
Sexual Dimorphisms, Anti-Hormonal Therapy and Cardiac Arrhythmias


 ,
,
Abstract
:1. Introduction
2. Sex Differences in QT Duration and TdP Risk
3. Role of Estrogen and Progesterone during the Menstrual Cycle, and Hormonal Substitution in Menopause in QTc Variation
4. QTc Variation in Hypoestrogenic and Hyperandrogenic States in Women
5. Exogenous Hormonal Therapy and its Effect on QTc Variation
5.1. Exogenous Hormonal Therapy in Women
5.2. Exogenous Hormonal Therapy in Men
6. Role of Sex and Inherited Channelopathies
6.1. Inherited Channelopathies in Women
6.2. Inherited Channelopathies in Men
6.3. Atrial Fibrillation and Sex Hormones
7. QTc Variation in Transgender Individuals
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Curtis, M.J.; Hancox, J.C.; Farkas, A.; Wainwright, C.L.; Stables, C.L.; Saint, D.A.; Clements-Jewery, H.; Lambiase, P.D.; Billman, G.E.; Janse, M.J.; et al. The Lambeth Conventions (II): Guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol. Ther. 2013, 139, 213–248. [Google Scholar] [CrossRef] [PubMed]
- Funck-Brentano, C.; Jaillon, P. Rate-corrected QT interval: Techniques and limitations. Am. J. Cardiol. 1993, 72, B17–B22. [Google Scholar] [CrossRef]
- Roden, D.M. Drug-induced prolongation of the QT interval. N. Engl. J. Med. 2004, 350, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Locati, E.T.; Bagliani, G.; Padeletti, L. Normal ventricular repolarization and QT interval: Ionic background, modifiers, and measurements. Card Electrophysiol. Clin. 2017, 9, 487–513. [Google Scholar] [CrossRef] [PubMed]
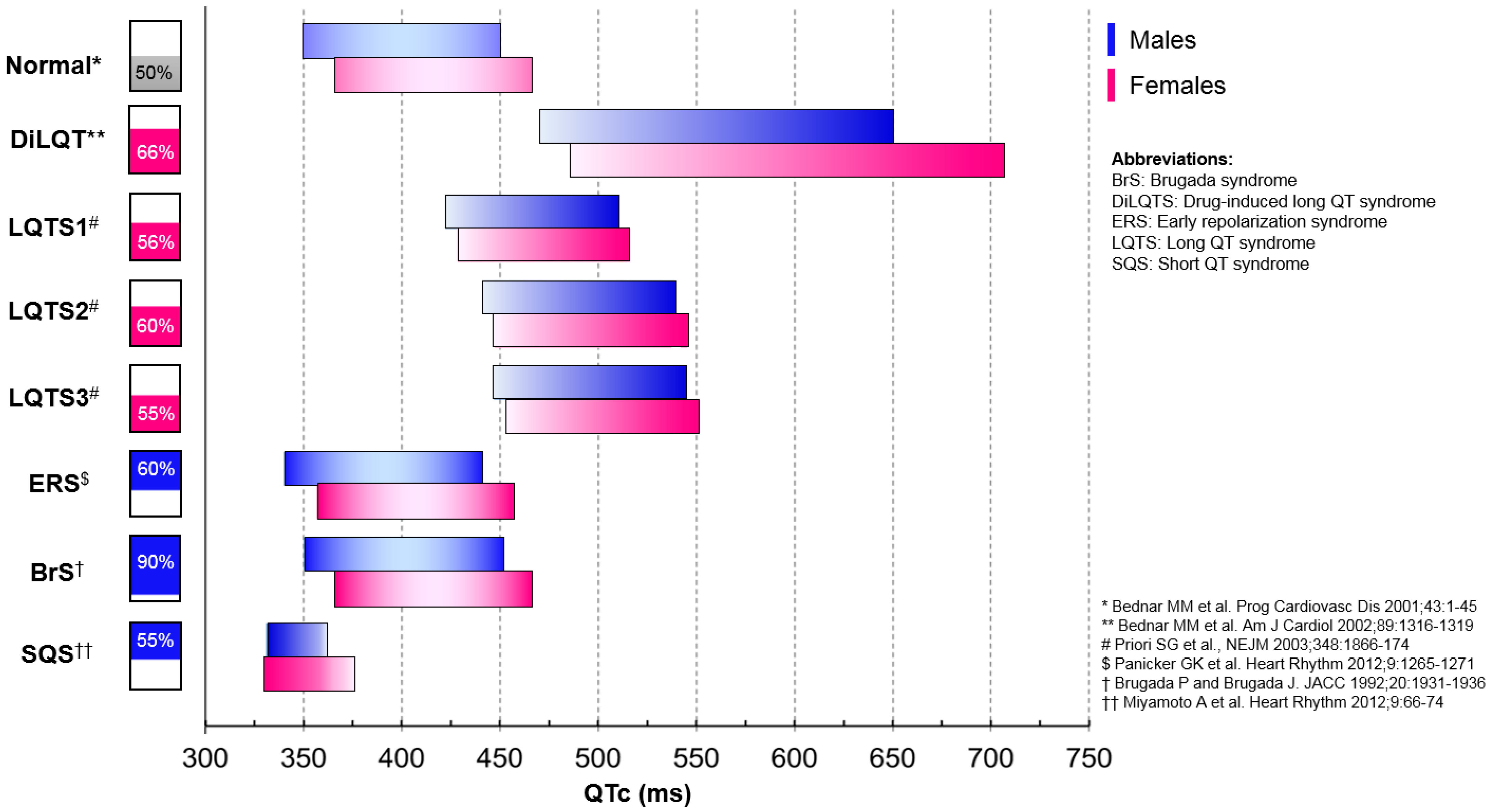
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk Stratification in the Long-QT Syndrome. N. Engl. J. Med. 2003, 348, 1866–1874. [Google Scholar] [CrossRef]
- Sauer, A.J.; Moss, A.J.; McNitt, S.; Peterson, D.R.; Zareba, W.; Robinson, J.L.; Qi, M.; Goldenberg, I.; Hobbs, J.B.; Ackerman, M.J.; et al. Long QT syndrome in adults. J. Am. Coll. Cardiol. 2007, 49, 329–337. [Google Scholar] [CrossRef]
- Locati, E.T.; Bagliani, G.; Cecchi, F.; Johny, H.; Lunati, M.; Pappone, C. Arrhythmias due to inherited and acquired abnormalities of ventricular repolarization. Card. Electrophysiol. Clin. 2019, 11, 345–362. [Google Scholar] [CrossRef]
- Barber, M.; Nguyen, L.S.; Wassermann, J.; Spano, J.-P.; Funck-Brentano, C.; Salem, J.-E. Cardiac arrhythmia considerations of hormone cancer therapies. Cardiovasc. Res. 2019, 115, 878–894. [Google Scholar] [CrossRef]
- Tester, D.J.; Ackerman, M.J. Genetics of Long QT Syndrome. Methodist DeBakey Cardiovasc. J. 2014, 10, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Mazzanti, A.; Maragna, R.; Vacanti, G.; Monteforte, N.; Bloise, R.; Marino, M.; Braghieri, L.; Gambelli, P.; Memmi, M.; Pagan, E.; et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with long QT syndrome. J. Am. Coll. Cardiol. 2018, 71, 1663–1671. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Yan, G.-X. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 2015, 12, 1852–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of ventricular fibrillation episodes in brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Koncz, I.; Gurabi, Z.; Patocskai, B.; Panama, B.K.; Szél, T.; Hu, D.; Barajas-Martínez, H.; Antzelevitch, C. Mechanisms underlying the development of the electrocardiographic and arrhythmic mani-festations of early repolarization syndrome. J. Mol. Cell Cardiol. 2014, 68, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, G.K.; Manohar, D.; Karnad, D.R.; Salvi, V.; Kothari, S.; Lokhandwala, Y. Early repolarization and short QT interval in healthy subjects. Heart Rhythm. 2012, 9, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Salem, J.-E.; Dureau, P.; Bachelot, A.; Germain, M.; Voiriot, P.; LeBourgeois, B.; Trégouët, D.-A.; Hulot, J.-S.; Funck-Brentano, C. Association of oral contraceptives with drug-induced QT interval prolongation in healthy nonmenopausal women. JAMA Cardiol. 2018, 3, 877–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, J.-E.; Waintraub, X.; Courtillot, C.; Shaffer, C.M.; Gandjbakhch, E.; Maupain, C.; Moslehi, J.J.; Badilini, F.; Haroche, J.; Gougis, P.; et al. Hypogonadism as a reversible cause of torsades de pointes in men. Circulation 2018, 138, 110–113. [Google Scholar] [CrossRef]
- Albert, C.M.; Chae, C.U.; Grodstein, F.; Rose, L.M.; Rexrode, K.M.; Ruskin, J.N.; Stampfer, M.J.; Manson, J.E. Prospective study of sudden cardiac death among women in the United States. Circulation 2003, 107, 2096–2101. [Google Scholar] [CrossRef]
- Rienstra, M.; Van Veldhuisen, D.J.; Hagens, V.E.; Ranchor, A.V.; Veeger, N.J.; Crijns, H.J.; Van Gelder, I.C.; RACE Investigators. Gender-related differences in rhythm control treatment in persistent atrial fibrillation: Data of the Rate Control Versus Electrical Cardioversion (RACE) study. J. Am. Coll. Cardiol. 2005, 46, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.R.; Fromm, B.S.; Steinman, R.T.; Meissner, M.D.; Lehmann, M.H. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 1993, 270, 2590–2597. [Google Scholar] [CrossRef]
- Yap, Y.G.; Camm, A.J. Drug induced QT prolongation and torsades de pointes. Heart Br. Card. Soc. 2003, 89, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, S. Drugs for men and women—How important is gender as a risk factor for TdP? Pharmacol. Ther. 2008, 119, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bazett, H.C. An analysis of the time-relations of electrocardiograms. Ann. Noninvasive Electrocardiol. 1997, 2, 177–194. [Google Scholar] [CrossRef]
- Kumar, N.; Saini, D.; Froelicher, V. A gender-based analysis of high school athletes using computerized electrocardiogram meas-urements. PLoS ONE 2013, 8, e53365. [Google Scholar]
- Hii, J.T.; Wyse, D.G.; Gillis, A.M.; Duff, H.J.; Solylo, M.A.; Mitchell, L.B. Precordial QT interval dispersion as a marker of torsade de pointes. Disparate effects of class Ia antiarrhythmic drugs and amiodarone. Circulation 1992, 86, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surawicz, B.; Parikh, S.R. Prevalence of male and female patterns of early ventricular repolarization in the normal ECG of males and females from childhood to old age. J. Am. Coll. Cardiol. 2002, 40, 1870–1876. [Google Scholar] [CrossRef] [Green Version]
- Griffet, V.; Finet, G.; Di Filippo, S.; Lantelme, P.; Caignault, J.R.; Guérard, S. Athlete’s heart in the young: Electrocardiographic and echocardiographic patterns in 107 French athletes. Ann. Cardiol. Angeiol. 2013, 62, 116–121. [Google Scholar] [CrossRef]
- Bessem, B.; de Bruijn, M.C.; Nieuwland, W. Gender differences in the electrocardiogram screening of athletes. J. Sci. Med. Sport 2017, 20, 213–217. [Google Scholar] [CrossRef]
- Mason, J.W.; Ramseth, D.J.; Chanter, D.O.; Moon, T.E.; Goodman, D.B.; Mendzelevski, B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J. Electrocardiol. 2007, 40, 228–234. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Kinirons, M.T.; Swift, C.G.; Jackson, S.H.D. Impact of age on QT interval and QT dispersion in healthy subjects: A regression analysis. Age Ageing 2003, 32, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Rabkin, S.W.; Cheng, X.-B.J.; Thompson, D.J. Detailed analysis of the impact of age on the QT interval. J. Geriatr. Cardiol. 2016, 13, 740–748. [Google Scholar] [PubMed]
- Rautaharju, P.M.; Zhang, Z.-M.; Haisty, W.K.; Gregg, R.E.; Warren, J.; Horacek, M.B.; Kucharska-Newton, A.M.; Rosamond, W.; Soliman, E.Z. Race- and sex-associated differences in rate-adjusted QT, QTpeak, ST elevation and other regional measures of repolarization: The Atherosclerosis Risk in Communities (ARIC) Study. J. Electrocardiol. 2014, 47, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Rijnbeek, P.R.; Van Herpen, G.; Bots, M.L.; Man, S.; Verweij, N.; Hofman, A.; Hillege, H.; Numans, M.E.; Swenne, C.A.; Witteman, J.C.; et al. Normal values of the electrocardiogram for ages 16–90 years. J. Electrocardiol. 2014, 47, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.; Johannesen, L.; Galeotti, L.; Strauss, D.G. Mechanisms of sex and age differences in ventricular repolarization in humans. Am. Heart J. 2014, 168, 749–756.e3. [Google Scholar] [CrossRef]
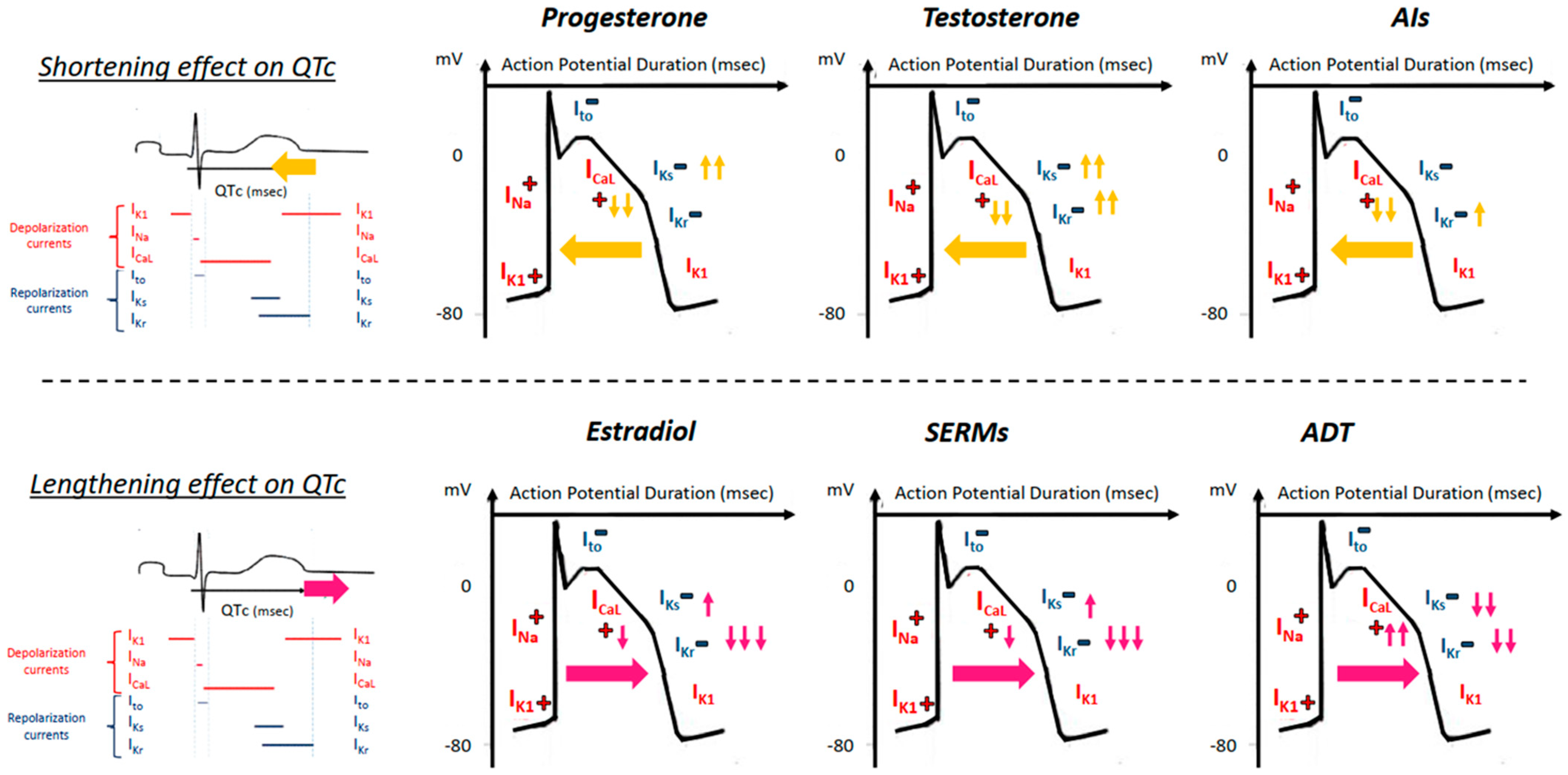
- Salem, J.-E.; Alexandre, J.; Bachelot, A.; Funck-Brentano, C. Influence of steroid hormones on ventricular repolarization. Pharmacol. Ther. 2016, 167, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Abehsira, G.; Bachelot, A.; Badilini, F.; Koehl, L.; Lebot, M.; Favet, C.; Touraine, P.; Funck-Brentano, C.; Salem, J.-E. Complex influence of gonadotropins and sex steroid hormones on QT interval duration. J. Clin. Endocrinol. Metab. 2016, 101, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.H.; Ehlert, F.A.; Kruse, J.T.; Parker, M.A.; Goldberger, J.J.; Kadish, A.H. Gender-specific differences in the QT interval and the effect of autonomic tone and menstrual cycle in healthy adults. Am. J. Cardiol. 1997, 79, 178–181. [Google Scholar] [CrossRef]
- Hulot, J.-S.; Démolis, J.-L.; Rivière, R.; Strabach, S.; Christin-Maitre, S.; Funck-Brentano, C. Influence of endogenous oestrogens on QT interval duration. Eur. Heart J. 2003, 24, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, I.; Kilborn, M.J.; Liu, X.-K.; Pezzullo, J.C.; Woosley, R.L. Drug-induced QT prolongation in women during the menstrual cycle. JAMA 2001, 285, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Dogan, M.; Yiginer, O.; Uz, O.; Kucuk, U.; Degirmencioglu, G.; Isilak, Z.; Uzun, M.; Davulcu, E. The effects of female sex hormones on ventricular premature beats and repolarization parameters in physiological menstrual cycle. Pacing Clin. Electrophysiol. PACE 2016, 39, 418–426. [Google Scholar] [CrossRef]
- Gowda, R.M.; Khan, I.A.; Punukollu, G.; Vasavada, B.C.; Sacchi, T.J.; Wilbur, S.L. Female preponderance in ibutilide-induced torsade de pointes. Int. J. Cardiol. 2004, 95, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Endres, S.; Mayuga, K.A.; De Cristofaro, A.; Taneja, T.; Goldberger, J.J.; Kadish, A. Menstrual cycle and ST height. Ann. Noninvasive Electrocardiol. 2004, 9, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Ooie, T.; Takahashi, N.; Taniguchi, Y.; Anan, F.; Yonemochi, H.; Saikawa, T. Influence of menstrual cycle on QT interval dynamics. Pacing Clin. Electrophysiol. 2006, 29, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Shugg, T.; Egly, C.; Stamatkin, C.W.; Patil, A.S.; Tisdale, J.E.; Overholser, B.R. Progesterone metabolites inhibit the human ether-a-go-go-related gene and predict QT interval length. J. Clin. Pharmacol. 2019, 60, 648–659. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Yu, D.J.; Soong, T.W.; Dawe, G.S.; Bian, J.S. Progesterone impairs human ether-a-go-go-related gene (HERG) trafficking by disruption of in-tracellular cholesterol homeostasis. J. Biol. Chem. 2011, 286, 22186–22194. [Google Scholar] [CrossRef] [Green Version]
- Carnethon, M.; Anthony, M.S.; Cascio, W.E.; Folsom, A.R.; Rautaharju, P.M.; Liao, D.; Evans, G.W.; Heiss, G. A prospective evaluation of the risk of QT prolongation with hormone replacement therapy: The atherosclerosis risk in communities study. Ann. Epidemiol. 2003, 13, 530–536. [Google Scholar] [CrossRef]
- Larsen, J.A.; Tung, R.; Sadananda, R.; Goldberger, J.J.; Horvath, G.; Parker, M.A.; Kadish, A. Effects of hormone replacement therapy on QT interval. Am. J. Cardiol. 1998, 82, 993–995. [Google Scholar] [CrossRef]
- Yildirir, A.; Aybar, F.; Kabakci, M.G.; Yarali, H.; Akgul, E.; Bukulmez, O.; Tokgozoglu, S.L.; Gurgan, T.; Oto, A. Hormone replacement therapy shortens QT dispersion in healthy postmenopausal women. Ann. Noninvasive Electrocardiol. 2001, 6, 193–197. [Google Scholar] [CrossRef]
- Gökçe, M.; Karahan, B.; Yilmaz, R.; Örem, C.; Erdöl, C.; Özdemir, Ş. Long term effects of hormone replacement therapy on heart rate variability, QT interval, QT dispersion and frequencies of arrhytmia. Int. J. Cardiol. 2005, 99, 373–379. [Google Scholar] [CrossRef]
- Haseroth, K.; Seyffart, K.; Wehling, M.; Christ, M. Effects of progestin–estrogen replacement therapy on QT-dispersion in postmenopausal women. Int. J. Cardiol. 2000, 75, 161–165. [Google Scholar] [CrossRef]
- Kadish, A.H.; Greenland, P.; Limacher, M.C.; Frishman, W.H.; Daugherty, S.A.; Schwartz, J.B. Estrogen and progestin use and the QT interval in postmenopausal women. Ann. Noninvasive Electrocardiol. 2004, 9, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, J.E.; Jaynes, H.A.; Overholser, B.R.; Sowinski, K.M.; Flockhart, D.A.; Kovacs, R.J. Influence of oral progesterone administration on Drug-Induced QT interval lengthening: A randomized, double-blind, placebo-controlled crossover study. JACC Clin. Electrophysiol. 2016, 2, 765–774. [Google Scholar] [CrossRef]
- Tisdale, J.E.; Jaynes, H.A.; Overholser, B.R.; Sowinski, K.M.; Kovacs, R.J. Progesterone pretreatment reduces the incidence of drug-induced torsades de pointes in atrioventricular node-ablated isolated perfused rabbit hearts. J. Cardiovasc. Electrophysiol. 2019, 30, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Eshre Guideline Group on POI; Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; de Muinck Keizer-Schrama, S.; Hogervorst, E.; et al. ESHRE Guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. Oxf. Engl. 2016, 31, 926–937. [Google Scholar]
- FSHR Gene—GeneCards|FSHR Protein|FSHR Antibody. Available online: http://www.genecards.org/cgi-bin/carddisp.pl?gene=FSHR&keywords=FSHR (accessed on 10 November 2015).
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.G.; Nielsen, J.C.; Trolle, C.; Gravholt, C.H.; Andersen, N.H. Prolonged QT interval and cardiac arrest after a single dose of amiodarone in a woman with Turner’s syndrome. Clin. Case Rep. 2017, 5, 154–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrahill, N.J.; Yetman, A.T.; Danford, D.A.; Starr, L.J.; Sanmann, J.N.; Robinson, J.A. The QT interval in patients with the turner syndrome. Am. J. Cardiol. 2021, 140, 118–121. [Google Scholar] [CrossRef]
- Dalla Pozza, R.; Bechtold, S.; Kääb, S.; Buckl, M.; Urschel, S.; Netz, H.; Schwarz, H.P. QTc interval prolongation in children with Ulrich-Turner syndrome. Eur. J. Pediatr. 2006, 165, 831–837. [Google Scholar] [CrossRef]
- Atıcı, A.; Panç, C.; Karaayvaz, E.B.; Demirkıran, A.; Kutlu, O.; Kaşalı, K.; Kekeç, E.; Sarı, L.; Sarı, Z.N.A.; Bilge, A.K. Evaluation of the Tp-Te interval, Tp-Te/QTc ratio, and QT dispersion in patients with Turner syndrome. Anatol. J. Cardiol. 2018, 20, 93–99. [Google Scholar] [CrossRef]
- Bondy, C.A.; Ceniceros, I.; Van, P.L.; Bakalov, V.K.; Rosing, D.R. Prolonged rate-corrected QT interval and other electrocardiogram abnormalities in girls with turner syndrome. Pediatrics 2006, 118, e1220–e1225. [Google Scholar] [CrossRef]
- Noordman, I.D.; Duijnhouwer, A.L.; Coert, M.; Bos, M.; Kempers, M.; Timmers, H.J.L.M.; Fejzic, Z.; Velden, J.A.E.M.V.D.; Kapusta, L. No QTc Prolongation in girls and women with turner syndrome. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Trolle, C.; Mortensen, K.H.; Pedersen, L.N.; Berglund, A.; Jensen, H.K.; Andersen, N.H.; Gravholt, C.H. Long QT interval in Turner syndrome—A high prevalence of LQTS gene mutations. PLoS ONE 2013, 8, e69614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ouyang, P.; Post, W.S.; Dalal, D.; Vaidya, D.; Blasco-Colmenares, E.; Soliman, E.Z.; Tomaselli, G.F.; Guallar, E. Sex-steroid hormones and electrocardiographic QT-interval duration: Findings from the third national health and nutrition examination survey and the multi-ethnic study of atherosclerosis. Am. J. Epidemiol. 2011, 174, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; Andersen, M.; Azziz, R.; et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil. Steril. 2018, 110, 364–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazi, E.; Gencer, M.; Hanci, V.; Temiz, A.; Altun, B.; Gungor, A.N.C.; Demirel, M.; Kirilmaz, B. Relationship between QT dispersion with sex hormones and insulin in young women with polycystic ovary syndrome: An observational study. AKD Anatol. J. Cardiol. 2013, 13, 772–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtovec, B.; Meden-Vrtovec, H.; Jensterle, M.; Radovancevic, B. Testosterone-related shortening of QTc interval in women with polycystic ovary syndrome. J. Endocrinol. Investig. 2008, 31, 653–655. [Google Scholar] [CrossRef]
- Akdag, S.; Cim, N.; Yildizhan, R.; Akyol, A.; Ozturk, F.; Babat, N. Two markers in predicting the cardiovascular events in patients with polycystic ovary syndrome: Increased P-wave and QT dispersion. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3508–3514. [Google Scholar]
- Rostamtabar, M.; Esmaeilzadeh, S.; Tourani, M.; Rahmani, A.; Baee, M.; Shirafkan, F.; Saleki, K.; Mirzababayi, S.S.; Ebrahimpour, S.; Nouri, H.R. Pathophysiological roles of chronic low-grade inflammation mediators in poly-cystic ovary syndrome. J. Cell Physiol. 2021, 236, 824–838. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Acampa, M.; Laghi-Pasini, F.; Bertolozzi, I.; Finizola, F.; Vanni, F.; Natale, M.; Bisogno, S.; Cevenini, G.; Cartocci, A.; et al. Cardiac arrest risk during acute infections: Systemic inflammation directly prolongs QTc interval via cytokine-mediated effects on potassium channel expression. Circ. Arrhythm. Electrophysiol. 2020, 13, e008627. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Boutjdir, M.; Capecchi, P.L. COVID-19, arrhythmic risk, and inflammation: Mind the gap! Circulation 2020, 142, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. Systemic inflammation and arrhythmic risk: Lessons from rheumatoid arthritis. Eur. Heart J. 2016, 38, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Yiginer, O.; Dogan, M.; Gün, I.; Kutlu, H.T.; Degirmencioglu, G.; Guliyev, I.; Tokatli, A.; Kilicaslan, F.; Tokatlı, A. The effects of supraphysiological oestrogen levels on ventricular repolarisation parameters. Kardiol. Pol. 2018, 76, 974–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelot, A.; Grouthier, V.; Courtillot, C.; Dulon, J.; Touraine, P. Management of endocrine disease: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Update on the management of adult patients and prenatal treatment. Eur. J. Endocrinol. 2017, 176, R167–R181. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.; Shufelt, C.; Iribarren, C.; Lyon, L.L.; Merz, C.N.B. Oral Contraceptive Use and the ECG: Evidence of an Adverse QT Effect on Corrected QT Interval. Ann. Noninvasive Electrocardiol. 2013, 18, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Salem, J.-E.; Germain, M.; Hulot, J.-S.; Voiriot, P.; LeBourgeois, B.; Waldura, J.; Trégouët, D.-A.; Charbit, B.; Funck-Brentano, C. GENomE wide analysis of sotalol-induced IKr inhibition during ventricular REPOLarization, “GENEREPOL study”: Lack of common variants with large effect sizes. PLoS ONE 2017, 12, e0181875. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Chun, Y.W.; Stroud, D.M.; Mosley, J.D.; Knollmann, B.C.; Hong, C.; Roden, D.M. Screening for acute IKr block is insufficient to detect torsades de pointes liability: Role of late sodium current. Circulation 2014, 130, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, J.; Moslehi, J.J.; Bersell, K.R.; Funck-Brentano, C.; Roden, D.M.; Salem, J.E. Anticancer drug-induced cardiac rhythm disorders: Current knowledge and basic un-derlying mechanisms. Pharmacol. Ther. 2018, 189, 89–103. [Google Scholar] [CrossRef]
- Yang, T.; Meoli, D.F.; Moslehi, J.; Roden, D.M. Inhibition of the α-subunit of phosphoinositide 3-kinase in heart increases late sodium current and is arrhythmogenic. J. Pharmacol. Exp. Ther. 2018, 365, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, J.; Sasano, T.; Kodama, M.; Li, M.; Ebana, Y.; Harada, N.; Honda, S.-I.; Nakaya, H.; Furukawa, T. Aromatase knockout mice reveal an impact of estrogen on drug-induced alternation of murine electrocardiography parameters. J. Toxicol. Sci. 2015, 40, 339–348. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [Green Version]
- Grouthier, V.; Lebrun-Vignes, B.; Glazer, A.M.; Touraine, P.; Funck-Brentano, C.; Pariente, A.; Courtillot, C.; Bachelot, A.; Roden, D.M.; Moslehi, J.; et al. Increased long QT and torsade de pointes reporting on tamoxifen compared with aromatase inhibitors. Heart 2018, 104, 1859–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussaarts, K.G.A.M.; Berger, F.A.; Binkhorst, L.; De Hoop, E.O.-; Van Leeuwen, R.W.F.; Van Alphen, R.J.; Van Stein, D.M.; De Groot, N.M.S.; Mathijssen, R.H.J.; Van Gelder, T. The risk of QTc-interval prolongation in breast cancer patients treated with tamoxifen in combination with serotonin reuptake inhibitors. Pharm. Res. 2019, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charbit, B.; Christin-Maitre, S.; Démolis, J.-L.; Soustre, E.; Young, J.; Funck-Brentano, C. Effects of testosterone on ventricular repolarization in hypogonadic men. Am. J. Cardiol. 2009, 103, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Gagliano-Jucá, T.; Travison, T.G.; Kantoff, P.W.; Nguyen, P.L.; Taplin, M.-E.; Kibel, A.S.; Huang, G.; Bearup, R.; Schram, H.; Manley, R.; et al. Androgen deprivation therapy is associated with prolongation of QTc interval in men with prostate cancer. J. Endocr. Soc. 2018, 2, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basaria, S.; Travison, T.G.; Alford, D.; Knapp, P.E.; Teeter, K.; Cahalan, C.; Eder, R.; Lakshman, K.; Bachman, E.; Mensing, G.; et al. Effects of testosterone replacement in men with opioid-induced androgen deficiency: A randomized controlled trial. Pain 2015, 156, 280–288. [Google Scholar] [CrossRef]
- Basaria, S.; Harman, S.M.; Travison, T.G.; Hodis, H.; Tsitouras, P.; Budoff, M.; Pencina, K.M.; Vita, J.; Dzekov, C.; Mazer, N.A.; et al. Effects of testosterone administration for 3 years on subclinical atherosclerosis progression in older men with low or low-normal testosterone levels: A randomized clinical trial. JAMA 2015, 314, 570–581. [Google Scholar] [CrossRef]
- Gauthaman, K.; Ganesan, A.P. The hormonal effects of Tribulus terrestris and its role in the management of male erectile dysfunction—An evaluation using primates, rabbit and rat. Phytomedicine 2008, 15, 44–54. [Google Scholar] [CrossRef]
- Rahnema, C.D.; Lipshultz, L.I.; Crosnoe, L.E.; Kovac, J.R.; Kim, E.D. Anabolic steroid–induced hypogonadism: Diagnosis and treatment. Fertil. Steril. 2014, 101, 1271–1279. [Google Scholar] [CrossRef]
- Salem, J.E.; Yang, T.; Moslehi, J.J.; Waintraub, X.; Gandjbakhch, E.; Bachelot, A.; Hidden-Lucet, F.; Hulot, J.S.; Knollmann, B.C.; Lebrun-Vignes, B.; et al. Androgenic effects on ventricular repolarization: A translational study from the international pharmacovigilance database to iPSC-cardiomyocytes. Circulation 2019, 140, 1070–1080. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Bertolozzi, I.; Acampa, M.; Cantara, S.; Castagna, M.G.; Pieragnoli, L.; D’Errico, A.; Rossi, M.; Bisogno, S.; El-Sherif, N.; et al. Androgen deprivation therapy for prostatic cancer in patients with torsades de pointes. Front. Pharmacol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Gheorghe, A.C.D.; Ciobanu, A.; Hodorogea, A.S.; Radavoi, G.D.; Jinga, V.; Nanea, I.T.; Gheorghe, G.S. Evolution of electrocardiographic repolarization parameters during antiandrogen therapy in patients with prostate cancer and hypogonadism. Cardiovasc. Toxicol. 2020, 20, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccirillo, G.; Moscucci, F.; Pofi, R.; D’Alessandro, G.; Minnetti, M.; Isidori, A.M.; Francomano, D.; Lenzi, A.; Puddu, P.E.; Alexandre, J.; et al. Changes in left ventricular repolarization after short-term testosterone replacement therapy in hypogonadal males. J. Endocrinol. Investig. 2019, 42, 1051–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, J.-E.; Bretagne, M.; Lebrun-Vignes, B.; Waintraub, X.; Gandjbakhch, E.; Hidden-Lucet, F.; Gougis, P.; Bachelot, A.; Funck-Brentano, C. Clinical characterization of men with long QT syndrome and torsades de pointes associated with hypogonadism: A review and pharmacovigilance study. Arch. Cardiovasc. Dis. 2019, 112, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Muensterman, E.T.; Jaynes, H.A.; Sowinski, K.M.; Overholser, B.R.; Shen, C.; Kovacs, R.J.; Tisdale, J.E. Effect of transdermal testosterone and oral progesterone on drug-induced QT interval lengthening in older men: A randomized, double-blind, placebo-controlled crossover-design study. Circulation 2019, 140, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Di Diego, J.M.; Cordeiro, J.M.; Goodrow, R.J.; Fish, J.M.; Zygmunt, A.C.; Pérez, G.J.; Scornik, F.S.; Antzelevitch, C. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 2002, 106, 2004–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, D.M. Predicting drug-induced QT prolongation and torsades de pointes. J. Physiol. 2016, 594, 2459–2468. [Google Scholar] [CrossRef] [Green Version]
- Er, F.; Michels, G.; Brandt, M.C.; Khan, I.F.Y.; Haase, H.; Eicks, M.; Lindner, M.; Hoppe, U.C. Impact of testosterone on cardiac L-type calcium channels and Ca2+ sparks: Acute actions antagonize chronic effects. Cell Calcium 2007, 41, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ciobotaru, A.; Bopassa, J.C.; Toro, L.; Stefani, E.; Eghbali, M. Estrogen contributes to gender differences in mouse ventricular repolarization. Circ. Res. 2009, 105, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, J.; Kodama, M.; Clancy, C.E.; Furukawa, T. Sex hormonal regulation of cardiac ion channels in drug-induced QT syndromes. Pharmacol. Ther. 2016, 168, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Kurokawa, J. Progesterone regulates cardiac repolarization through a nongenomic pathway: An in vitro patch-clamp and computational modeling study. Circulation 2007, 116, 2913–2922. [Google Scholar] [CrossRef]
- Bai, C.-X.; Kurokawa, J.; Tamagawa, M.; Nakaya, H.; Furukawa, T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation 2005, 112, 1701–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vink, A.S.; Clur, S.-A.B.; Wilde, A.A.M.; Blom, N.A. Effect of age and gender on the QTc-interval in healthy individuals and patients with long-QT syndrome. Trends Cardiovasc. Med. 2018, 28, 64–75. [Google Scholar] [CrossRef]
- Locati, E.H.; Zareba, W.; Moss, A.J.; Schwartz, P.J.; Vincent, G.M.; Lehmann, M.H.; Towbin, J.A.; Priori, S.G.; Napolitano, C.; Robinson, J.L.; et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the International LQTS Registry. Circulation 1998, 97, 2237–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anneken, L.; Baumann, S.; Vigneault, P.; Biliczki, P.; Friedrich, C.; Xiao, L.; Girmatsion, Z.; Takac, I.; Brandes, R.P.; Kissler, S.; et al. Estradiol regulates human QT-interval: Acceleration of cardiac repolarization by enhanced KCNH2 membrane trafficking. Eur. Heart J. 2016, 37, 640–650. [Google Scholar] [CrossRef]
- Odening, K.E.; Koren, G.; Kirk, M. Normalization of QT interval duration in a long QT syndrome patient during pregnancy and the postpartum period due to sex hormone effects on cardiac repolarization. Heart Case Rep. 2016, 2, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odening, K.E.; Choi, B.R.; Liu, G.X.; Hartmann, K.; Ziv, O.; Chaves, L.; Schofield, L.; Centracchio, J.; Zehender, M.; Peng, X.; et al. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while pro-gesterone is protective. Heart Rhythm. 2012, 9, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Giudicessi, J.R.; Brost, B.C.; Traynor, K.D.; Ackerman, M.J. Potential depot medroxyprogesterone acetate-triggered torsades de pointes in a case of congenital type 2 long QT syndrome. Heart Rhythm. 2012, 9, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Obeyesekere, M.N.; Antzelevitch, C.; Krahn, A.D. Management of ventricular arrhythmias in suspected channelopathies. Circ. Arrhythmia Electrophysiol. 2015, 8, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.J.; Tikkanen, J.T.; Porthan, K.; Oikarinen, L.; Jula, A.; Kenttä, T.; Salomaa, V.; Huikuri, H.V. Relationship between testosterone level and early repolarization on 12-lead electrocardiograms in men. J. Am. Coll. Cardiol. 2013, 62, 1633–1634. [Google Scholar] [CrossRef]
- Osborn, J.J. Experimental hypothermia: Respiratory and blood ph changes in relation to cardiac function. Am. J. Physiol. Content 1953, 175, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, K.; Nakagawa, M.; Taniguchi, Y.; Nagano, Y.; Teshima, Y.; Yufu, K.; Takahashi, N.; Nomura, T.; Satoh, F.; Mimata, H.; et al. Gender differences in the ST segment: Effect of androgen-deprivation therapy and possible role of testosterone. Circ. J. 2010, 74, 2448–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hünük, B. The impact of testosterone levels on J-wave patterns observed in healthy Turkish males. Eur. Res. J. 2019, 6, 438–448. [Google Scholar] [CrossRef]
- Noseworthy, P.A.; Tikkanen, J.T.; Porthan, K.; Oikarinen, L.; Pietilä, A.; Harald, K.; Peloso, G.M.; Merchant, F.M.; Jula, A.; Väänänen, H.; et al. The early repolarization pattern in the general population: Clinical correlates and heritability. J. Am. Coll. Cardiol. 2011, 57, 2284–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, J.W.; Moser, C.B.; Murabito, J.M.; Sullivan, L.M.; Wang, N.; Ellinor, P.T.; Vasan, R.S.; Benjamin, E.J.; Coviello, A.D. Association of sex hormones, aging, and atrial fibrillation in men: The Framingham Heart Study. Circ. Arrhythm. Electrophysiol. 2014, 7, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Oni, O.A.; Gupta, K.; Sharma, M.; Sharma, R.; Singh, V.; Parashara, D.; Kamalakar, S.; Dawn, B.; Chen, G.; et al. Normalization of testosterone levels after testosterone replacement therapy is associated with decreased incidence of atrial fibrillation. J. Am. Heart. Assoc. 2017, 6, e004880. [Google Scholar] [CrossRef]
- Chamberlain, A.M.; Agarwal, S.K.; Ambrose, M.; Folsom, A.R.; Soliman, E.Z.; Alonso, A. Metabolic syndrome and incidence of atrial fibrillation among blacks and whites in the Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 2010, 159, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Tsuneda, T.; Yamashita, T.; Kato, T.; Sekiguchi, A.; Sagara, K.; Sawada, H.; Aizawa, T.; Fu, L.-T.; Fujiki, A.; Inoue, H. Deficiency of testosterone associates with the substrate of atrial fibrillation in the rat model. J. Cardiovasc. Electrophysiol. 2009, 20, 1055–1060. [Google Scholar] [CrossRef]
- Tsai, W.C.; Lee, T.I.; Chen, Y.C.; Kao, Y.H.; Lu, Y.Y.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Testosterone replacement increases aged pulmonary vein and left atrium arrhythmogenesis with enhanced adrenergic activity. Int. J. Cardiol. 2014, 176, 110–118. [Google Scholar] [CrossRef]
- Hembree, W.C.; Cohen-Kettenis, P.T.; Gooren, L.; Hannema, S.E.; Meyer, W.J.; Murad, M.H.; Rosenthal, S.M.; Safer, J.D.; Tangpricha, V.; T’Sjoen, G.G. Endocrine treatment of gen-der-dysphoric/gender-incongruent persons: An endocrine society clinical practice guideline. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2017, 23, 1437. [Google Scholar]
- Wamboldt, R.; Haseeb, S.; Waddington, A.; Baranchuk, A. Cardiac arrhythmias secondary to hormone therapy in trans women. Expert Rev. Cardiovasc. Ther. 2019, 17, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Sichrovsky, T.; Mittal, S. Brugada syndrome unmasked by use of testosterone in a transgender male: Gender trumps sex as a risk factor. J. Innov. Card. Rhythm. Manag. 2019, 10, 3526–3529. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Amoabeng, D.; Doshi, R.; Adalja, D.; Kumar, A.; Desai, R.; Islam, R.; Gullapalli, N. Burden of arrythmias in transgender patients hospitalized for gender-affirming surgeries. J. Arrhythmia 2020, 36, 797–800. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Men | |
| Testosterone | QTc shortening in normal men and in hypogonadal men receiving testosterone QTc lengthening in hypogonadal men |
| Estradiol | No influence within physiological ranges Possible QTc lengthening in transwomen |
| Progesterone | No influence within physiological ranges |
| Gonadotropins | QTc lengthening |
| Androgen deprivation therapy | QTc lengthening |
| Anabolic steroid | QTc shortening for exogenous testosterone, tribulus terrestris Variable effects on QTc for other exogenous anabolic-androgenic steroids |
| Women | |
| Progesterone | QTc shortening |
| Estradiol | QTc lengthening particularly when progesterone levels are low |
| Testosterone | No influence within physiologic ranges QTc shortening within supraphysiological testosterone levels |
| Gonadotropins | QTc lengthening |
| Contraceptive pills | QTc shortening with first and second contraceptive generation QTc lengthening with anti-androgenic contraceptive pills |
| Hormone replacement therapy | QTc lengthening with HRT with estrogens alone No influence with HRT combining estrogens and progestins |
| Selective estrogen receptor modulators | QTc lengthening |
| Aromatase inhibitors | QTc shortening |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grouthier, V.; Moey, M.Y.Y.; Gandjbakhch, E.; Waintraub, X.; Funck-Brentano, C.; Bachelot, A.; Salem, J.-E. Sexual Dimorphisms, Anti-Hormonal Therapy and Cardiac Arrhythmias. Int. J. Mol. Sci. 2021, 22, 1464. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031464
Grouthier V, Moey MYY, Gandjbakhch E, Waintraub X, Funck-Brentano C, Bachelot A, Salem J-E. Sexual Dimorphisms, Anti-Hormonal Therapy and Cardiac Arrhythmias. International Journal of Molecular Sciences. 2021; 22(3):1464. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031464
Chicago/Turabian StyleGrouthier, Virginie, Melissa Y. Y. Moey, Estelle Gandjbakhch, Xavier Waintraub, Christian Funck-Brentano, Anne Bachelot, and Joe-Elie Salem. 2021. "Sexual Dimorphisms, Anti-Hormonal Therapy and Cardiac Arrhythmias" International Journal of Molecular Sciences 22, no. 3: 1464. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031464