. Biol. Chem. 2006, 281, 3604–3613. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Holekamp, N.M.; Bouck, N.; Volpert, O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am. J. Ophthalmol. 2002, 134, 220–227. [Google Scholar] [CrossRef]
- Duh, E.J.; Yang, H.S.; Suzuma, I.; Miyagi, M.; Youngman, E.; Mori, K.; Katai, M.; Yan, L.; Suzuma, K.; West, K.; et al. Pigment epithelium-derived factor suppresses ischemia-induced retinal neovascularization and VEGF-induced migration and growth. Investig. Ophthalmol. Vis. Sci. 2002, 43, 821–829. [Google Scholar]
- Macular Photocoagulation Study Group. Argon laser photocoagulation for senile macular degeneration. Results of a randomized clinical trial. Arch. Ophthalmol. 1982, 100, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Macular Photocoagulation Study Group. Krypton laser photocoagulation for neovascular lesions of age-related macular degeneration. Results of a randomized clinical trial. Macular Photocoagulation Study Group. Arch. Ophthalmol. 1990, 108, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Schlötzer-Schrehardt, U.; Viestenz, A.; Naumann, G.O.; Laqua, H.; Michels, S.; Schmidt-Erfurth, U. Dose-related structural effects of photodynamic therapy on choroidal and retinal structures of human eyes. Graefe‘s Arch. Clin. Exp. Ophthalmol. 2002, 240, 748–757. [Google Scholar]
- Donati, G.; Kapetanios, A.D.; Pournaras, C.J. Principles of treatment of choroidal neovascularization with photodynamic therapy in age-related macular degeneration. Semin. Ophthalmol. 1999, 14, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Schmidt-Erfurth, U.; Sickenberg, M.; Pournaras, C.J.; Laqua, H.; Barbazetto, I.; Zografos, L.; Piguet, B.; Donati, G.; Lane, A.M.; et al. Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: Results of a single treatment in a phase 1 and 2 study. Arch. Ophthalmol. 1999, 117, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Erfurth, U.; Miller, J.W.; Sickenberg, M.; Laqua, H.; Barbazetto, I.; Gragoudas, E.S.; Zografos, L.; Piguet, B.; Pournaras, C.J.; Donati, G.; et al. Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: Results of retreatments in a phase 1 and 2 study. Arch. Ophthalmol. 1999, 117, 1177–1187. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Adamis, A.P.; Cunningham, E.T., Jr.; Goldbaum, M.; Guyer, D.R.; Katz, B.; Patel, M. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 1508.e1-25. [Google Scholar]
- Clearkin, L.; Ramasamy, B.; Wason, J.; Tiew, S. Anti-VEGF intervention in neovascular AMD: Benefits and risks restated as natural frequencies. BMJ Open Ophthalmol. 2019, 4, e000257. [Google Scholar] [CrossRef]
- Stefanini, F.R.; Badaró, E.; Falabella, P.; Koss, M.; Farah, M.E.; Maia, M. Anti-VEGF for the management of diabetic macular edema. J. Immunol. Res. 2014, 2014, 632307. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.; Alivernini, G.; Bolletta, E.; Parodi, M.B.; Perri, P. Anti-VEGF Therapy for Retinal Vein Occlusions. Curr. Drug Targets 2016, 17, 328–336. [Google Scholar] [CrossRef]
- El Annan, J.; Carvounis, P.E. Current management of vitreous hemorrhage due to proliferative diabetic retinopathy. Int. Ophthalmol. Clin. 2014, 54, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Singh, R.P. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context 2018, 7, 212532. [Google Scholar] [CrossRef] [PubMed]
- Simha, A.; Aziz, K.; Braganza, A.; Abraham, L.; Samuel, P.; Lindsley, K.B. Anti-vascular endothelial growth factor for neovascular glaucoma. Cochrane Database Syst. Rev. 2020, 2, Cd007920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cp, J.; Al, G.; Jd, L. Combination of intravitreal bevacizumab and peripheral photocoagulation: An alternative treatment in eales disease. Med. Hypothesis Discov. Innov. Ophthalmol. 2013, 2, 30–34. [Google Scholar]
- Li, S.; Deng, G.; Liu, J.; Ma, Y.; Lu, H. The effects of a treatment combination of anti-VEGF injections, laser coagulation and cryotherapy on patients with type 3 Coat’s disease. BMC Ophthalmol. 2017, 17, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Zarbin, M.A.; Bhagat, N. Anti-VEGF for Management of Neovascularization of Iris and Neovascular Glaucoma. J. Vitreoretinal Dis. 2018, 2, 194–199. [Google Scholar] [CrossRef]
- Slabaugh, M.; Salim, S. Use of Anti-VEGF Agents in Glaucoma Surgery. J. Ophthalmol. 2017, 2017, 1645269. [Google Scholar] [CrossRef] [PubMed]
- Mak, R.K.; Chan, T.C.; Marcet, M.M.; Choy, B.N.; Shum, J.W.; Shih, K.C.; Wong, I.Y.; Ng, A.L. Use of anti-vascular endothelial growth factor in the management of pterygium. Acta Ophthalmol. 2017, 95, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.D.; Cernichiaro-Espinosa, L.A.; Berrocal, A.M. Management of Retinopathy of Prematurity--Use of Anti-VEGF Therapy. Asia Pac. J. Ophthalmol. (Phila) 2018, 7, 56–62. [Google Scholar]
- Ghasemi Falavarjani, K.; Parvaresh, M.-M.; Modarres, M.; Hashemi, M.; Samiy, N. Intravitreal bevacizumab for pseudophakic cystoid macular edema; a systematic review. J. Ophthalmic. Vis. Res. 2012, 7, 235–239. [Google Scholar]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27 (Suppl. 1), i33–i39. [Google Scholar] [CrossRef]
- Park, D.J.; Thomas, N.J.; Yoon, C.; Yoon, S.S. Vascular endothelial growth factor a inhibition in gastric cancer. Gastric. Cancer 2015, 18, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanke, C.D.; Rankin, C.; Corless, C.; Eary, J.F.; Mulder, K.; Okuno, S.H.; George, S.; Heinrich, M. S0502: A SWOG Phase III Randomized Study of Imatinib, With or Without Bevacizumab, in Patients With Untreated Metastatic or Unresectable Gastrointestinal Stromal Tumors. Oncologist 2015, 20, 1353–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Vervenne, W.L.; Bennouna, J.; Humblet, Y.; Gill, S.; Van Laethem, J.L.; Verslype, C.; Scheithauer, W.; Shang, A.; Cosaert, J.; et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2009, 27, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Korpanty, G.; Smyth, E.; Carney, D.N. Update on anti-angiogenic therapy in non-small cell lung cancer: Are we making progress? J. Thorac. Dis. 2011, 3, 19–29. [Google Scholar]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. Ranibizumab versus Verteporfin for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.C.; Busbee, B.G.; Regillo, C.D.; Wieland, M.R.; Van Everen, S.A.; Li, Z.; Rubio, R.G.; Lai, P. Twenty-four-Month Efficacy and Safety of 0.5 mg or 2.0 mg Ranibizumab in Patients with Subfoveal Neovascular Age-Related Macular Degeneration. Ophthalmology 2014, 121, 2181–2192. [Google Scholar] [CrossRef] [Green Version]
- Wykoff, C.C.; Croft, D.E.; Brown, D.M.; Wang, R.; Payne, J.F.; Clark, L.; Abdelfattah, N.S.; Sadda, S.R. Prospective Trial of Treat-and-Extend versus Monthly Dosing for Neovascular Age-Related Macular Degeneration: TREX-AMD 1-Year Results. Ophthalmology 2015, 122, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Antoszyk, A.N.; Tuomi, L.; Chung, C.Y.; Singh, A. Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration (FOCUS): Year 2 results. Am. J. Ophthalmol. 2008, 145, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Schauwvlieghe, A.M.; Dijkman, G.; Hooymans, J.M.; Verbraak, F.D.; Hoyng, C.B.; Dijkgraaf, M.G.; Peto, T.; Vingerling, J.R.; Schlingemann, R.O. Comparing the Effectiveness of Bevacizumab to Ranibizumab in Patients with Exudative Age-Related Macular Degeneration. The BRAMD Study. PLoS ONE 2016, 11, e0153052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, K.; Pedersen, T.R.; Sandvik, L.; Bragadóttir, R. Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology 2015, 122, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L., 3rd. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: Two-year results. Ophthalmology 2012, 119, 1388–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodjikian, L.; Souied, E.H.; Mimoun, G.; Mauget-Faÿsse, M.; Behar-Cohen, F.; Decullier, E.; Huot, L.; Aulagner, G. Ranibizumab versus Bevacizumab for Neovascular Age-related Macular Degeneration: Results from the GEFAL Noninferiority Randomized Trial. Ophthalmology 2013, 120, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Harding, S.P.; Rogers, C.A.; Downes, S.M.; Lotery, A.J.; Culliford, L.A.; Reeves, B.C. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013, 382, 1258–1267. [Google Scholar] [CrossRef]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef]
- Dugel, P.U.; Koh, A.; Ogura, Y.; Jaffe, G.J.; Schmidt-Erfurth, U.; Brown, D.M.; Gomes, A.V.; Warburton, J.; Weichselberger, A.; Holz, F.G. HAWK and HARRIER: Phase 3, Multicenter, Randomized, Double-Masked Trials of Brolucizumab for Neovascular Age-Related Macular Degeneration. Ophthalmology 2020, 127, 72–84. [Google Scholar] [CrossRef]
- Li, X.; Xu, G.; Wang, Y.; Xu, X.; Liu, X.; Tang, S.; Zhang, F.; Zhang, J.; Tang, L.; Wu, Q.; et al. Safety and efficacy of conbercept in neovascular age-related macular degeneration: Results from a 12-month randomized phase 2 study: AURORA study. Ophthalmology 2014, 121, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Song, Y.; Xu, G.; Ye, J.; Wu, Z.; Liu, X.; Dong, X.; Zhang, M.; Xing, Y.; Zhu, S.; et al. Conbercept for Treatment of Neovascular Age-related Macular Degeneration: Results of the Randomized Phase 3 PHOENIX Study. Am. J. Ophthalmol. 2019, 197, 156–167. [Google Scholar] [CrossRef]
- Danzig, C.; Quezada, C.; Basu, K.; Grzeschik, S.; Sahni, J.; Silverman, D.; Osborne, A. Efficacy and safety of faricimab every 16 or 12 weeks for neovascular age-related macular degeneration: STAIRWAY phase 2 results. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1212. [Google Scholar]
- Lien, S.; Lowman, H.B. Therapeutic anti-VEGF antibodies. Handb. Exp. Pharm. 2008, 131–150. [Google Scholar] [CrossRef]
- Blick, S.K.; Keating, G.M.; Wagstaff, A.J. Ranibizumab. Drugs 2007, 67, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- van Asten, F.; Michels, C.T.J.; Hoyng, C.B.; van der Wilt, G.J.; Klevering, B.J.; Rovers, M.M.; Grutters, J.P.C. The cost-effectiveness of bevacizumab, ranibizumab and aflibercept for the treatment of age-related macular degeneration-A cost-effectiveness analysis from a societal perspective. PLoS ONE 2018, 13, e0197670. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef]
- Stewart, M.W.; Grippon, S.; Kirkpatrick, P. Aflibercept. Nat. Rev. Drug Discov. 2012, 11, 269–270. [Google Scholar] [CrossRef]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem. Pharm. 2019, 168, 341–351. [Google Scholar] [CrossRef]
- Fernandes, C.F.C.; Pereira, S.D.S.; Luiz, M.B.; Zuliani, J.P.; Furtado, G.P.; Stabeli, R.G. Camelid Single-Domain Antibodies As an Alternative to Overcome Challenges Related to the Prevention, Detection, and Control of Neglected Tropical Diseases. Front. Immunol. 2017, 8, 653. [Google Scholar] [CrossRef]
- Gaudreault, J.; Gunde, T.; Floyd, H.S.; Ellis, J.; Tietz, J.; Binggeli, D.; Keller, B.; Schmidt, A.; Escher, D. Preclinical Pharmacology and Safety of ESBA1008, a Single-chain Antibody Fragment, Investigated as Potential Treatment for Age Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3025. [Google Scholar]
- Plyukhova, A.A.; Budzinskaya, M.V.; Starostin, K.M.; Rejdak, R.; Bucolo, C.; Reibaldi, M.; Toro, M.D. Comparative Safety of Bevacizumab, Ranibizumab, and Aflibercept for Treatment of Neovascular Age-Related Macular Degeneration (AMD): A Systematic Review and Network Meta-Analysis of Direct Comparative Studies. J. Clin. Med. 2020, 9, 1522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yu, D.; Yang, C.; Xia, Q.; Li, W.; Liu, B.; Li, H. The Pharmacology Study of a New Recombinant Human VEGF Receptor-Fc Fusion Protein on Experimental Choroidal Neovascularization. Pharm. Res. 2009, 26, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Wu, C.; Bian, A.-l.; Geng, S.; Dai, R.-p. Comparison of aqueous humor levels of PlGF and VEGF in proliferative diabetic retinopathy before and after intravitreal conbercept injection. Diabetes Res. Clin. Pract. 2020, 162, 108083. [Google Scholar] [CrossRef] [PubMed]
- Regula, J.T.; Lundh von Leithner, P.; Foxton, R.; Barathi, V.A.; Cheung, C.M.G.; Bo Tun, S.B.; Wey, Y.S.; Iwata, D.; Dostalek, M.; Moelleken, J.; et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol. Med. 2016, 8, 1265–1288. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Ripasudil: First global approval. Drugs 2014, 74, 2211–2215. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Q.; Zhang, Y.; Gao, X.; Li, H.; Yuan, Z. Ripasudil alleviated the inflammation of RPE cells by targeting the miR-136-5p/ROCK/NLRP3 pathway. BMC Ophthalmol. 2020, 20, 134. [Google Scholar] [CrossRef]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-M.; Estcourt, M.J.; Wikstrom, M.; Himbeck, R.P.; Barnett, N.L.; Brankov, M.; Tee, L.B.G.; Dunlop, S.A.; Degli-Esposti, M.A.; Rakoczy, E.P. rAAV.sFlt-1 Gene Therapy Achieves Lasting Reversal of Retinal Neovascularization in the Absence of a Strong Immune Response to the Viral Vector. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4279–4287. [Google Scholar] [CrossRef]
- Rakoczy, E.P.; Magno, A.L.; Lai, C.M.; Pierce, C.M.; Degli-Esposti, M.A.; Blumenkranz, M.S.; Constable, I.J. Three-Year Follow-Up of Phase 1 and 2a rAAV.sFLT-1 Subretinal Gene Therapy Trials for Exudative Age-Related Macular Degeneration. Am. J. Ophthalmol. 2019, 204, 113–123. [Google Scholar] [CrossRef]
- Constable, I.J.; Pierce, C.M.; Lai, C.-M.; Magno, A.L.; Degli-Esposti, M.A.; French, M.A.; McAllister, I.L.; Butler, S.; Barone, S.B.; Schwartz, S.D.; et al. Phase 2a Randomized Clinical Trial: Safety and Post Hoc Analysis of Subretinal rAAV.sFLT-1 for Wet Age-related Macular Degeneration. EBioMedicine 2016, 14, 168–175. [Google Scholar] [CrossRef] [Green Version]
- EXPLORE: A Phase II Study to Evaluate the Safety and Efficacy of Two Doses of GT005. Available online: https://clinicaltrials.gov/ct2/show/NCT04437368 (accessed on 15 October 2020).
- RGX-314 Gene Therapy for Neovascular AMD Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT03066258 (accessed on 15 October 2020).
- Safety and Tolerability Study of AAV2-sFLT01 in Patients with Neovascular Age-Related Macular Degeneration (AMD). Available online: https://clinicaltrials.gov/ct2/show/NCT01024998 (accessed on 15 October 2020).
- Study of AdGVPEDF.11D in Neovascular Age-Related Macular Degeneration (AMD). Available online: https://clinicaltrials.gov/ct2/show/NCT00109499 (accessed on 15 October 2020).
- A Follow-up Study to Evaluate the Safety of RetinoStat® in Patients with Age-Related Macular Degeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT01678872 (accessed on 15 October 2020).
- Safety and Efficacy Study of rAAV.sFlt-1 in Patients with Exudative Age-Related Macular Degeneration. Available online: https://ClinicalTrials.gov/show/NCT01494805 (accessed on 15 October 2020).
- AAVCAGsCD59 for the Treatment of Wet AMD. Available online: https://ClinicalTrials.gov/show/NCT03585556 (accessed on 15 October 2020).
- ADVM-022 Intravitreal Gene Therapy for Wet AMD. Available online: https://ClinicalTrials.gov/show/NCT03748784 (accessed on 15 October 2020).
- Regenxbio Inc. Long-Term Follow-Up Study of RGX-314. Available online: https://ClinicalTrials.gov/show/NCT03999801: 2019 (accessed on 15 October 2020).
- Figueroa, A.G.; Boyd, B.M.; Christensen, C.A.; Javid, C.G.; McKay, B.S.; Fagan, T.C.; Snyder, R.W. Levodopa Positively Affects Neovascular Age-Related Macular Degeneration. Am. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
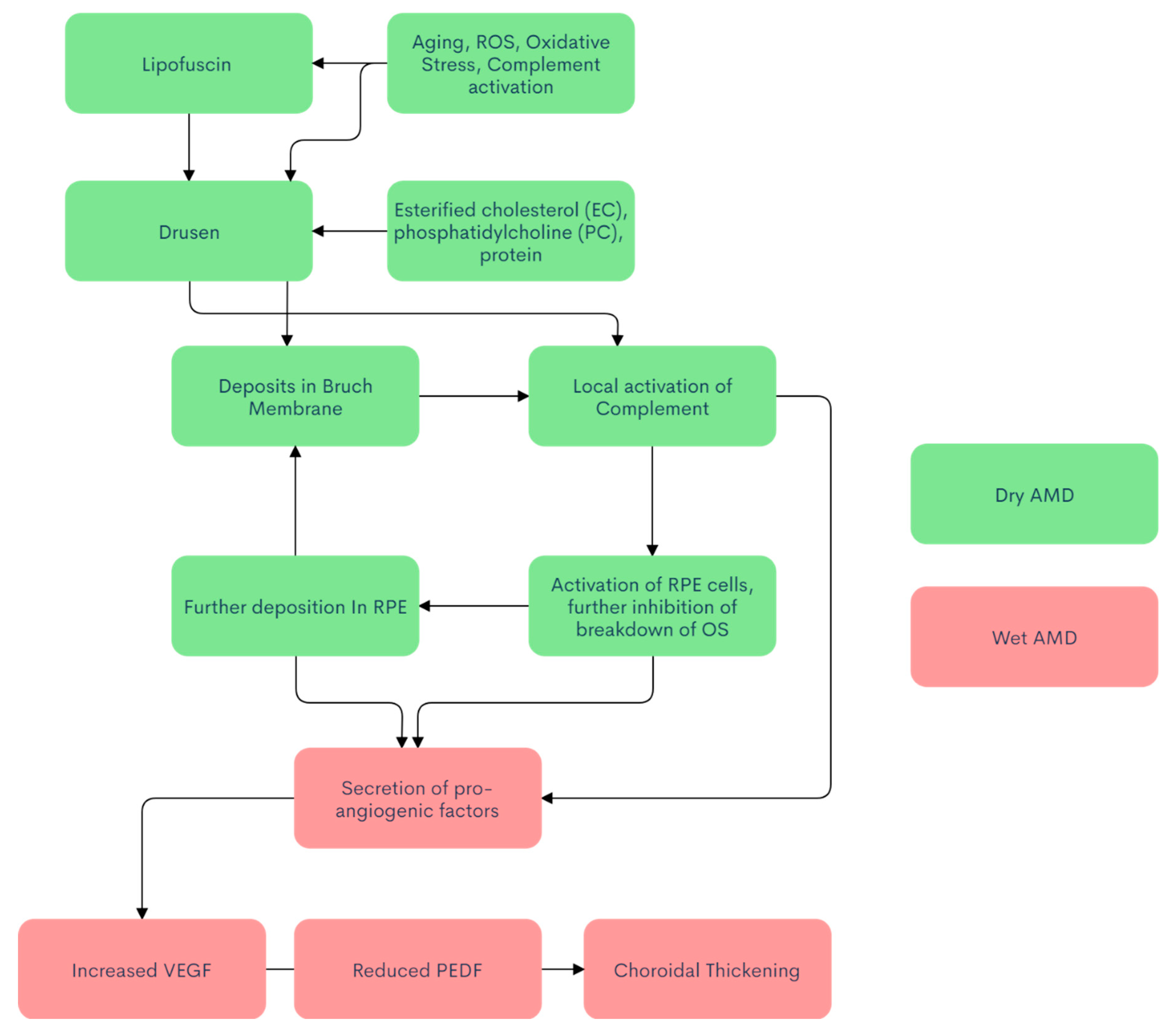



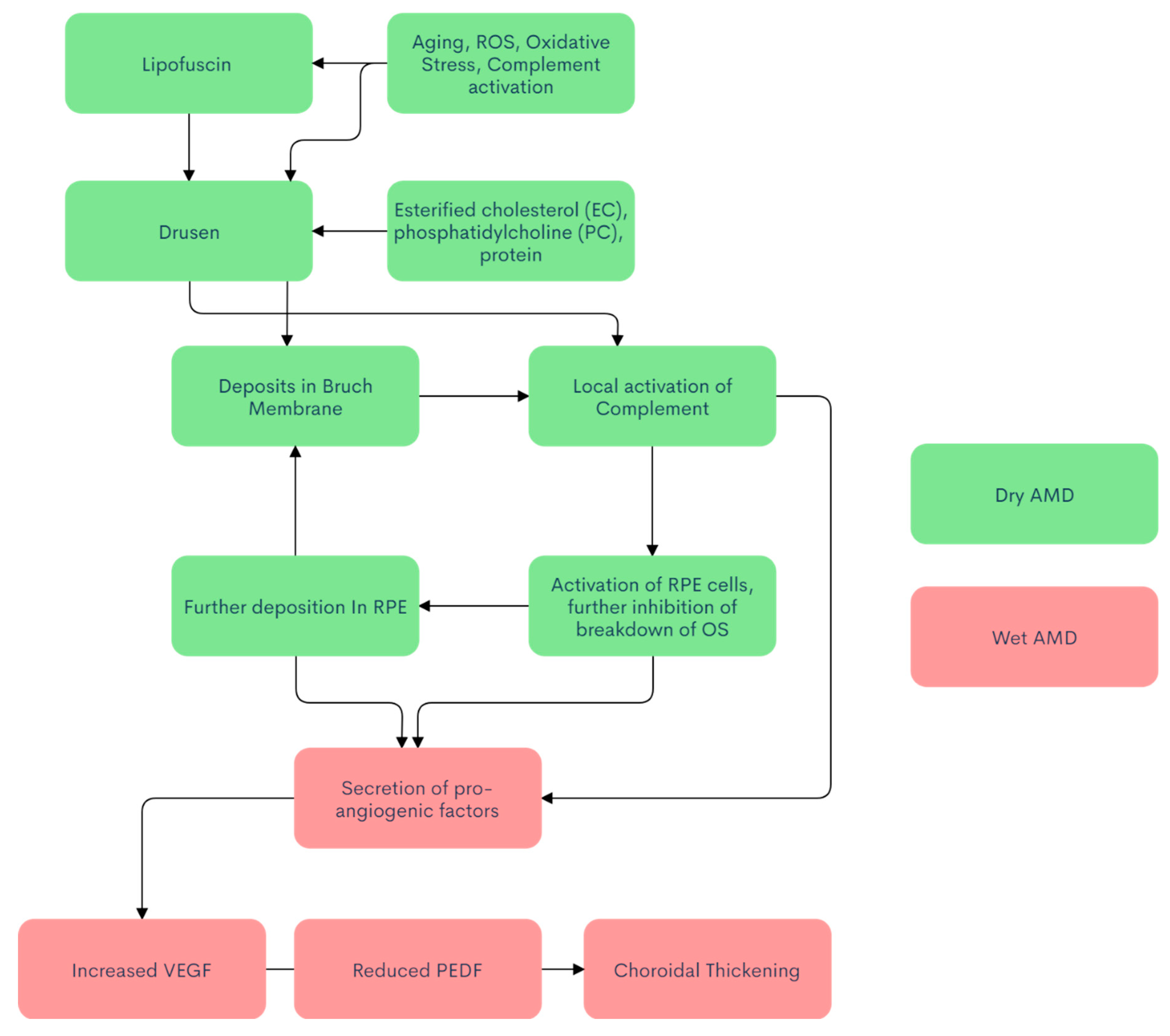{kind=link}
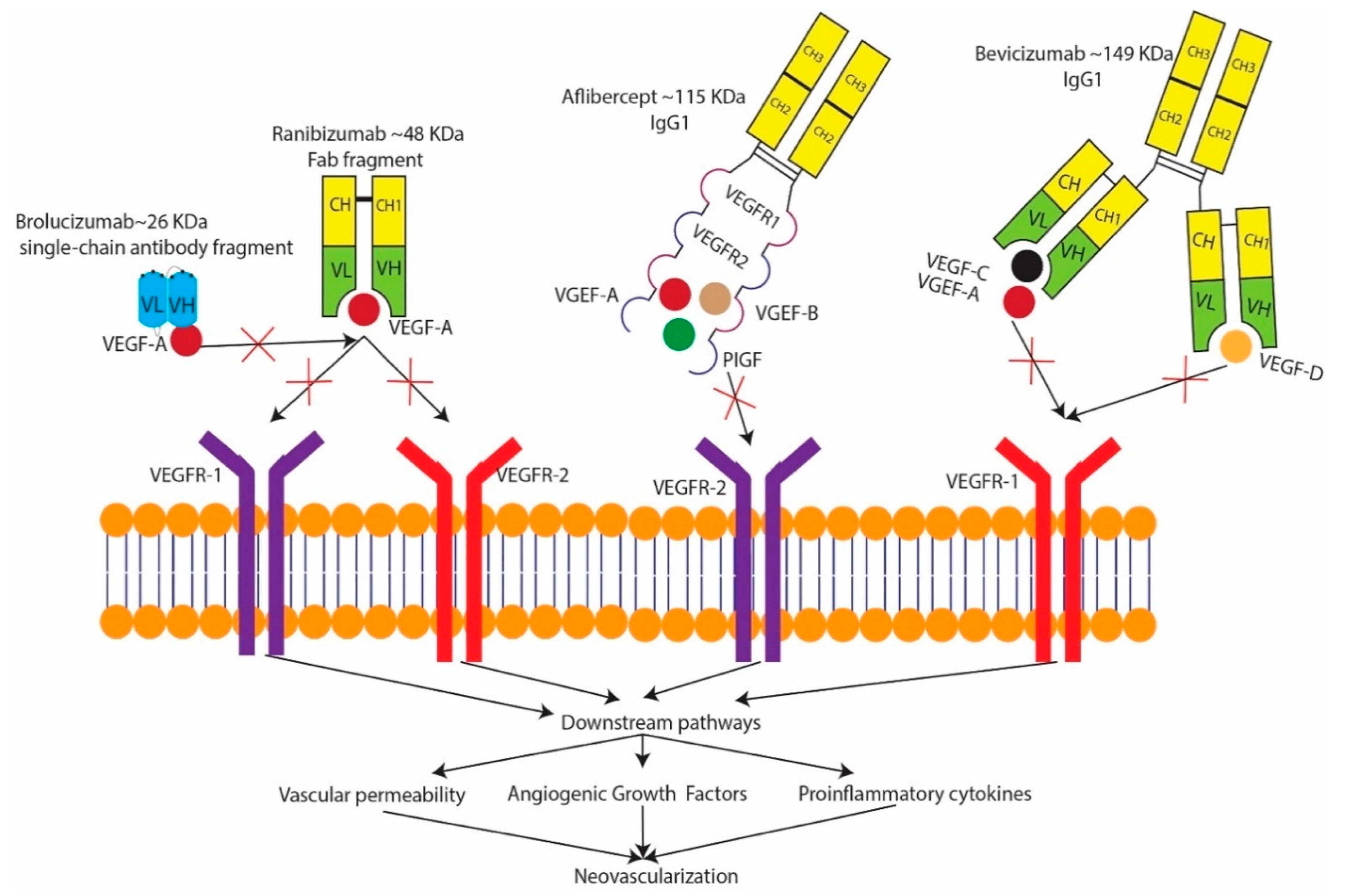
{kind=link}
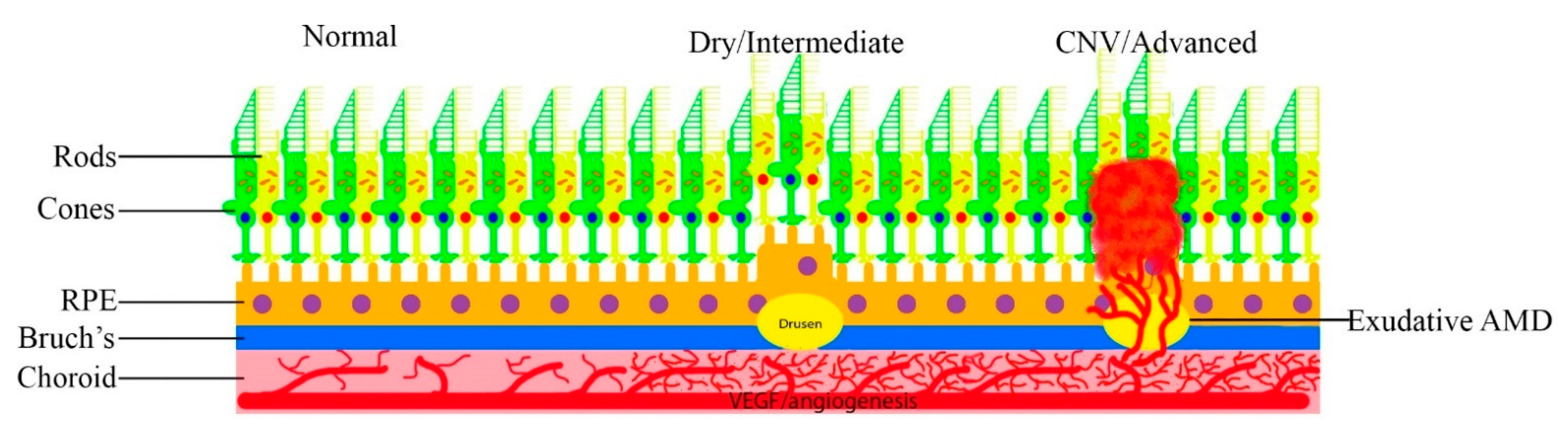
{kind=link}
{kind=link}
| Age (Years) | Predicted Prevalence % (nvAMD) | Range |
|---|---|---|
| 50 | 0.04 | 0.02–0.07 |
| 55 | 0.08 | 0.05–0.13 |
| 60 | 0.17 | 0.11–0.25 |
| 65 | 0.34 | 0.23–0.49 |
| 70 | 0.7 | 0.48–0.97 |
| 75 | 1.4 | 0.99–1.90 |
| 80 | 2.79 | 1.99–3.79 |
| 85 | 5.48 | 3.91–7.45 |
| 90 | 10.49 | 7.45–14.37 |
| Study | Participants (n=) | Population | nvAMD Prevalence |
|---|---|---|---|
| Beaver Dam Eye Study [25] | 4771 | White | 1.2% Total |
| National Health and Nutrition Examination Survey III [26] | 7888 | White | 0.30% |
| Black | 0.10% | ||
| Mexican-Americans | 0.10% | ||
| The Baltimore eye Study [20] | 4361 | White | 0.60% |
| Black | 0.10% | ||
| The Los Angles Latino Eye Study [27] | 5875 | Mexican-Americans | 0.29% |
| Proyecto [24] | 2776 | Mexican-Americans | 0.14% |
| The Salisbury Eye Evaluation Project [21] | 2008 | White | 1.70% |
| Black | 1.00% | ||
| Hisayama Study [28] | 1486 | Japanese | 0.67% |
| Factor | Characteristics | Increased Risk | Suggested Method of Action |
|---|---|---|---|
| Smoking | Current smokers | 4.55 fold nvAMD * [57] | Direct oxidation of RPE [4] |
| Depletion of antioxidants [58] | |||
| Former smokers | 1.54 fold nvAMD * [57] | Activation of Complement [59] | |
| * compared to having never smoked | Atherosclerotic vascular changes [60] | ||
| Alcohol | Heavy drinker (daily alcoholic beverages >4) | RR 6.51 nvAMD * [37] | Oxidative stress [61] |
| * compared to subjects with 0 g Alcohol/day, adjusted for age and gender | |||
| BMI | Obesity (≥30 kg/m2) | 32% increase incidence of late AMD [55] | Oxidative stress [62] |
| Diet | Mediterranean diet | −26% lower risk of progression to advanced (nvAMD or GA) AMD [2] | Certain foods are retino-protective or rich in anti-oxidants [63,64] |
| Western diet | +56% increase in early AMD | ||
| +270% increase advanced AMD [2] |
| Study Authors | Diet Rich vs. Poor in Nutrient | Dose Required for Reduction | Odds Ratio | Reduction in AMD |
|---|---|---|---|---|
| Aoki et al., (2016) [72] | Zinc | ≥10.2 (mg/d) | 0.10 1 | nvAMD = 60–90% [2] |
| Vitamin D | ≥27.5 (μgram/d) | 0.40 1 | ||
| α-tocopherol | ≥10.6 (mg/d) | 0.20 1 | ||
| Vitamin C | ≥184.9 (mg/d) | 0.40 1 | ||
| Omega−3 fatty acids | ≥3.9 (g/d) | 0.20 1 | ||
| β-carotene | ≥6.04 (mg/d) | 0.20 1 | ||
| Hogg et al., (2017) [73] | MDS * score | Quartile 4 (high) | 0.53 2 0.61 3 | nvAMD = 47% [2] nvAMD = 39% [2] |
| SanGiovanni et al., (2007) [74] | Total fish intake | >2 servings/week | nvAMD = 39% [2] | |
| Chiu et al., (2014) [66] | Oriental vs. Western diet | Diet quintile | ||
| Oriental pattern score | Quintile 5 (high) | 0.38 4 | Advanced AMD = 62% [2] | |
| Western pattern score | Quintile 5 (high) | 3.7 5 | Advanced AMD = 270% increase [2] |
| Study Name | Drug | Control | Treatment | Follow up | Change in ETDRS | |
|---|---|---|---|---|---|---|
| Treatment 1 | Control 2 | |||||
| MARINA [218] | Ranibizumab | Sham injections q 4 weeks | Ranibizumab 0·5 mg q 4 weeks | 1 year | +7.2 | –10.4 |
| ANCHOR [219] | Ranibizumab | Sham injection q 4 weeks plus PDT | Ranibizumab 0·5 mg q 4 weeks+ sham PDT | 1 year | +11.3 | –9.5 |
| HARBOR [220] | Ranibizumab | Ranibizumab 0·5 q 4 weeks | Ranibizumab 0·5 mg q 4 weeks × 3 months then as required | 2 years | +7.9 | +9.1 |
| TREX [221] | Ranibizumab | Ranibizumab 0·5 q 4 weeks | Ranibizumab 0·5 mg q 4 weeks × 3 months until disease inactive then extension per protocol | 1 year | +10.5 | +9.2 |
| FOCUS [222] | Ranibizumab | Sham injections+PDT | Ranibizumab+PDT 0.5 mg q 4 weeks, PDT = day one + quarterly if needed | 2 years | 4.6 | −7.8 |
| BRAMD [223] | Bevacizumab | Ranibizumab 0·5 mg q 4 weeks | Bevacizumab 1·25 mg q 4 weeks | 1 year | +5.1 | +6.4 |
| LUCAS [224] | Bevacizumab | Ranibizumab 0·5 mg q 4 weeks until nvAMD is inactive, then extension by 2 weeks (maximum of 12 weeks) | Bevacizumab 1·25 mg q 4 weeks until nvAMD is inactive, then extension by 2 weeks (maximum of 12 weeks) | 1 year | +7.9 | +8.2 |
| CATT [225] | Bevacizumab | Ranibizumab 0·5 mg q 4 weeks | Bevacizumab 1·25 mg q 4 weeks | 1 year | +7.8 | +8.8 |
| GEFAL [226] | Bevacizumab | Ranibizumab 0·5 mg q 4 weeks × 3 months, then as needed | Bevacizumab 0·5 mg q 4 weeks × 3 months, then as needed | 1 year | +4.8 | +2.9 |
| IVAN [227] | Bevacizumab | Ranibizumab 0·5 mg q 4 weeks | Bevacizumab 1·25 mg q 4 weeks | 2 yearss | +4.1 | +4.9 |
| VIEW 1 [228] | Aflibercept | Ranibizumab 0·5 mg q 4 weeks | Aflibercept 2 mg every 2 months | 1 year | +7.9 | +8.1 |
| VIEW 2 [228] | Aflibercept | Ranibizumab 0·5 mg q 4 weeks | Aflibercept 2 mg every 2 months | 1 year | +8.9 | +9.4 |
| HAWK [229] | Brolucizumab | Aflibercept 2 mg q 8 weeks | Brolucizumab 3 mg q 12 weeks Or Brolucizumab 6 mg q12 weeks | 96 weeks | +5.6 +5.9 | +5.3 |
| HARRIER [229] | Brolucizumab | Aflibercept 2 mg q 8 weeks | Brolucizumab 6 mg q12 weeks | 96 weeks | +6.1 | +6.6 |
| AURORA [230] | Conbercept | Conbercept 0·5 mg q 4 weeks × 3 months, then monthly or as required | Conbercept 2 mg q 4 weeks × 3 months, then monthly or as required | 1 year | +15.4 | +9.3 |
| PHOENIX [231] | Conbercept | Sham injections q 4 weeks × 3 months then conbercept 0.5 mg q 4 weeks × 3 months | Conbercept 0.5 mg q 4 weeks × 3 months, then quarterly | 1 year | +9.98 | +8.81 |
| STAIRWAY [232] | Farcizumab | 0.5 mg ranibizumab q 4 weeks × 52 weeks | Farcizumab 6 mg q 16 weeks | 52 weeks | +11.4 | +9.6 |
| Or | ||||||
| Farcizumab 6 mg q 12 weeks | +10.1 | |||||
| Trial Name | Targeted Gene | Vector | Sponsor |
|---|---|---|---|
| NCT01494805 [257] | sFLT01 | AAV-2 | Adverum Biotechnologies, Inc. (Redwoodcity, CA, USA) |
| NCT03585556 [258] | sCD59 | AAV-2 | Hemera Biosciences (Waltham, MA, USA) |
| NCT01024998 [254] | sFLT-1 | AAV-2 | Sanofi Genzyme (Cambridge, MA, USA) |
| NCT03748784 [259] | Aflibercept | AAV-2 | Adverum Biotechnologies, Inc. (Redwoodcity, CA, USA) |
| NCT03066258 [253] | Anti-VEGF Fab | AAV-8 | Regenxbio Inc.(Rockville, MD, USA) |
| NCT01678872 [256] | Endostatin/Angiostatin | EIAV | Oxford BioMedica (Oxford, UK) |
| NCT00109499 [255] | PEDF | AAV5 | GenVec (Gaithersburg, MD, USA) |
| NCT03999801 [260] | Monoclonal antibody fragment | AAV-8 | Regenxbio Inc. (Rockville, MD, USA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. Int. J. Mol. Sci. 2021, 22, 1170. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031170
Pugazhendhi A, Hubbell M, Jairam P, Ambati B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. International Journal of Molecular Sciences. 2021; 22(3):1170. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031170
Chicago/Turabian StylePugazhendhi, Arunbalaji, Margaret Hubbell, Pooja Jairam, and Balamurali Ambati. 2021. "Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy" International Journal of Molecular Sciences 22, no. 3: 1170. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031170