
Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium

Department of Internal Medicine I, University Hospital Jena, 07747 Jena, Thüringen, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(3), 1498; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031498
Submission received: 15 January 2021
/
Revised: 27 January 2021
/
Accepted: 28 January 2021
/
Published: 2 February 2021
(This article belongs to the Special Issue Bioenergetics, Mitochondrial Dynamics and Cardiac Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Heart failure remains the most common cause of death in the industrialized world. In spite of new therapeutic interventions that are constantly being developed, it is still not possible to completely protect against heart failure development and progression. This shows how much more research is necessary to understand the underlying mechanisms of this process. In this review, we give a detailed overview of the contribution of impaired mitochondrial dynamics and energy homeostasis during heart failure progression. In particular, we focus on the regulation of fatty acid metabolism and the effects of fatty acid accumulation on mitochondrial structural and functional homeostasis.
1. Introduction
Heart failure is manifested by the disability of the heart to maintain its pumping efficiency. In 2016, cardiovascular disease was responsible for 840,768 deaths in the US [1]. Therefore, it remains the most common cause of death in Western countries. The causes of heart failure are diverse; however, all can contribute to cardiomyocyte death and ultimately lead to cardiac dysfunction [1]. In addition to the symptoms typically observed in heart failure [2], metabolic alterations have been reported in heart failure patients [3,4,5]. Such changes have attracted increasing attention as novel biomarkers [6,7] as well as for drug targets [8,9]. Hage et al. recently identified different metabolic profiles between new-onset HFpEF and HFrEF patients [10]. Interestingly, Haase et al. demonstrated that the altered metabolic alterations in aortic stenosis patients could be partially reversed after transaortic valve replacement [11]. Among all the metabolites analyzed, fatty acids and fatty-acid-associated metabolites are predominantly affected. The changes in cardiac metabolism, especially the fatty acid β-oxidation, have always been a major research focus in the study of the development and progression of heart failure [12,13].
2. Metabolic Plasticity of the Developing Heart
The adult heart is a metabolic omnivore and relies mainly on fatty acid β-oxidation for energy production under physiological conditions [13]. This reliance is essential for the proliferation and maturation of cardiac myocytes, but it is not the primary energy source in early developmental stages [14]. During cardiac differentiation of pluripotent stem cells and early postnatal stages, glycolysis is utilized as the main source of ATP generation [15,16]. It was shown in 1991 that the glycolytic rate in a 1-day-old rabbit heart was higher than that in a 7-day-old heart, in which increased palmitate oxidation was observed [17]. Both in vitro [5,6] and in vivo [7] data demonstrate that there is a metabolic switch from cytoplasm-localized glycolysis to mitochondria-localized fatty acid β-oxidation. Some of the main regulators of such a switch are peroxisome proliferator-activated receptors (PPARs) [18,19]. PPARs belong to the class II nuclear hormone receptors and contain three isoforms: PPARα, PPARβ/δ, and PPARγ. They have diverse cellular functions and are involved in different metabolic and developmental signaling pathways [20,21,22]. PPARα is involved in transcriptional regulation of fatty acid β-oxidation related genes, such as carnitine palmitoyltransferase 1 (CPT1) [23], clusters of differentiation 36 (CD36) [24] and diacyl-glycerol acyltransferase (DGAT) [25]. Cao et al. showed that the metabolic switch from glycolysis to fatty acid β-oxidation in mice at an early post-natal stage is promoted by PPARα [14]. Despite being an important metabolic mediator in the early post-natal stage, the role of PPARα in the adult myocardium is more ambivalent. It has already been shown that PPARα −/− mice are characterized by reduced cardiac metabolism without significant effect on cardiac function at baseline level [26,27]. Additionally, Kalliora et al. demonstrated that the activation of PPARα/γ by the dual agonist tesaglitazar led to cardiotoxicity. This is mediated by the reduction and inhibition of cardiac PGC1α (PPARGC1A), a key regulator in mitochondrial metabolism and homeostasis [19]. The finding illustrates the diverse effects of PPARα during different developmental stages. Nevertheless, PPARα activation is directly linked to fatty acid β-oxidation and, therefore, mitochondrial metabolism. The maintenance of the mitochondria homeostasis is therefore crucial for not only cell vitality but also for the heart’s functional integrity.
3. Mitochondrial Metabolism and Mitochondria-Associated Disorders
Mitochondria are the main sites for ATP generation in all mammalian organisms. The electron transport chain (ETC) or respiratory chain converts inter alia NADH into ATP via its 5 subunits [28,29]. Briefly, Complex I is responsible for the removal of two electrons from NADH and transfers them to ubiquinone to create ubiquinol [30,31]. Complex II is also required for the generation of ubiquinol but from succinate instead of NADH [32]. Both complexes generate hydronium ions, which are pumped into the inner mitochondria membrane space to generate a proton gradient. Complex III provides the substrates for complex I and II by converting ubiquinol back to ubiquinone [33]. Complex III gets reduced during this process, and the hydronium ion gradient is further increased inside of the mitochondria. Complex IV oxidizes complex III and allows it to maintain its biological function [34,35,36]. Complex V uses all the generated hydronium ions to convert ADP to ATP. Complex V is an ion channel that transports the hydronium ion out of the mitochondria. During this process, complex V is moved in a rotational direction, which causes the oxidative phosphorylation of ADP to ATP [37,38]. Dysfunction in each respective complex leads to different diseases called mitochondrial respiratory chain disorders (MRCDs) [39,40]. The Kearns-Sayre syndrome [41], Pearson Syndrome [42], and Leigh syndrome [43] are MRCDs associated with the development of heart failure.
The necessary substrate i.e., NADH, is primarily provided by the tricarboxylic acid cycle (TCA cycle). Shortly, the TCA cycle is localized in the inner mitochondria matrix and converts initially supplied acetyl-CoA into different carboxylic acids. During these reactions, three NADH are generated inter alia, which are one of the basic substrates for the respiratory chain. While the TCA generates NADH mainly from acetyl-CoA, it is also possible to produce intermediate molecules like α-ketoglutarate from other sources e.g., glutamate [44]. Glutamate is an amino acid involved in various signaling pathways in a tissue-dependent manner [45]. In mitochondrial metabolism, glutamate is predominantly catalyzed by glutamate dehydrogenase (GDH) into α-ketoglutarate, which is then converted in the TCA to enable continuous NADH production [46]. Disruption of the glutamate metabolism is associated with diseases such as hyperinsulinism/hyperammonemia syndrome (HI/HA) [47], olivopontocerebellar degeneration [48], or retinal diseases [49].
One of the major ways to provide acetyl-CoA for the TCA is via pyruvate conversion. Pyruvate is the conjugated base of pyruvic acid and is catalyzed by pyruvate dehydrogenase to acetyl-CoA [50] or by pyruvate carboxylase to oxaloacetate [51]. Both molecules participate in the TCA cycle localized in the mitochondria matrix [52,53]. Pyruvate is initially produced at the end of glycolysis, and a dysregulated glycolytic pathway is associated with diseases such as hemolytic anemia [54].
The second way to provide acetyl-CoA for the TCA cycle is fatty acid oxidation, which serves as the most important mechanism in cardiac metabolism and is also known as β-oxidation [55]. In mitochondria, acetyl-CoA can also be generated via β-oxidation of acyl-CoA [56]. During fatty acid β-oxidation, free fatty acids are transported from the blood into the cells via fatty acid transport proteins (FATPs) e.g., CD36 [57,58,59]. The long chain fatty acid CoA ligase 1 (ACSL1) then converts the fatty acid under the utilization of CoA and ATP into acyl-CoA [60,61,62]. This intermediate molecule is attached to carnitine, creating acylcarnitine in the process, and is regulated by carnitine palmitoyltransferase I (CPTI) [63,64]. Afterwards, acylcarnitine is transported by carnitine–acylcarnitine translocase (CACT) [65] into the inner mitochondria matrix, where acyl-CoA is separated from the acylcarnitine by carnitine palmitoyltransferase II (CPTII) [66,67]. The initial carnitine is transported back into the cytoplasm again for the next acyl-CoA to bind. In the inner mitochondria matrix, acyl-CoA is further cleaved by continuous thiolysis [68], a process in which an acetyl-CoA molecule is split from the original acyl-CoA by 3-ketoacyl-CoA thiolase [69,70]. This process continues until the complete acyl-CoA is converted into acetyl-CoA. One thiolysis cycle generates five molecules of ATP, and thus making fatty acid β-oxidation the most efficient method for energy production.
An additional method of acetyl-CoA generation is via ketone body oxidation. Ketone bodies are metabolic products of fatty acid β-oxidation and are characterized by their keto group (C=O), with acetone as the simplest embodiment. They are primarily synthesized in the liver from acetyl-CoA when the plasma glucose level is low. The primarily circulating ketone body β-hydroxybutyrate (βOHB) is transported via the bloodstream into the heart. Inside the heart, monocarboxylate transporters (MCT1) transfer βOHB inside the mitochondria, where it is oxidized by βOHB dehydrogenase (BHD1) into acetoacetate. This intermediate molecule is further processed into acetyl-CoA for the TCA cycle [71]. Although ketone bodies do not contribute significantly to myocardial ATP synthesis, increased circulating levels of βOHB and ketone body uptake have been observed in heart failure patients [72,73,74,75,76]. However, Ho et al. demonstrated that increased ketone oxidation in hypertrophied hearts is not accompanied by improved cardiac efficiency [77]. Further research is necessary to understand if ketone bodies simply serve as an alternative energy source for the failing heart or if increased ketone body oxidation has long-term benefits [78,79,80].
The complexity and diversity of the involved molecules and signaling pathways show how regulation and function of mitochondria homeostasis are intertwined, especially the fatty acid metabolism for cardiac muscle cells [13,81]. The shift from physiological fatty acid β-oxidation of healthy mitochondria to lipotoxic fatty acid accumulation, which subsequently leads to mitochondrial damage and apoptosis, is associated with different cardiac dysfunctions [12]. Therefore, a better understanding of how mitochondrial fatty acid metabolism is impaired and its associated consequences are crucial for the development of new therapeutic interventions to treat heart failure patients.
4. Lipotoxicity and Mitochondrial Homeostasis
Numerous pathogenic factors are associated with the development and progression of heart failure, including accumulated adipose tissue and fatty acids. In recent years, the epicardial adipose tissue (EAT) has gained much attention due to its localization in the heart and versatile functions [82,83,84]. Under physiological conditions, the EAT fulfills cardioprotective functions e.g., by the secretion of anti-inflammatory and anti-atherosclerotic cytokines such as adiponectin and adrenomedullin [85,86]. EAT acts as an inflammatory mediator by recruiting activated macrophages, secreting proinflammatory cytokines, and increasing transport of fatty acids [87]. Although further research is necessary to determine the definitive role of EAT in heart failure development, it emphasizes the relevance of fatty acids in lipotoxic cardiomyopathy. Fatty acid accumulation, which arises from genetic disorders [88] or lifestyle [89], is associated with a chronic inflammatory state. The accumulation is a consequence of insufficient fatty acid β-oxidation caused by impaired mitochondrial function and occurs in advanced stages of heart failure. An elevated fatty acid level further promotes inflammation and mitochondrial damage. The inflammation is characterized by elevated production of inflammatory cytokines such as TNFα, IL-1ß, and IL-6 [90,91].
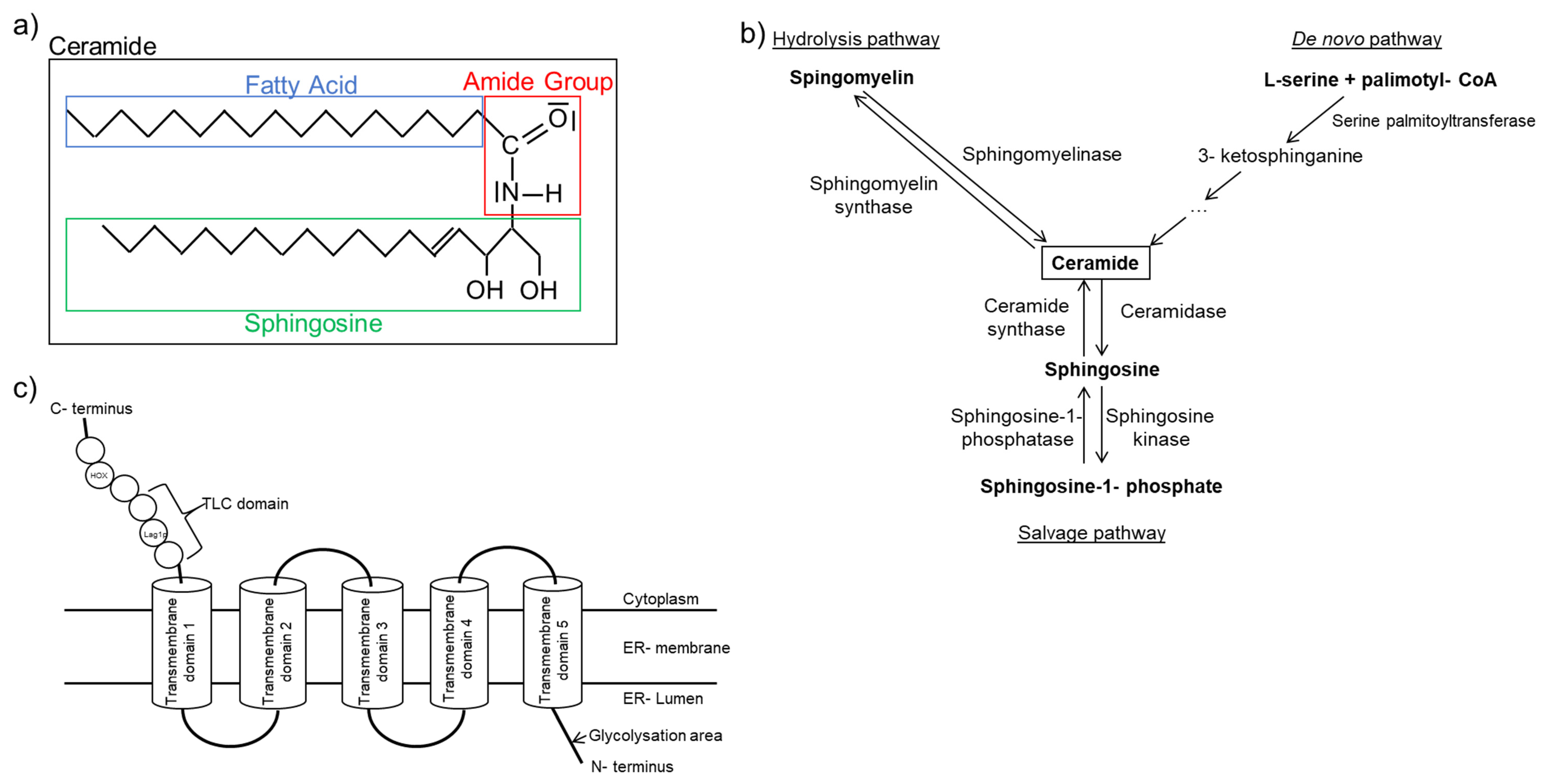
One category of fatty acid species that has a strong association with inflammation is ceramide. Ceramides contain a sphingosine backbone with an attached fatty acid connected via the amide group. The attached fatty acid is of various length, and ceramides of different fatty acid length have diverse effects in cells (Figure 1a). Long chain (C > 14) and very long chain (C > 20) ceramides are increased in heart failure patients and are linked to a pro-inflammatory state [92].
Ceramides are synthesized via three separate pathways (Figure 1b). The salvage pathway is characterized by the constant catabolism of various complex sphingolipids by different enzymes in lysosomes [102]. The de novo pathway synthesizes ceramides from L-serine and palmitoyl-CoA. This enzymatic reaction is regulated by serine-palmitoyltransferase (SPT), which is the rate-limiting enzyme in this distinctive signaling pathway [103,104]. The generated ceramides differ in chain length based on the attached fatty acid. Knockout of serine-palmitoyltransferase long chain base subunit 2 (SPTLC2), one of the long chain subunits of SPT, is also responsible for its biological function, leading to dilated cardiomyopathy in older mice [105]. However, an improved cardiac function is observed after myocardial infarction due to reduced very long chain ceramides [106]. Furthermore, inhibition of SPT with myriocin [107] mitigates inflammation after ischemia/reperfusion following transient coronary occlusion in mice [108]. Myriocin treatment also improves mitochondrial function by increasing mRNA expression of metabolic related genes, including PPARα, PPARγ, CPT1a, and CPT1b after I/R, thus reducing tissue damage [109]. These findings imply that the SPT may serve as a potential target for heart failure treatment.
Ceramides can also be synthesized via direct attachment of a fatty acid to sphingosine. This reaction is catalyzed by one of six isoforms of ceramide synthase (CerS) [93] (Figure 1c). The human gene was first identified in 1991 and was initially labeled as upstream of growth and differentiation-1 [110]. Later, more isoforms were identified, and they were classified as ceramide synthases [96,98,111,112,113]. CerS displays tissue-dependent expression patterns, and different CerS have preference towards different fatty acid substrates [94]. For example, CerS2 shows ubiquitous expression in most tissues and synthesizes very long chain ceramides. It plays an important role as a tumor suppressor [114]. In recent years, CerS2 gained increased attention for its contribution to very long chain ceramide formation and cardiotoxicity. Law et al. showed that the overexpression of CerS2 led to increased very long chain ceramide accumulation and subsequently mitochondrial damage and autophagy in cardiomyocytes [115]. Similar effects were also observed for CerS5. Russo et al. demonstrated that lipotoxic cardiac hypertrophy caused by myristate (C14:0) is reduced in CerS5 knockdown cardiomyocytes, as characterized by the reduction of autophagy promoting genes [116]. CerS6, but not CerS5, is associated with mitochondrial fragmentation and the development of insulin resistance in obese mice [117]. Furthermore, knockout of CerS6 improves mitochondrial structure [118]. CerS3 and CerS4 are mostly associated with skin barriers and their function [119,120], while CerS1 is mostly involved in diseases associated with the nervous system [121]. Despite the origin of proinflammatory ceramides differing, the outcomes are the same. The lipotoxicity-associated damages in mitochondrial structure and function result in apoptosis and autophagy [122,123,124].
The mitochondrial structural integrity is essential for its function and viability. Under physiological conditions, the number of mitochondria differs between different cell types [125], and they display a broad net-like structure within the cell. The structural integrity is regulated by different mitochondrial fusion and fission proteins and there is a constant cycle between mitochondrial synthesis and degradation, also known as “mitochondrial dynamism”. Mitofusin 1 (MFN1) and Mitofusin 2 (MFN2) are two GTPases localized at the outer mitochondria membrane [126,127,128] which are responsible for outer membrane fusion as well as mediating the connection with endoplasmatic reticulum (ER) [129]. Owing to the important regulatory role in mitochondria structure, a problem with either MFN1 or MFN2 or both impairs physiological development [130]. Knockout of MFN1 in murine cardiomyocytes leads to an increased number of spherical mitochondria, while mitochondria function remains mostly intact [131]. Nevertheless, the translational inhibition of MFN1 via miR-140 promotes mitochondrial fission followed by apoptosis [132]. Furthermore, it has been shown that the interaction with β II protein kinase C (βIIPKC) leads to MFN1 inactivation and promotes mitochondrial degradation in rat cardiomyocytes during heart failure [133]. Homozygous MFN2 knockout is lethal due to severe disruption in the physiological cell layer of placental trophoblasts. Furthermore, the conditional knockout of both genes at the same time has also been shown to be lethal [130]. Specific deletion of MFN1 and MFN2 in adult cardiomyocytes caused mitochondrial fragmentation and subsequently dilated cardiomyopathy [134]. The knocking out of MFN2 in old cardiomyocytes alone only caused a mild form of cardiomyopathy but showed better recovery after reperfusion injury [135]. Interestingly, Hall et al. showed that the animals with both MFN1 and MFN2 knockout did not develop cardiomyopathy at the age of 8–10 weeks and were cardioprotective against acute myocardial infarction. Such protection was rendered by reduction of ROS due to reduced Ca2+ overload [136]. These results indicate that MFN1 and MFN2 seem to have age-dependent effects on mitochondrial function and cardiac vitality as ablation of both genes appears to have negligible impact in young animals. This implies that further research is necessary to better understand how mitochondrial structure is regulated other than by MFN1 and MFN2. Hu et al. recently showed that obese diabetic mice are characterized by reduced MFN2 expression and disrupted mitochondria. Reconstitution of depleted MFN2 with an adenoviral vector encoding MFN2 inhibited mitochondrial fission [137]. Additionally, transient knockdown of MFN2 in the murine skeletal muscle resulted in reduced Ca2+ uptake and reduced tether frequency [138].
Another protein involved in mitochondria structural integrity is dynamin-like 120 kDa protein, mitochondrial (OPA1). OPA1 is responsible for the inner membrane fusion and, therefore, cristae morphogenesis and function [139,140]. Next to optic atrophy, homozygous OPA1 mutation in humans is accompanied by hypertrophic cardiomyopathy [141]. Lipotoxicity generally causes mitochondrial damage, which is characterized by the downregulation of mitochondrial fusion-associated genes and the potential upregulation of fission-related genes to enhance mitochondrial degradation.
The center of mitochondrial fission is the dynamin-related protein 1 (DRP1), which belongs to the family of dynamins and is also a GTPase [142,143]. For the fission of mitochondria by DRP1, certain adaptor proteins and their interaction with the GTPase are necessary. Mitochondrial dynamics protein 49 (aka MIEF2) and 51 (aka MIEF1) are localized at the outer membrane of the mitochondria and recruit DRP1 [144,145]. Upon recruitment, DRP1 and MID49/51 encircle the mitochondria, which leads to the fission of the cell organelle via nucleotide-driven allostery [146,147,148,149]. Chen et al. showed that reduction of DRP1 by siDRP1 attenuated palmitate-induced apoptosis, therefore linking lipotoxicity to mitochondrial homeostasis [150]. It has been shown that DRP1 activity is affected by several post-translational modifications [151]. Kashatus et al. demonstrated that DRP1 phosphorylation by Erk2 led to mitochondrial fission [152]. Furthermore, a DRP1-mediated effect can be partially regulated by the outer mitochondria membrane-associated E3 ubiquitin ligase MARCH5 [153]. In addition, SUMOylation has also been identified as a post-translational modifier of DRP1 [154]. Lipotoxicity-induced ubiquitination could therefore affect DPR1 activity and promote mitochondrial fission [155]. Additional recruitment partners for DRP1 are mitochondrial fission factor (Mff) and fission, mitochondrial 1 (FIS1). Both Mff and FIS1 are also located at the outer mitochondria membrane and are able to recruit DRP1. However, Mff can also work independently of FIS1 [156]. While FIS1 is highly involved in mitochondrial degradation [157,158], it seems not to be indispensable for the general process of mitochondrial fission [156,159].
Disruption of mitochondrial structure is directly associated with mitochondrial function [160]. Zhao et al. demonstrated that the knockdown of MFN2 in Kunming white mice embryos led to reduced ATP content and increased apoptosis at the blastocyst stage, proving the importance of intact mitochondrial homeostasis during early embryonal development [161]. Furthermore, MFN2 ablation is associated with cardiac myopathy [113] and impaired insulin signaling in murine hepatic cells [162]. Furthermore, decreased glucose and fatty acid β-oxidation [163] and reduced mitochondrial ATP production are observed in skeletal muscle cells [164,165,166]. Reduction of mitochondrial fusion-related genes not only decreases mitochondrial function but also increases expression of fission-related genes. DRP1 is essential for healthy mitochondrial dynamism and cardiac knockout of DRP1 leads to mitochondria enlargement caused by impaired physiological mitophagy. This ultimately leads to cardiac cell death [167,168,169]. Nevertheless, it has been shown that DRP1 inhibition with mitochondrial division inhibitor-1 or P110 reduces in vitro cell death induced by simulated ischemia/reperfusion [141]. Furthermore, reduction in the myocardial infarct size has also been observed in vivo [170,171]. Similar results are observed for FIS1/DRP1 interaction during septic cardiomyopathy [172]. The authors showed that treatment of LPS induced septic mice with P110 reduced mortality rate, which was accompanied by improved mitochondrial structure and function. Furthermore, Hu et al. demonstrated that a high-fat diet led to increased DRP1 acetylation and the subsequent mitochondrial fission resulted in cardiotoxicity and cell death [173]. Although knocking out DRP1 is detrimental for embryonic development, its inhibition shortly after cardiomyopathy appears to be beneficial. These results show that the mitochondria structure is irreversibly connected to mitochondria function and that impairment of structural integrity leads to reduced ATP generation and cell death [174]. Collectively, these findings provide evidence showing that mitochondria homeostasis and vitality are not only involved in lipotoxicity-mediated heart failure but also in different forms of cardiomyopathies.
Mitochondrial homeostasis is directly linked to calcium (Ca2+) flux. Ca2+ signaling is essential for myocardial contractility, but Ca2+ overload is detrimental for cells. It has been shown that verapamil, a Ca2+ channel blocker, reduces Ca2+-induced cardiotoxicity and improves survival in Sprague Dawley rats [175]. Further studies have determined the role of Ca2+ level in heart failure [176,177,178,179], and an improved intracellular Ca2+ level has been proven to be beneficial for heart failure patients [180,181].
In addition to type 2 ryanodine receptor (RyR2) [182,183] and sarco/endoplasmic reticulum Ca2+ATPase (SERCA) [184,185], the B cell lymphoma 2 family (Bcl-2) has been identified as an important regulator of Ca2+ homeostasis and mitochondria-mediated apoptosis. The Bcl-2 family contains different proteins with different functions for apoptosis regulation [186]. Bcl-2 is localized in the ER and is actively transported to the outer mitochondrial membrane to inhibit apoptosis [187,188]. The Bcl-2 family members Bax and Bak promote apoptosis due to mitochondria membrane permeabilization and cytochrome C release [189]. The dual role of the Bcl-2 protein family in apoptosis has also been observed for Ca2+ homeostasis.
Ca2+ efflux from the ER is in part regulated by the inositol trisphosphate receptor (IP3R) [190]. Ca2+ uptake at the outer mitochondria membrane is regulated by voltage dependant anion channel (VDAC) [191]. It is then transposed to the inner membrane by mitochondrial calcium uniporter (MCU) [192]. The ER is connected to the outer mitochondria membrane through mitochondria-associated ER membranes (MAMs). MAMs are primarily connected via MFN2 and S100B [193,194] to enable Ca2+ transmission. It has been shown that different IP3R isoforms are located at MAMs and are in close proximity to VDAC. The juxtaposition facilitates Ca2+ transport from the ER into the outer mitochondrial membrane [195]. Subsequently, Ca2+ is transported into the inner mitochondrial membrane by MCU to activate the pyruvate dehydrogenase complex for ATP synthesis. Reduced Ca2+ transport into the mitochondria due to decreased IP3R-VDAC interaction is associated with mitochondrial dysfunction [196]. Interactions of Bcl-2 and Mcl-1 with all IP3R isoforms have been observed and are accompanied by increased Ca2+ release and reduced apoptosis [197]. Furthermore, Vander Heiden et al. demonstrated that recombinant Bcl-x(L) kept VDAC in an open configuration to maintain metabolic flux and prevent cytochrome c release [198].
The Bcl-2 family protein that plays an adverse role in Ca2+ transport is the BCL2/adenovirus E1B 19kDa protein-interacting protein 3 (BNIP3). BNIP3 is proapoptotic and induces autophagy. It has been shown that BNIP3 overexpression resulted in increased Ca2+ leakage from the ER and mitochondrial uptake. Mitochondrial Ca2+ accumulation leads to caspase-independent cell death due to the opening of the mitochondrial permeability transition pore (mPTP) [199,200]. Palmitate-overload-dependent lipotoxicity is associated with increased Ca2+ flux from the ER into the mitochondria and potentially involves BNIP3 [201]. Furthermore, BNIP3 contributes to mitochondria fragmentation and fission by the binding of OPA1, subsequently leading to Bax/Bak-dependent apoptosis [202,203,204]. BNIP3-mediated mitochondrial fragmentation is also associated with maladaptive mitochondrial autophagy (mitophagy) and cardiac hypertrophy [205,206,207].
Mitophagy is a form of macroautophagy, where defective proteins and cell organelles e.g., depolarized mitochondria, are phagocytized and subsequently degraded. The degraded products provide substrates for energy metabolism and prevent the accumulation of harmful proteins. Therefore, macrophagy plays a major role in the regulation of cell survival and shows protective as well as maladaptive effects in the cardiovascular system [208,209,210]. Ma et al. demonstrated that BNIP3 overexpression leads to autophagosome accumulation and reduced lysosomal clearance that subsequently results in cell death [211]. Furthermore, it is found that BNIP3-mediated mitophagy is regulated by oxidative stress in myocardial ischemia and reperfusion [212]. Additionally, Sebastián et al. showed that the lack of MFN2 in old mice induced BNIP3-mediated mitophagy in a ROS-dependent manner [213]. Increased ROS production, in part caused by proinflammatory ceramides, could therefore trigger the proapoptotic effects of BNIP3-induced mitophagy [214,215]. Further examinations are necessary to verify the proposed interconnection.
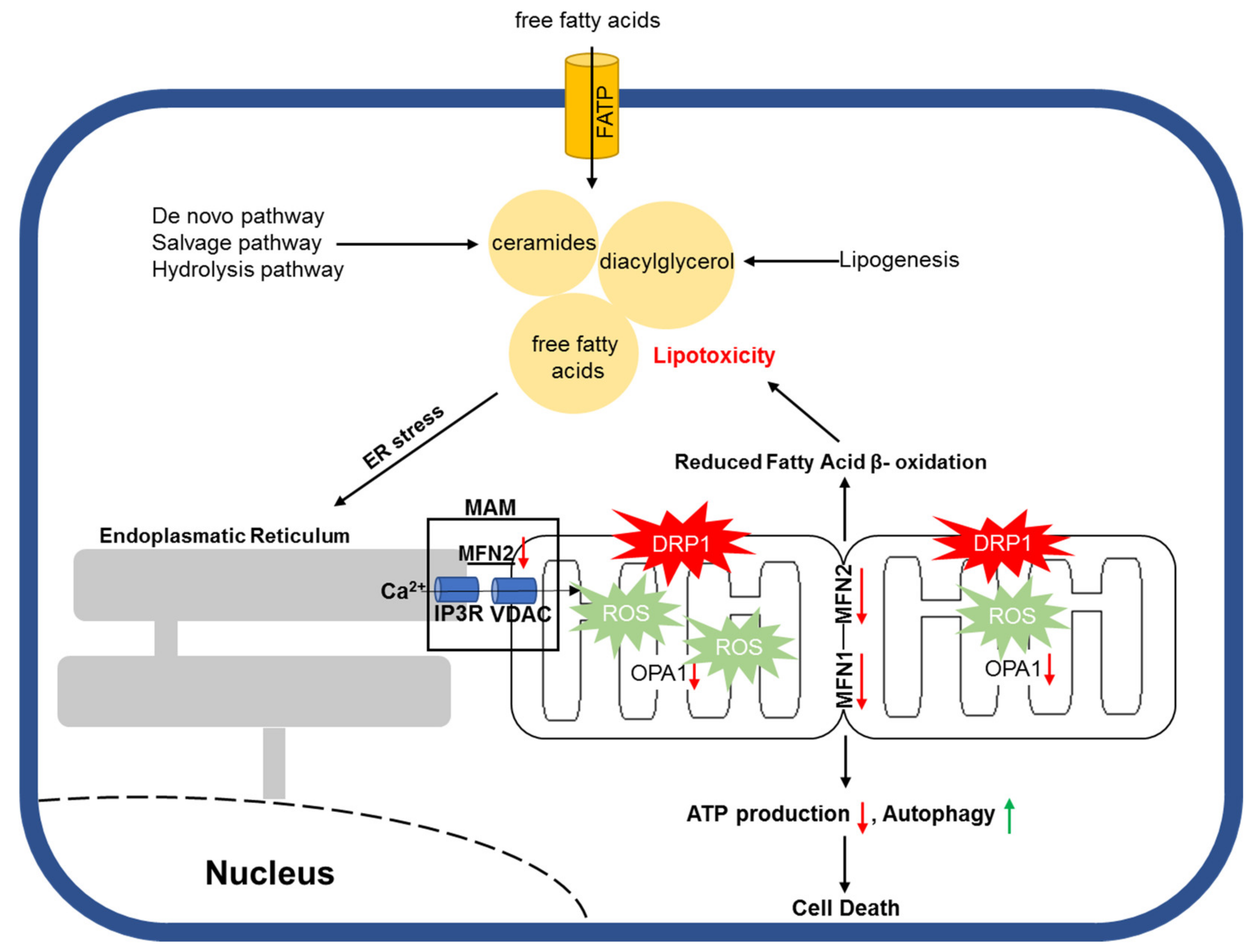
One way by which accumulated toxic lipids impair ER and mitochondrial homeostasis is by increased reactive oxygen species (ROS). Under physiological conditions, ROS are important second messengers with a short half-life and are involved in various signaling pathways for cell survival [216,217,218]. However, excessive ROS production is negatively associated with cell vitality due to enhanced cellular damage and apoptosis [219,220] (Figure 2). Joseph et al. showed that NADPH oxidase 2 (NOX2) contributed to the formation of mitochondrial superoxides during LPS-mediated sepsis and that NOX2 inhibition with apocynin improved cardiac contractility in vivo [221]. In addition, Nakamura et al. proved that diabetic cardiomyopathy is associated with mitochondrial ROS accumulation as a result of increased p53/SCO2 activation following lipid accumulation in murine cardiomyocytes [222]. Furthermore, Tsushima et al. demonstrated that lipotoxicity caused by ACSL1 overexpression or palmitate or oleate treatment could lead to increased mitochondrial ROS production, which was accompanied by reduced mitochondrial ATP synthesis and enhanced mitochondrial fission in neonatal rat ventricular cardiomyocytes (NRVCs) [223]. Different lines of studies provide evidence that fatty acid accumulation is associated with increased mitochondrial ROS production, which is followed by impaired mitochondrial structure and function both in vitro and in vivo. Such an effect also appears to be cell type independent.
It is observed that lipid accumulation is a consequence of impaired fatty acid β-oxidation [13]. The cause of such impairment varies and includes obesity, diabetes, and cardiovascular disease [224,225,226]. However, Ji et al. demonstrated that SPTLC2 knockout mice show preserved cardiac function after myocardial infarction (MI) due to reduced proinflammatory ceramides [106]. Similar results were found by SPT inhibition with myriocin [109].
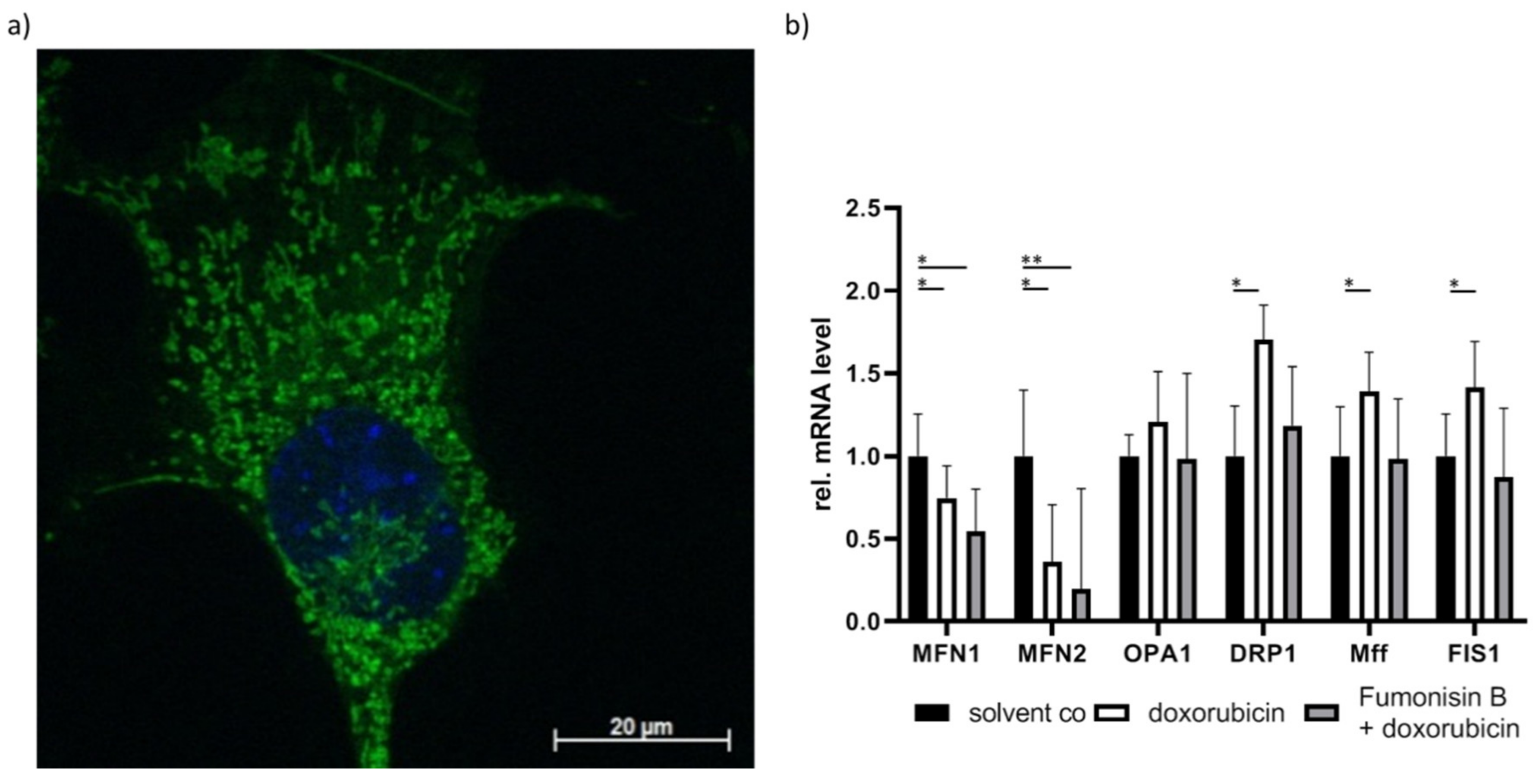
These results are also observed in our own research. We treated murine cardiomyocytes with doxorubicin and observed increased long chain and very long chain ceramides, which were accompanied by impaired mitochondrial distribution (Figure 3a). To determine if ceramides are a contributor to this effect, the cells were pre-treated with the unspecific CerS inhibitor fumonisin B. In addition to ceramide reduction, we also observed improved mitochondrial homeostasis (Figure 3b). To further investigate the role of ceramides in cardiotoxicity, CerS2-overexpressed cells were pre-treated with fumonisin B. We observed similar effects in CerS2-overexpressed cells. Therefore, ceramide accumulation could be an important contributor to doxorubicin-mediated cardiotoxicity (unpublished data). This indicates that lipotoxicity may play a causative role in cardiotoxicity. This also implicates that mitochondrial impairment arises from insufficient fatty acid β-oxidation, which then leads to fatty acid accumulation and promotes further mitochondrial damage. Further research is necessary to determine if cardiomyopathy is caused by toxic lipid accumulation, impaired fatty acid β-oxidation, or a combination of both.
5. Additional and Alternative Mitochondria Regulation
Mitochondria homeostasis is not affected by lipotoxicity alone. Other factors that influence myocardial metabolism also contribute to heart failure development. PKCδ has been found to lead to mitochondrial dysfunction by inducing ROS production in different disease models, including LPS-induced sepsis [227] and cardiac reperfusion [228,229]. Furthermore, it also promotes mitochondria-mediated apoptosis [230]. Sirtuin 1 (SIRT1), a histone deacetylase, is shown to be cardioprotective during I/R by activating the MAPK pathway and reducing ROS level [231]. Furthermore, it deacetylates and activates FoxO1 to promote cardioprotective autophagy during starvation [232]. Cardioprotective autophagy is reduced in diabetic cardiomyopathy [233]. SIRT1 positively regulates PGC-1α by maintaining and improving mitochondrial function [234]. SIRT3 is also involved in the maintenance of mitochondrial function [235] and is negatively regulated by miR-195 [236]. SIRT3 mitigates cardiac hypertrophy [237] and regulates acetylation and activation of 84 metabolism-associated proteins [238]. Krüppel-like factors (KLFs) belong to the family of zinc finger proteins and regulate the transcription of diverse genes. KLF4 is one of KLF isoforms that regulates physiological estrogen-related receptor/PPARγ coactivator 1 (ERR/PGC-1) functionality and is involved in the transcriptional regulation of autophagy [239]. KLF5 interacts with PPARα by directly binding on its promotor and is therefore directly involved in fatty acid β-oxidation [240]. On the other hand, KLF15 binds and activates the E1a-binding protein (p300) to regulate lipid flux [241]. In addition, the G protein-coupled receptor kinase 2 (GRK2) is negatively associated with cardiomyocyte survival after I/R by inducing mitochondrial dysfunction following translocation to the mitochondria [242,243].
6. Conclusions
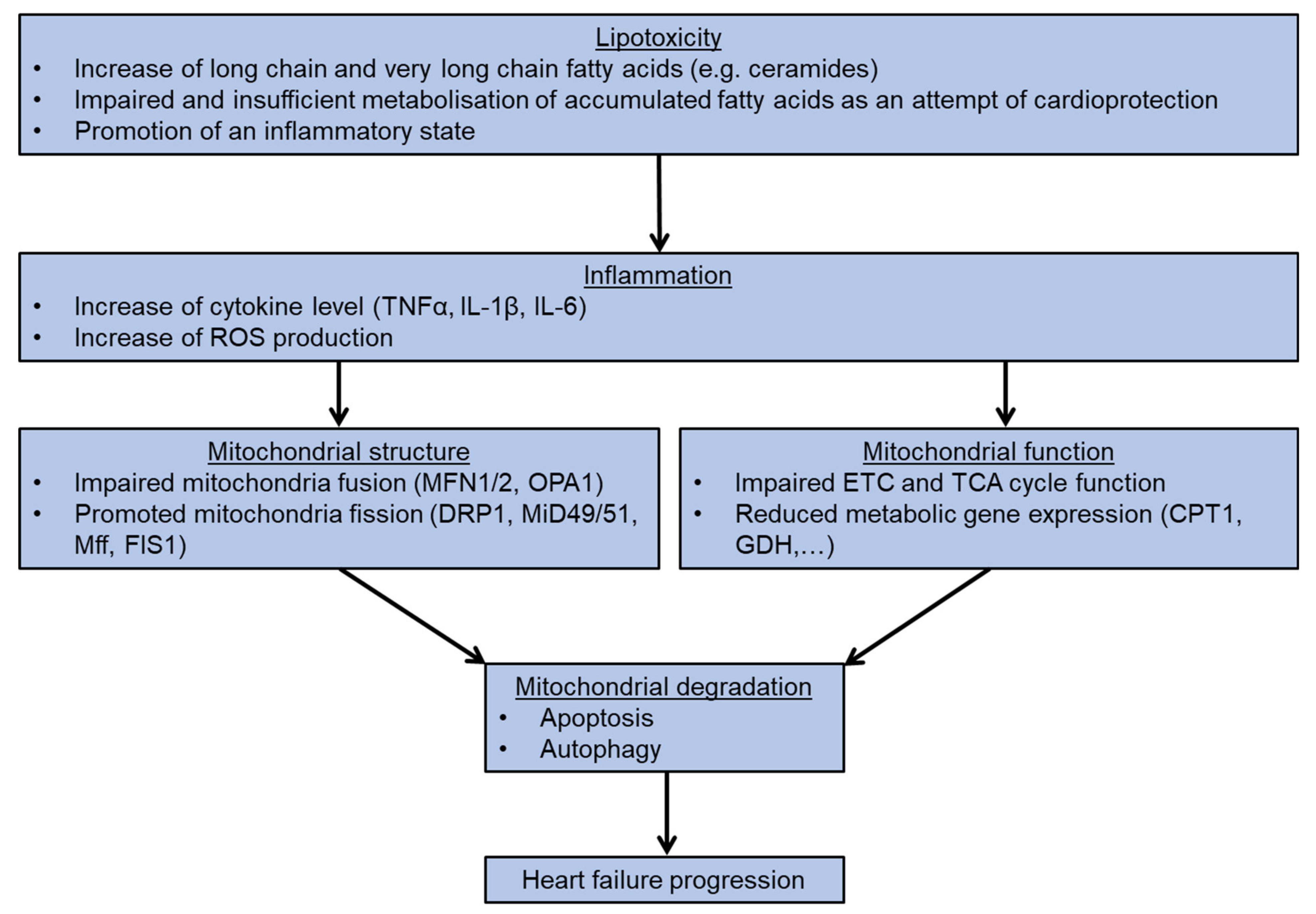
In this review, we elaborate on the importance of mitochondria homeostasis in different forms of cardiomyopathies, in particular the effects of fatty acid accumulation and subsequent impairment of mitochondria structure and function. Both are essential for the development of cardiotoxicity and mitochondria-mediated cell death. The integrity of mitochondrial homeostasis is regulated by various factors, including the amount of long chain fatty acid levels e.g., ceramides, the mitochondrial ROS level, the status of mitochondrial fusion, and the fission and expression of metabolic genes. All these factors serve as valid and promising targets for the development of new therapeutic interventions for heart failure patients. (Figure 4).
Author Contributions
T.K. performed the literature research, wrote the original manuscript and created the figures. J.M.F.W. revised the manuscript. P.C.S. provided supervision, feedback and funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Acknowledgments
P.C.S. is supported by a grant from the Else- Kröner- Fresenius Foundation, Germany.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Scally, C.; Rudd, A.; Mezincescu, A.; Wilson, H.; Srivanasan, J.; Horgan, G.; Broadhurst, P.; Newby, D.E.; Henning, A.; Dawson, D.K. Persistent Long-Term Structural, Functional, and Metabolic Changes After Stress-Induced (Takotsubo) Cardiomyopathy. Circulation 2018, 137, 1039–1048. [Google Scholar] [CrossRef]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz-Siemion, M.; Ciborowski, M.; Ptaszynska-Kopczynska, K.; Szpakowicz, A.; Lisowska, A.; Jasiewicz, M.; Waszkiewicz, E.; Kretowski, A.; Musial, W.J.; Kaminski, K.A. LC-MS-based serum fingerprinting reveals significant dysregulation of phospholipids in chronic heart failure. J. Pharm. Biomed. Anal. 2018, 154, 354–363. [Google Scholar] [CrossRef]
- Nadar, S.K.; Shaikh, M.M. Biomarkers in Routine Heart Failure Clinical Care. Card. Fail. Rev. 2019, 5, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Albert, C.L.; Tang, W.H.W. Metabolic Biomarkers in Heart Failure. Heart Fail. Clin. 2018, 14, 109–118. [Google Scholar] [CrossRef]
- Lund, A.; Nordrehaug, J.E.; Slettom, G.; Solvang, S.H.; Pedersen, E.K.; Midttun, O.; Ulvik, A.; Ueland, P.M.; Nygard, O.; Giil, L.M. Plasma kynurenines and prognosis in patients with heart failure. PloS ONE 2020, 15, e0227365. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Li, L.; Chai, N.; Chen, Y.; Wu, F.; Zhang, W.; Wang, L.; Shi, S.; Zhang, L.; et al. Spermine and spermidine reversed age-related cardiac deterioration in rats. Oncotarget 2017, 8, 64793–64808. [Google Scholar] [CrossRef] [Green Version]
- Hage, C.; Lofgren, L.; Michopoulos, F.; Nilsson, R.; Davidsson, P.; Kumar, C.; Ekstrom, M.; Eriksson, M.J.; Lynga, P.; Persson, B.; et al. Metabolomic Profile in Patients with Heart Failure With Preserved Ejection Fraction Versus Patients With Heart Failure With Reduced Ejection Fraction. J. Card. Fail. 2020. [Google Scholar] [CrossRef]
- Haase, D.; Baz, L.; Bekfani, T.; Neugebauer, S.; Kiehntopf, M.; Mobius-Winkler, S.; Franz, M.; Schulze, P.C. Metabolomic profiling of patients with high gradient aortic stenosis undergoing transcatheter aortic valve replacement. Clin. Res. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, T.; Liccardo, D.; LaCanna, R.; Zhang, X.; Lu, R.; Finck, B.N.; Leigh, T.; Chen, X.; Drosatos, K.; Tian, Y. Fatty Acid Oxidation Promotes Cardiomyocyte Proliferation Rate but Does Not Change Cardiomyocyte Number in Infant Mice. Front. Cell Dev. Biol. 2019, 7, 42. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Yan, Y.; Terashvili, M.; Wells, C.; Horikoshi, H.; Fujita, S.; Bosnjak, Z.J.; Bai, X. Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells 2019, 8, 1095. [Google Scholar] [CrossRef] [Green Version]
- Bekhite, M.M.; Gonzalez Delgado, A.; Menz, F.; Kretzschmar, T.; Wu, J.M.F.; Bekfani, T.; Nietzsche, S.; Wartenberg, M.; Westermann, M.; Greber, B.; et al. Longitudinal metabolic profiling of cardiomyocytes derived from human-induced pluripotent stem cells. Basic Res. Cardiol. 2020, 115, 37. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Spafford, M.A.; Marsh, D.R. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol. 1991, 261, H1698–H1705. [Google Scholar] [CrossRef]
- Kelly, D.P. PPARs of the heart: Three is a crowd. Circ. Res. 2003, 92, 482–484. [Google Scholar] [CrossRef] [Green Version]
- Kalliora, C.; Kyriazis, I.D.; Oka, S.I.; Lieu, M.J.; Yue, Y.; Area-Gomez, E.; Pol, C.J.; Tian, Y.; Mizushima, W.; Chin, A.; et al. Dual peroxisome-proliferator-activated-receptor-alpha/gamma activation inhibits SIRT1-PGC1alpha axis and causes cardiac dysfunction. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. B.B.A. 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Poulsen, L.; Siersbaek, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Pol, C.J.; Lieu, M.; Drosatos, K. PPARs: Protectors or Opponents of Myocardial Function? PPAR Res. 2015, 2015, 835985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, O.; Kuriki, C.; Fukui, Y.; Motojima, K. Dual promoter structure of mouse and human fatty acid translocase/CD36 genes and unique transcriptional activation by peroxisome proliferator-activated receptor alpha and gamma ligands. J. Biol. Chem. 2002, 277, 15703–15711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Fujii, H.; Takahashi, T.; Kodama, M.; Aizawa, Y.; Ohta, Y.; Ono, T.; Hasegawa, G.; Naito, M.; Nakajima, T.; et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J. Biol. Chem. 2000, 275, 22293–22299. [Google Scholar] [CrossRef] [Green Version]
- Luptak, I.; Balschi, J.A.; Xing, Y.; Leone, T.C.; Kelly, D.P.; Tian, R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005, 112, 2339–2346. [Google Scholar] [CrossRef] [Green Version]
- Rich, P.R.; Marechal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell. Dev. Biol. 2018, 76, 179–190. [Google Scholar] [CrossRef]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell. Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell. Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, G. Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. B.B.A. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B.; Schneyder, B.; Mell, O.; Stroh, S.; Reimann, A. Respiratory chain proteins. Rev. Neurol. 1991, 147, 436–442. [Google Scholar]
- Shimada, S.; Shinzawa-Itoh, K.; Baba, J.; Aoe, S.; Shimada, A.; Yamashita, E.; Kang, J.; Tateno, M.; Yoshikawa, S.; Tsukihara, T. Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J. 2017, 36, 291–300. [Google Scholar] [CrossRef]
- Rich, P.R. Mitochondrial cytochrome c oxidase: Catalysis, coupling and controversies. Biochem. Soc. Trans. 2017, 45, 813–829. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Neupane, P.; Bhuju, S.; Thapa, N.; Bhattarai, H.K. ATP Synthase: Structure, Function and Inhibition. Biomol. Concepts 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Berardo, A.; Musumeci, O.; Toscano, A. Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol. 2011, 30, 9–15. [Google Scholar]
- Menezes, M.J.; Riley, L.G.; Christodoulou, J. Mitochondrial respiratory chain disorders in childhood: Insights into diagnosis and management in the new era of genomic medicine. B.B.A 2014, 1840, 1368–1379. [Google Scholar] [CrossRef]
- Shemesh, A.; Margolin, E. Kearns Sayre Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Farruggia, P.; Di Marco, F.; Dufour, C. Pearson syndrome. Expert. Rev. Hematol. 2018, 11, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, J.; Ananthakrishnan, R.; Korson, M.; Goldenthal, M.J.; Perez-Atayde, A. Cardiac mitochondrial dysfunction in Leigh syndrome. Pediatr. Cardiol. 1996, 17, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Casimir, M.; Carobbio, S.; Maechler, P. Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. B.B.A. 2008, 1777, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Keshava Prasad, T.S.; Pandey, A.; Somani, B.L.; Gowda, H. A pathway map of glutamate metabolism. J. Cell. Commun. Signal. 2016, 10, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.C.; Daniel, R.M. L-glutamate dehydrogenases: Distribution, properties and mechanism. Comp. Biochem. Physiol. B 1993, 106, 767–792. [Google Scholar] [CrossRef]
- Kelly, A.; Stanley, C.A. Disorders of glutamate metabolism. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 287–295. [Google Scholar] [CrossRef]
- Plaitakis, A.; Berl, S.; Yahr, M.D. Abnormal glutamate metabolism in an adult-onset degenerative neurological disorder. Science 1982, 216, 193–196. [Google Scholar] [CrossRef]
- Ishikawa, M. Abnormalities in glutamate metabolism and excitotoxicity in the retinal diseases. Scientifica (Cairo) 2013, 2013, 528940. [Google Scholar] [CrossRef] [Green Version]
- Leong, H.S.; Brownsey, R.W.; Kulpa, J.E.; Allard, M.F. Glycolysis and pyruvate oxidation in cardiac hypertrophy--why so unbalanced? Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 2003, 135, 499–513. [Google Scholar] [CrossRef]
- Adina-Zada, A.; Zeczycki, T.N.; St Maurice, M.; Jitrapakdee, S.; Cleland, W.W.; Attwood, P.V. Allosteric regulation of the biotin-dependent enzyme pyruvate carboxylase by acetyl-CoA. Biochem. Soc. Trans. 2012, 40, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cooper, D.E.; Cluntun, A.A.; Warmoes, M.O.; Zhao, S.; Reid, M.A.; Liu, J.; Lund, P.J.; Lopes, M.; Garcia, B.A.; et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 2018, 175, 502–513.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, P.; Fermo, E.; Glader, B.; Kanno, H.; Agarwal, A.; Barcellini, W.; Eber, S.; Hoyer, J.D.; Kuter, D.J.; Maia, T.M.; et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: Consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am. J. Hematol. 2019, 94, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Luiken, J.J.; Arumugam, Y.; Glatz, J.F.; Tandon, N.N. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J. Biol.Chem. 2000, 275, 14501–14508. [Google Scholar] [CrossRef] [Green Version]
- Luiken, J.J.; Koonen, D.P.; Coumans, W.A.; Pelsers, M.M.; Binas, B.; Bonen, A.; Glatz, J.F. Long-chain fatty acid uptake by skeletal muscle is impaired in homozygous, but not heterozygous, heart-type-FABP null mice. Lipids 2003, 38, 491–496. [Google Scholar] [CrossRef]
- Nickerson, J.G.; Alkhateeb, H.; Benton, C.R.; Lally, J.; Nickerson, J.; Han, X.X.; Wilson, M.H.; Jain, S.S.; Snook, L.A.; Glatz, J.F.; et al. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J. Biol. Chem. 2009, 284, 16522–16530. [Google Scholar] [CrossRef] [Green Version]
- Hisanaga, Y.; Ago, H.; Nakagawa, N.; Hamada, K.; Ida, K.; Yamamoto, M.; Hori, T.; Arii, Y.; Sugahara, M.; Kuramitsu, S.; et al. Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 2004, 279, 31717–31726. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.M.; Li, L.O.; Wu, P.C.; Koves, T.R.; Ilkayeva, O.; Stevens, R.D.; Watkins, S.M.; Muoio, D.M.; Coleman, R.A. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Met. 2010, 12, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.A.; Heath, S.H.; Hagen, T.M. Acetyl-L-carnitine supplementation reverses the age-related decline in carnitine palmitoyltransferase 1 (CPT1) activity in interfibrillar mitochondria without changing the L-carnitine content in the rat heart. Mech. Ageing Dev. 2012, 133, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. B.B.A. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Huertas, M.D.; et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef]
- Lehmann, D.; Motlagh, L.; Robaa, D.; Zierz, S. Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features. Int. J. Mol. Sci. 2017, 18, 82. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Phenotype of carnitine palmitoyltransferase II (CPT II) deficiency: A questionnaire-based survey. J. Clin. Neurosci. 2019, 59, 32–36. [Google Scholar] [CrossRef]
- Dumbrepatil, A.B.; Zegalia, K.A.; Sajja, K.; Kennedy, R.T.; Marsh, E.N.G. Targeting viperin to the mitochondrion inhibits the thiolase activity of the trifunctional enzyme complex. J. Biol. Chem. 2020, 295, 2839–2849. [Google Scholar] [CrossRef]
- Kiema, T.R.; Harijan, R.K.; Strozyk, M.; Fukao, T.; Alexson, S.E.; Wierenga, R.K. The crystal structure of human mitochondrial 3-ketoacyl-CoA thiolase (T1): insight into the reaction mechanism of its thiolase and thioesterase activities. Acta Crystallogr. D. Biol. Crystallogr. 2014, 70, 3212–3225. [Google Scholar] [CrossRef]
- Martines, A.M.F.; van Eunen, K.; Reijngoud, D.J.; Bakker, B.M. The promiscuous enzyme medium-chain 3-keto-acyl-CoA thiolase triggers a vicious cycle in fatty-acid beta-oxidation. PLoS Comput. Biol. 2017, 13, e1005461. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. B.B.A. Mol. Cell. Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Du, Z.; Shen, A.; Huang, Y.; Su, L.; Lai, W.; Wang, P.; Xie, Z.; Xie, Z.; Zeng, Q.; Ren, H.; et al. 1H-NMR-based metabolic analysis of human serum reveals novel markers of myocardial energy expenditure in heart failure patients. PloS ONE 2014, 9, e88102. [Google Scholar] [CrossRef] [PubMed]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Kruger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Toh, R.; Irino, Y.; Mori, T.; Nakajima, H.; Hara, T.; Honjo, T.; Satomi-Kobayashi, S.; Shinke, T.; Tanaka, H.; et al. beta-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 475, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.M.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2020, 2, 1248–1264. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef]
- McCommis, K.S.; Kovacs, A.; Weinheimer, C.J.; Shew, T.M.; Koves, T.R.; Ilkayeva, O.R.; Kamm, D.R.; Pyles, K.D.; King, M.T.; Veech, R.L.; et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat. Metab. 2020, 2, 1232–1247. [Google Scholar] [CrossRef]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Invest. 2001, 107, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, N.; Moreno-Villegas, Z.; Gonzalez-Bris, A.; Egido, J.; Lorenzo, O. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, A.; Hamilton, D.J.; Deng, T. Epicardial Fat in the Maintenance of Cardiovascular Health. Methodist Debakey Cardiovasc. J. 2017, 13, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Song, F.; Wu, C.; Hong, Y.X.; Li, G. The roles of epicardial adipose tissue in heart failure. Heart failure reviews 2020. [Google Scholar] [CrossRef]
- Turer, A.T.; Scherer, P.E. Adiponectin: Mechanistic insights and clinical implications. Diabetologia 2012, 55, 2319–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, S.A.; Baxter, G.F. Adrenomedullin: Regulator of systemic and cardiac homeostasis in acute myocardial infarction. Pharmacol. Ther. 2005, 105, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Fosshaug, L.E.; Dahl, C.P.; Risnes, I.; Bohov, P.; Berge, R.K.; Nymo, S.; Geiran, O.; Yndestad, A.; Gullestad, L.; Aukrust, P.; et al. Altered Levels of Fatty Acids and Inflammatory and Metabolic Mediators in Epicardial Adipose Tissue in Patients With Systolic Heart Failure. J. Card. Fail. 2015, 21, 916–923. [Google Scholar] [CrossRef]
- Alonso, R.; Perez de Isla, L.; Muniz-Grijalvo, O.; Diaz-Diaz, J.L.; Mata, P. Familial Hypercholesterolaemia Diagnosis and Management. Eur. Cardiol. 2018, 13, 14–20. [Google Scholar] [CrossRef]
- Cha, E.; Akazawa, M.K.; Kim, K.H.; Dawkins, C.R.; Lerner, H.M.; Umpierrez, G.; Dunbar, S.B. Lifestyle habits and obesity progression in overweight and obese American young adults: Lessons for promoting cardiometabolic health. Nurs. Health Sci. 2015, 17, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Trompier, D.; Andreoletti, P.; Le Borgne, F.; Demarquoy, J.; Lizard, G. Fatty acids - induced lipotoxicity and inflammation. Curr. Drug Metab. 2012, 13, 1358–1370. [Google Scholar] [CrossRef]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB life 2010, 62, 347–356. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spassieva, S.; Seo, J.G.; Jiang, J.C.; Bielawski, J.; Alvarez-Vasquez, F.; Jazwinski, S.M.; Hannun, Y.A.; Obeid, L.M. Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J. Biol. Chem. 2006, 281, 33931–33938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riebeling, C.; Allegood, J.C.; Wang, E.; Merrill, A.H., Jr.; Futerman, A.H. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J. Biol. Chem. 2003, 278, 43452–43459. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem. J. 2006, 398, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kihara, A.; Chiba, H.; Tojo, H.; Igarashi, Y. 2-Hydroxy-ceramide synthesis by ceramide synthase family: Enzymatic basis for the preference of FA chain length. J. Lipid Res. 2008, 49, 2356–2364. [Google Scholar] [CrossRef] [Green Version]
- Mesika, A.; Ben-Dor, S.; Laviad, E.L.; Futerman, A.H. A new functional motif in Hox domain-containing ceramide synthases: Identification of a novel region flanking the Hox and TLC domains essential for activity. J. Biol. Chem. 2007, 282, 27366–27373. [Google Scholar] [CrossRef] [Green Version]
- Kageyama-Yahara, N.; Riezman, H. Transmembrane topology of ceramide synthase in yeast. Biochem. J. 2006, 398, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Nishijima, M.; Akamatsu, Y. A temperature-sensitive mammalian cell mutant with thermolabile serine palmitoyltransferase for the sphingolipid biosynthesis. J. Biol. Chem. 1990, 265, 22137–22142. [Google Scholar] [CrossRef]
- Ikushiro, H.; Hayashi, H. Mechanistic enzymology of serine palmitoyltransferase. B.B.A. 2011, 1814, 1474–1480. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, J.R.; Hu, Y.; Khan, R.; Kim, S.J.; Bharadwaj, K.G.; Davidson, M.M.; Choi, C.S.; Shin, K.O.; Lee, Y.M.; et al. Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. J. Biol. Chem. 2012, 287, 18429–18439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight 2017, 2. [Google Scholar] [CrossRef]
- Campisi, G.M.; Signorelli, P.; Rizzo, J.; Ghilardi, C.; Antognetti, J.; Caretti, A.; Lazarevic, J.S.; Strettoi, E.; Novelli, E.; Ghidoni, R.; et al. Determination of the serine palmitoyl transferase inhibitor myriocin by electrospray and Q-trap mass spectrometry. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef] [Green Version]
- Reforgiato, M.R.; Milano, G.; Fabrias, G.; Casas, J.; Gasco, P.; Paroni, R.; Samaja, M.; Ghidoni, R.; Caretti, A.; Signorelli, P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 2016, 111, 12. [Google Scholar] [CrossRef]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J. Expression of growth/differentiation factor 1 in the nervous system: Conservation of a bicistronic structure. Proc. Natl. Acad. Sci. 1991, 88, 4250–4254. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H., Jr.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef] [Green Version]
- Weinmann, A.; Galle, P.R.; Teufel, A. LASS6, an additional member of the longevity assurance gene family. Int. J. Mol. Med. 2005, 16, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, J.Y.; Yan, W.; Wang, D.D.; Yang, S.J.; Zhou, S.Y.; Zhong, S.L.; Tang, J.H. Clinical and pathological significance of Homo sapiens ceramide synthase 2 (CerS-2) in diverse human cancers. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, B.A.; Liao, X.; Moore, K.S.; Southard, A.; Roddy, P.; Ji, R.; Szulc, Z.; Bielawska, A.; Schulze, P.C.; Cowart, L.A. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB Journal. 2018, 32, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Russo, S.B.; Baicu, C.F.; Van Laer, A.; Geng, T.; Kasiganesan, H.; Zile, M.R.; Cowart, L.A. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Invest. 2012, 122, 3919–3930. [Google Scholar] [CrossRef] [Green Version]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 2019, 177, 1536–1552.e23. [Google Scholar] [CrossRef]
- Jennemann, R.; Rabionet, M.; Gorgas, K.; Epstein, S.; Dalpke, A.; Rothermel, U.; Bayerle, A.; van der Hoeven, F.; Imgrund, S.; Kirsch, J.; et al. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum. Mol. Genet. 2012, 21, 586–608. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Ishikawa, J.; Naoe, A.; Yoshida, H.; Hachiya, A.; Fujimura, T.; Kitahara, T.; Takema, Y. Ceramide synthase 4 is highly expressed in involved skin of patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ogretmen, B. Ceramide stress in survival versus lethal autophagy paradox: Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Autophagy 2013, 9, 258–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosetti, B.; Brodesser, S.; Brunn, A.; Deckert, M.; Bluher, M.; Doehner, W.; Anker, S.D.; Wenzel, D.; Fleischmann, B.; Pongratz, C.; et al. A tissue-specific screen of ceramide expression in aged mice identifies ceramide synthase-1 and ceramide synthase-5 as potential regulators of fiber size and strength in skeletal muscle. Aging Cell 2020, 19, e13049. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.W. The Evolution of Per-cell Organelle Number. Front. Cell. Dev. Biol. 2016, 4, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Ngoh, G.A.; Dabkowski, E.R.; O’Connell, K.A.; Ribeiro, R.F., Jr.; Stanley, W.C.; Walsh, K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. 2012, 302, H167–H179. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, Y.; Jiao, J.; Wang, J.; Li, Y.; Qin, D.; Li, P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 2014, 34, 1788–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., II; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Hu, L.; Ding, M.; Tang, D.; Gao, E.; Li, C.; Wang, K.; Qi, B.; Qiu, J.; Zhao, H.; Chang, P.; et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics 2019, 9, 3687–3706. [Google Scholar] [CrossRef]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef] [Green Version]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Yoon, Y. Mitochondrial Membrane Dynamics-Functional Positioning of OPA1. Antioxidants 2018, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burte, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernandez-Resendiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [Green Version]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef] [Green Version]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Chen, C.Y.; Li, S.J.; Wang, C.Y.; Mersmann, H.J.; Ding, S.T. The impact of DRP1 on myocardial fibrosis in the obese minipig. Eur J. Clin. Investig. 2020, 50, e13204. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S. Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB life 2008, 60, 448–455. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherok, E.; Xu, S.; Li, S.; Das, S.; Meltzer, W.A.; Zalzman, M.; Wang, C.; Karbowski, M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell. 2017, 28, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Iniguez-Lluhi, J.A.; Stadler, J.; Chang, C.R.; Arnoult, D.; Keller, P.J.; Hong, Y.; Blackstone, C.; Feldman, E.L. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009, 23, 3917–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolis, M.; Bond, L.M.; Kampmann, M.; Pulimeno, P.; Chitraju, C.; Jayson, C.B.K.; Vaites, L.P.; Boland, S.; Lai, Z.W.; Gabriel, K.R.; et al. Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol. Cell 2019, 74, 32–44.e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Stojanovski, D.; Koutsopoulos, O.S.; Okamoto, K.; Ryan, M.T. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 2004, 117, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. B.B.A. 2008, 1777, 860–866. [Google Scholar] [CrossRef]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. B.B.A. 2013, 1833, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Zhang, Y.; Liu, Q.; Xiang, W. Mfn2 Affects Embryo Development via Mitochondrial Dysfunction and Apoptosis. PloS ONE 2015, 10, e0125680. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacin, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorzano, A.; Hernandez-Alvarez, M.I.; Sebastian, D.; Munoz, J.P. Mitofusin 2 as a driver that controls energy metabolism and insulin signaling. Antioxid. Redox Signal. 2015, 22, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Seidlmayer, L.K.; Mages, C.; Berbner, A.; Eder-Negrin, P.; Arias-Loza, P.A.; Kaspar, M.; Song, M.; Dorn, G.W., II; Kohlhaas, M.; Frantz, S.; et al. Mitofusin 2 Is Essential for IP3-Mediated SR/Mitochondria Metabolic Feedback in Ventricular Myocytes. Front. Physiol. 2019, 10, 733. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Hoke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Met. 2015, 21, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [Green Version]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, H.; Gutierrez Cortes, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Zaloga, G.P.; Malcolm, D.; Holaday, J.; Chernow, B. Verapamil reverses calcium cardiotoxicity. Ann. Emerg. Med. 1987, 16, 637–639. [Google Scholar] [CrossRef]
- Lou, Q.; Janardhan, A.; Efimov, I.R. Remodeling of calcium handling in human heart failure. Adv. Exp. Med. Biol. 2012, 740, 1145–1174. [Google Scholar] [CrossRef] [Green Version]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered myocardial calcium cycling and energetics in heart failure—A rational approach for disease treatment. Cell Met. 2015, 21, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Samuel, T.J.; Rosenberry, R.P.; Lee, S.; Pan, Z. Correcting Calcium Dysregulation in Chronic Heart Failure Using SERCA2a Gene Therapy. Int. J. Mol. Sci. 2018, 19, 1086. [Google Scholar] [CrossRef] [Green Version]
- Jensen, A.C.; Polcwiartek, C.; Sogaard, P.; Mortensen, R.N.; Davidsen, L.; Aldahl, M.; Eriksen, M.A.; Kragholm, K.; Torp-Pedersen, C.; Hansen, S.M. The Association Between Serum Calcium Levels and Short-Term Mortality in Patients with Chronic Heart Failure. Am. J. Med. 2019, 132, 200–208.e201. [Google Scholar] [CrossRef]
- Reiken, S.; Wehrens, X.H.; Vest, J.A.; Barbone, A.; Klotz, S.; Mancini, D.; Burkhoff, D.; Marks, A.R. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 2003, 107, 2459–2466. [Google Scholar] [CrossRef]
- Knoll, K.; Kurowski, V.; Schunkert, H.; Sager, H.B. Management of hypercalcaemia-induced heart failure using mechanical circulatory support. Eur. J. Cardiothorac. Surg. 2018, 54, 784–785. [Google Scholar] [CrossRef]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ. Res. 2014, 114, 1311–1319; discussion 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.M.; Hajjar, R.J. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.J.; Oh, J.G. SERCA2a: A prime target for modulation of cardiac contractility during heart failure. BMB Rep. 2013, 46, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Hanson, C.J.; Bootman, M.D.; Distelhorst, C.W.; Maraldi, T.; Roderick, H.L. The cellular concentration of Bcl-2 determines its pro- or anti-apoptotic effect. Cell Calcium 2008, 44, 243–258. [Google Scholar] [CrossRef] [Green Version]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell. Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef]
- Portier, B.P.; Taglialatela, G. Bcl-2 localized at the nuclear compartment induces apoptosis after transient overexpression. J. Biol. Chem. 2006, 281, 40493–40502. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zhang, S.; Fritz, N.; Ibarra, C.; Uhlen, P. Inositol 1,4,5-trisphosphate receptor subtype-specific regulation of calcium oscillations. Neurochem. Res. 2011, 36, 1175–1185. [Google Scholar] [CrossRef] [Green Version]
- Spat, A.; Szanda, G.; Csordas, G.; Hajnoczky, G. High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium 2008, 44, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, D.; Sancak, Y.; Mootha, V.K.; Clapham, D.E. MCU encodes the pore conducting mitochondrial calcium currents. Elife 2013, 2, e00704. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: The role of Ras. Mitochondrion 2009, 9, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.C.; Gomes, D.A.; Thompson, M.; Souto, N.C.; Goes, T.S.; Goes, A.M.; Rodrigues, M.A.; Gomez, M.V.; Nathanson, M.H.; Leite, M.F. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 2005, 280, 40892–40900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dia, M.; Gomez, L.; Thibault, H.; Tessier, N.; Leon, C.; Chouabe, C.; Ducreux, S.; Gallo-Bona, N.; Tubbs, E.; Bendridi, N.; et al. Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 2020, 115, 74. [Google Scholar] [CrossRef] [PubMed]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Li, X.X.; Gottleib, E.; Hill, R.B.; Thompson, C.B.; Colombini, M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001, 276, 19414–19419. [Google Scholar] [CrossRef] [Green Version]
- Vande Velde, C.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, L.; Liu, H.; Borowitz, J.L.; Isom, G.E. BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB J. 2009, 23, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Egnatchik, R.A.; Leamy, A.K.; Jacobson, D.A.; Shiota, M.; Young, J.D. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol. Metab. 2014, 3, 544–553. [Google Scholar] [CrossRef]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnaune-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, A.B. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 2011, 32, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Ann. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Yang, Y.; Li, T.; Li, Z.; Liu, N.; Yan, Y.; Liu, B. Role of Mitophagy in Cardiovascular Disease. Aging Dis. 2020, 11, 419–437. [Google Scholar] [CrossRef] [Green Version]
- Chi, C.; Riching, A.S.; Song, K. Lysosomal Abnormalities in Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 811. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Godar, R.J.; Liu, H.; Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012, 8, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. 2008, 295, H2025–H2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. Handb. Exp. Pharmacol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Li, P.L.; Gulbins, E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid. Redox Signal. 2018, 28, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015, 80, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Joseph, L.C.; Kokkinaki, D.; Valenti, M.C.; Kim, G.J.; Barca, E.; Tomar, D.; Hoffman, N.E.; Subramanyam, P.; Colecraft, H.M.; Hirano, M.; et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI insight 2017, 2. [Google Scholar] [CrossRef]
- Nakamura, H.; Matoba, S.; Iwai-Kanai, E.; Kimata, M.; Hoshino, A.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53 promotes cardiac dysfunction in diabetic mellitus caused by excessive mitochondrial respiration-mediated reactive oxygen species generation and lipid accumulation. Circ. Heart Fail. 2012, 5, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [Green Version]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 14, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazici, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Reyes, M.V.; Lakkadi, K.R.; Gowen, B.H.; Hasko, G.; Drosatos, K.; Morrow, J.P. PKCdelta causes sepsis-induced cardiomyopathy by inducing mitochondrial dysfunction. Am. J. Physiol. Heart Circ. 2020, 318, H778–H786. [Google Scholar] [CrossRef]
- Inagaki, K.; Chen, L.; Ikeno, F.; Lee, F.H.; Imahashi, K.; Bouley, D.M.; Rezaee, M.; Yock, P.G.; Murphy, E.; Mochly-Rosen, D. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 2003, 108, 2304–2307. [Google Scholar] [CrossRef] [Green Version]
- Churchill, E.N.; Szweda, L.I. Translocation of deltaPKC to mitochondria during cardiac reperfusion enhances superoxide anion production and induces loss in mitochondrial function. Arch. Biochem. Biophys. 2005, 439, 194–199. [Google Scholar] [CrossRef]
- Murriel, C.L.; Churchill, E.; Inagaki, K.; Szweda, L.I.; Mochly-Rosen, D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: A mechanism involving BAD and the mitochondria. J. Biol. Chem. 2004, 279, 47985–47991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becatti, M.; Taddei, N.; Cecchi, C.; Nassi, N.; Nassi, P.A.; Fiorillo, C. SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes. Cell. Mol. Life Sci. 2012, 69, 2245–2260. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koka, S.; Aluri, H.S.; Xi, L.; Lesnefsky, E.J.; Kukreja, R.C. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: Potential role of NO/SIRT1/PGC-1alpha signaling. Am. J. Physiol. Heart Circ. 2014, 306, H1558–H1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Mobius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J.; Zhang, Y.; Bu, P. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PloS ONE 2015, 10, e0118909. [Google Scholar] [CrossRef]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 36. [Google Scholar] [CrossRef]
- Liao, X.; Zhang, R.; Lu, Y.; Prosdocimo, D.A.; Sangwung, P.; Zhang, L.; Zhou, G.; Anand, P.; Lai, L.; Leone, T.C.; et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Invest. 2015, 125, 3461–3476. [Google Scholar] [CrossRef] [Green Version]
- Drosatos, K.; Pollak, N.M.; Pol, C.J.; Ntziachristos, P.; Willecke, F.; Valenti, M.C.; Trent, C.M.; Hu, Y.; Guo, S.; Aifantis, I.; et al. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circ. Res. 2016, 118, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Prosdocimo, D.A.; Anand, P.; Liao, X.; Zhu, H.; Shelkay, S.; Artero-Calderon, P.; Zhang, L.; Kirsh, J.; Moore, D.; Rosca, M.G.; et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J. Biol. Chem. 2014, 289, 5914–5924. [Google Scholar] [CrossRef] [Green Version]
- Sato, P.Y.; Chuprun, J.K.; Grisanti, L.A.; Woodall, M.C.; Brown, B.R.; Roy, R.; Traynham, C.J.; Ibetti, J.; Lucchese, A.M.; Yuan, A.; et al. Restricting mitochondrial GRK2 post-ischemia confers cardioprotection by reducing myocyte death and maintaining glucose oxidation. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, P.Y.; Chuprun, J.K.; Ibetti, J.; Cannavo, A.; Drosatos, K.; Elrod, J.W.; Koch, W.J. GRK2 compromises cardiomyocyte mitochondrial function by diminishing fatty acid-mediated oxygen consumption and increasing superoxide levels. J. Mol. Cell. Cardiol. 2015, 89, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Ceramide structure and synthesis. (a) Schematic diagram illustrating ceramide structure. (b) Simplified ceramide synthesis pathways with essential enzymes in the respective pathway. (c) Schematic presentation of ceramide synthase structure based on previous findings [93,94,95,96,97,98,99,100,101].
Figure 1.
Ceramide structure and synthesis. (a) Schematic diagram illustrating ceramide structure. (b) Simplified ceramide synthesis pathways with essential enzymes in the respective pathway. (c) Schematic presentation of ceramide synthase structure based on previous findings [93,94,95,96,97,98,99,100,101].

Figure 2.
Schematic illustration of lipotoxicity-mediated mitochondrial damage.

Figure 3.
Visualization of mitochondrial structure. (a) Impaired and disturbed mitochondrial distribution was stained with MitoTracker (green) and visualized with laser scanning microscopy in doxorubicin-treated HL-1 murine cardiomyocytes. Nucleus was stained with Hoechst (blue). (b) mRNA expression of mitochondrial fusion and fission-related genes. Cardiac cell line HL-1 cells treated with doxorubicin showed decreased MFN1 and MFN2 expression but increased DRP1, Mff, and FIS1 expression. Doxorubicin-dependent induction of ceramide level was attenuated upon treatment with the unspecific CerS inhibitor fumonisin B. The fission-related mRNA expression (DRP1, Mff, FIS1) returned to non-significant levels following fumonisin B treatment. Significance was calculated with t-test. * p < 0.05, ** p < 0.01.
Figure 3.
Visualization of mitochondrial structure. (a) Impaired and disturbed mitochondrial distribution was stained with MitoTracker (green) and visualized with laser scanning microscopy in doxorubicin-treated HL-1 murine cardiomyocytes. Nucleus was stained with Hoechst (blue). (b) mRNA expression of mitochondrial fusion and fission-related genes. Cardiac cell line HL-1 cells treated with doxorubicin showed decreased MFN1 and MFN2 expression but increased DRP1, Mff, and FIS1 expression. Doxorubicin-dependent induction of ceramide level was attenuated upon treatment with the unspecific CerS inhibitor fumonisin B. The fission-related mRNA expression (DRP1, Mff, FIS1) returned to non-significant levels following fumonisin B treatment. Significance was calculated with t-test. * p < 0.05, ** p < 0.01.

Figure 4.
Flowchart presents the process of heart failure development following lipotoxicity.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kretzschmar, T.; Wu, J.M.F.; Schulze, P.C. Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium. Int. J. Mol. Sci. 2021, 22, 1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031498
AMA Style
Kretzschmar T, Wu JMF, Schulze PC. Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium. International Journal of Molecular Sciences. 2021; 22(3):1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031498
Chicago/Turabian StyleKretzschmar, Tom, Jasmine M. F. Wu, and P. Christian Schulze. 2021. "Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium" International Journal of Molecular Sciences 22, no. 3: 1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031498
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.