
Therapeutic Potential of Regorafenib—A Multikinase Inhibitor in Pulmonary Hypertension

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
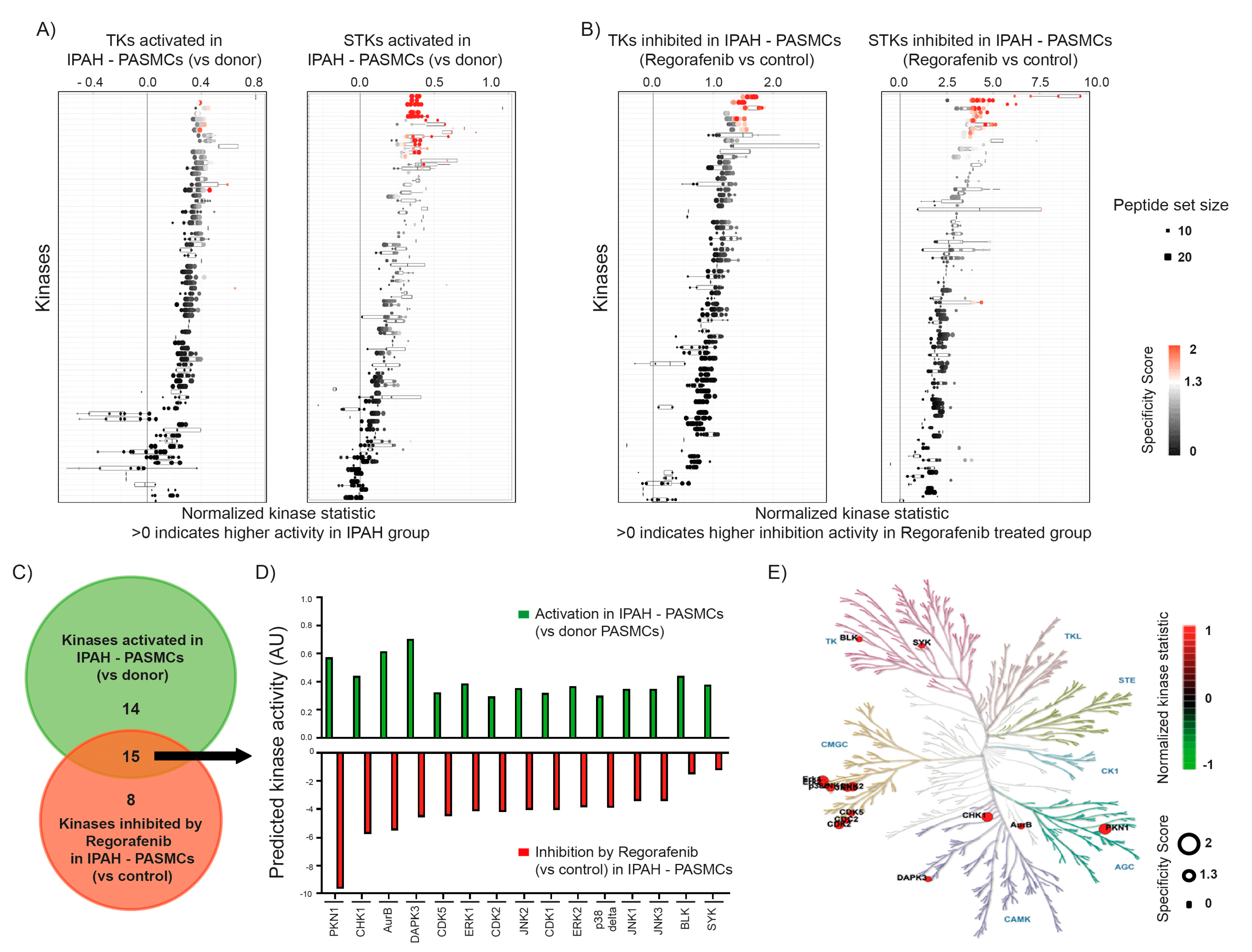
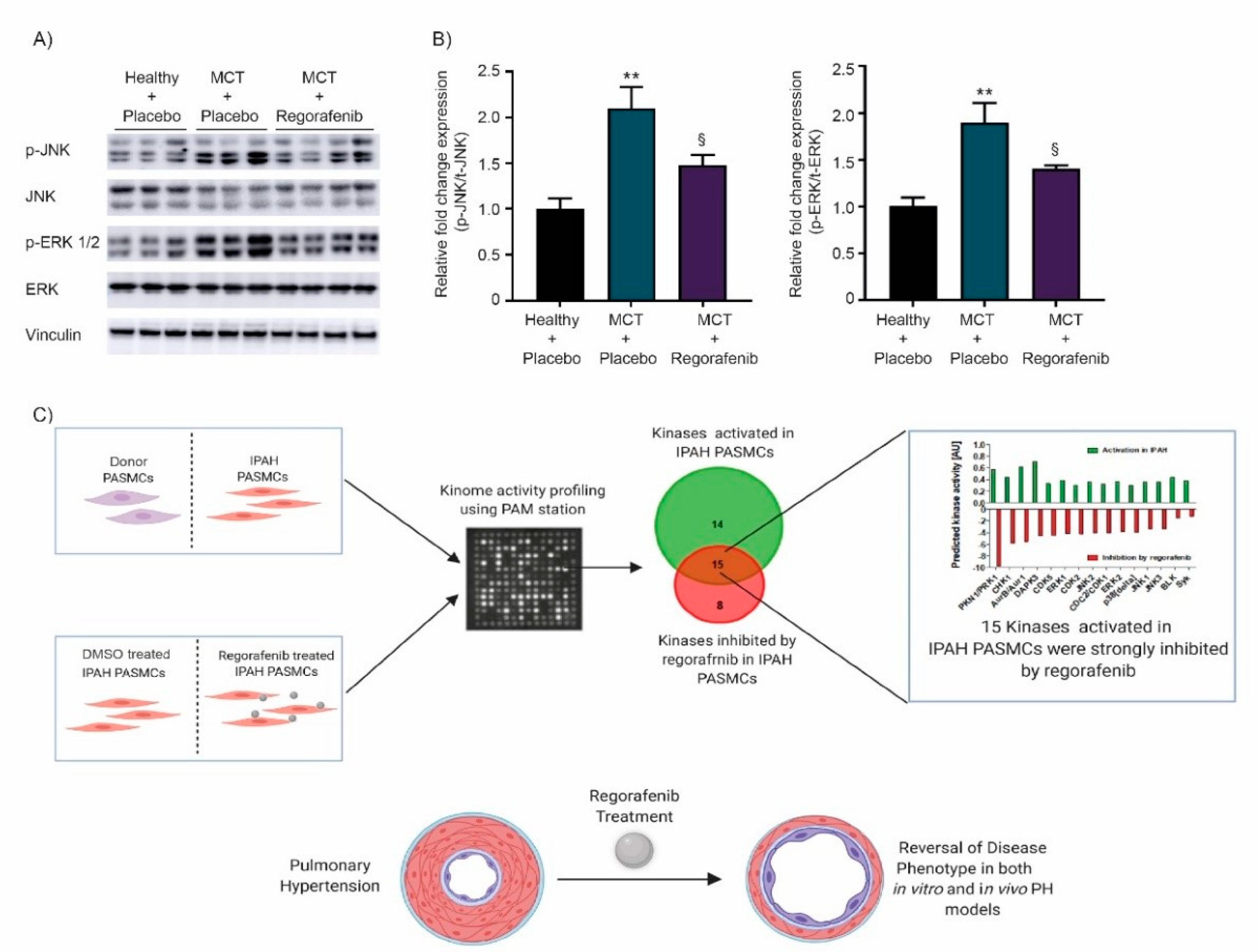
2.1. Effect of Regorafenib on Kinase Activity in Pulmonary Smooth Muscle Cells Isolated from Patients with Idiopathic Pulmonary Arterial Hypertension
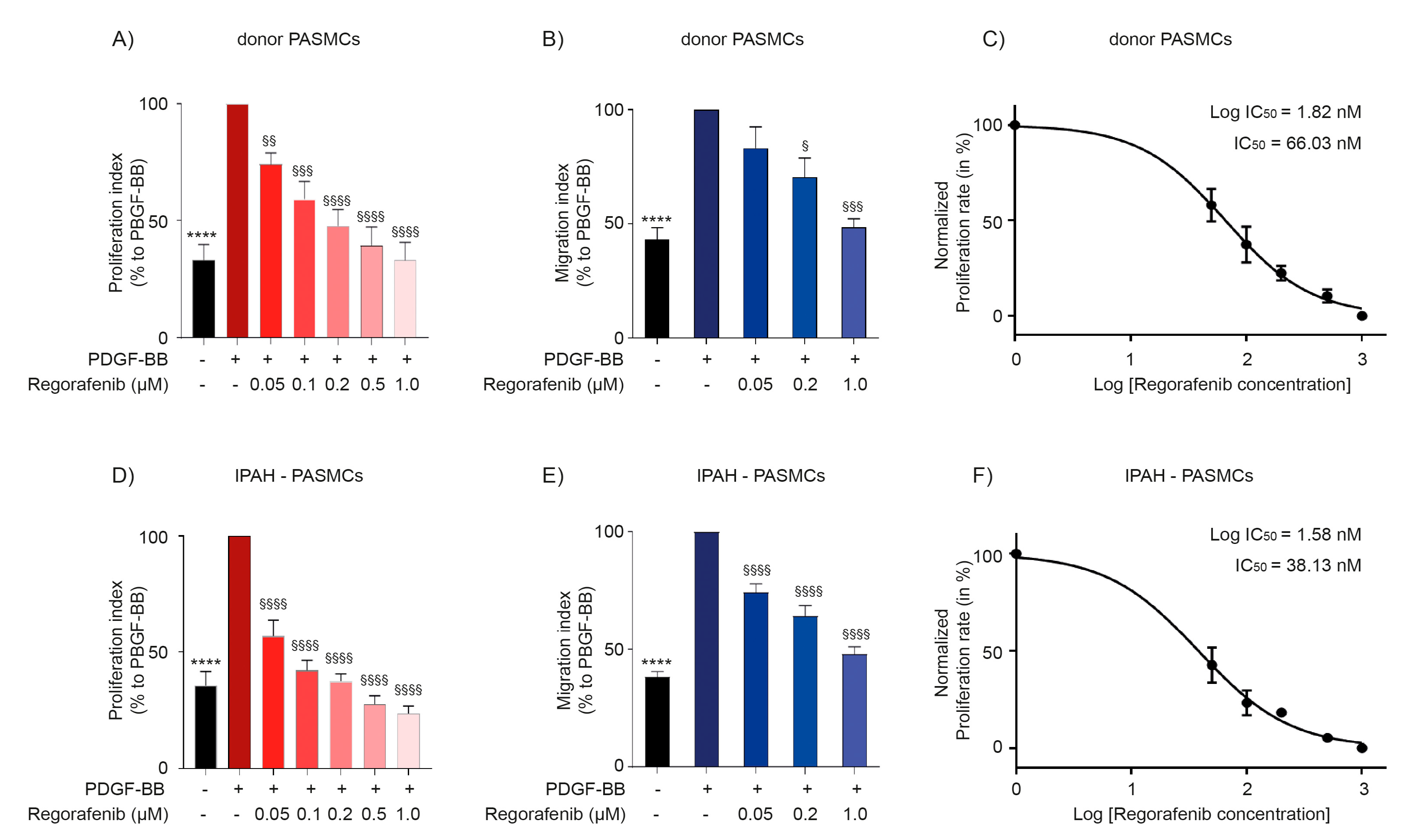
2.2. Regorafenib Inhibits Proliferation and Migration of Human Pulmonary Arterial Smooth Muscle Cells
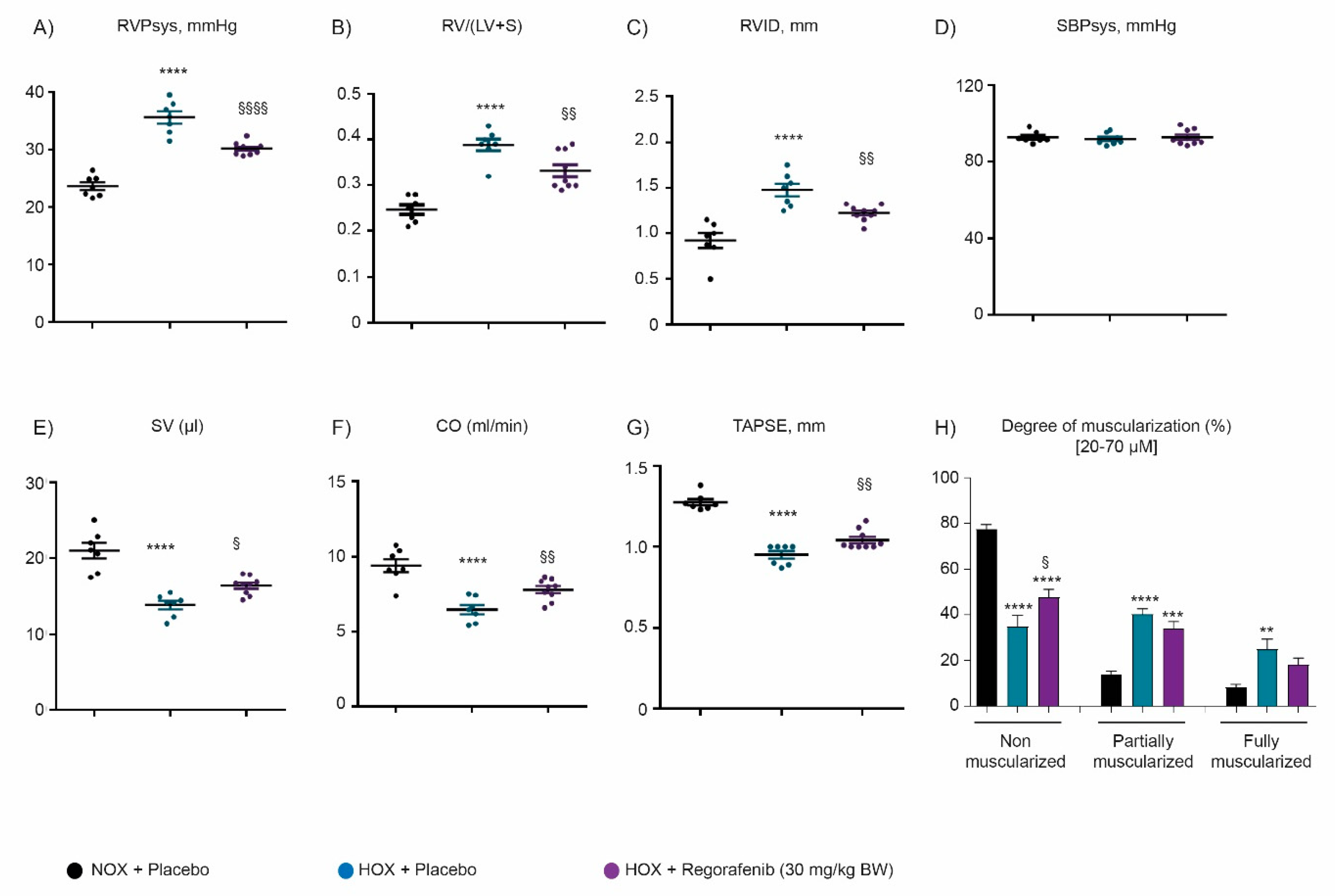
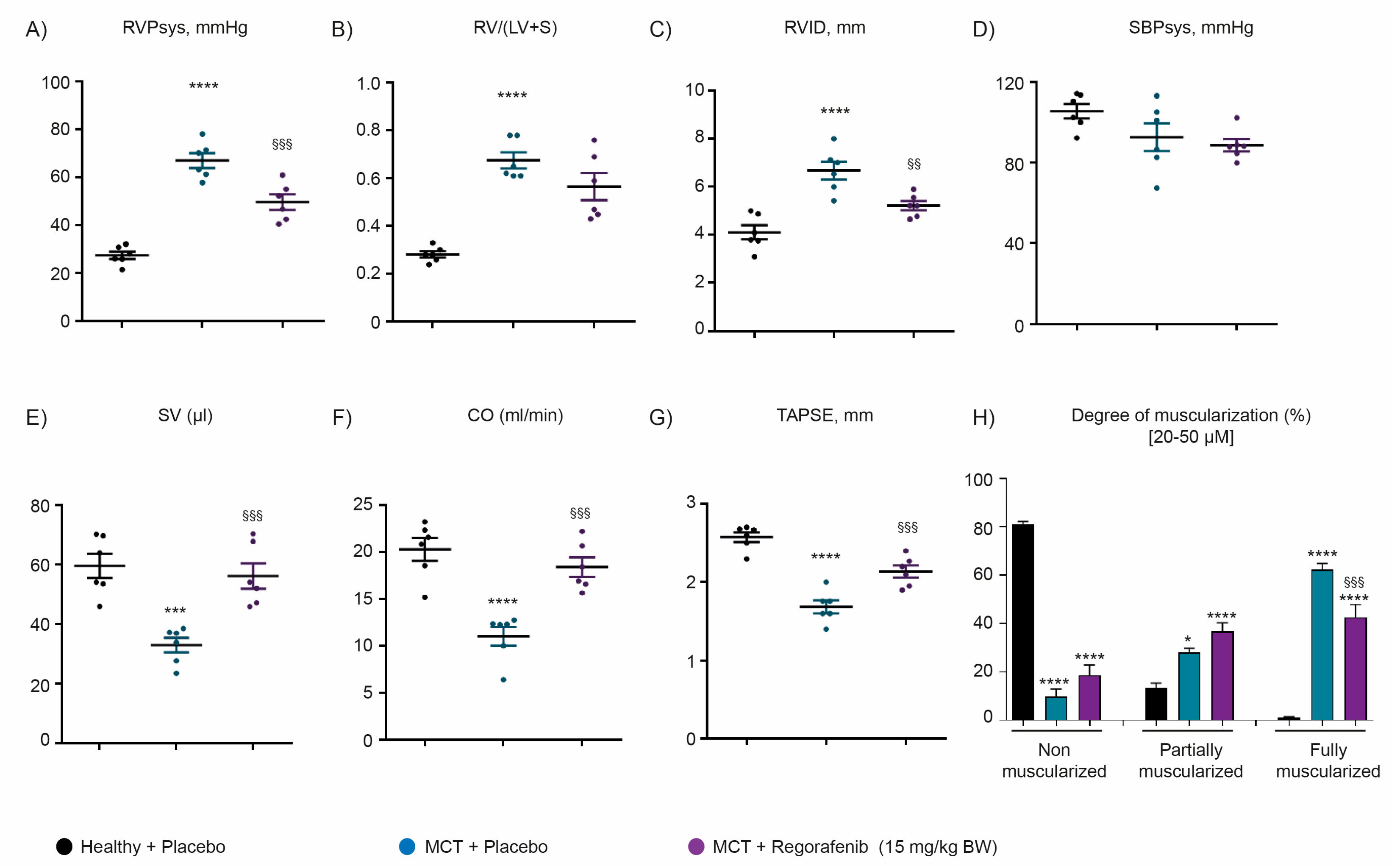
2.3. Regorafenib Improves Cardiac Function and Reverses Pulmonary Vascular Remodeling Both in Hypoxia- and Monocrotaline-Induced Pulmonary Hypertension
2.4. Regorafenib Attenuates the Activity of MAPK Signaling via ERK and JNK
3. Discussion
4. Materials and Methods
4.1. Drugs and Antibodies
4.2. In Vivo Experiments
4.3. Cell Culture Experiments
4.4. Proliferation Assays
4.5. Migration Assays
4.6. Kinome Analysis
4.7. Western Blot Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovacs, G.; Dumitrescu, D.; Barner, A.; Greiner, S.; Grünig, E.; Hager, A.; Köhler, T.; Kozlik-Feldmann, R.; Kruck, I.; Lammers, A.E.; et al. Definition, clinical classification and initial diagnosis of pulmonary hypertension: Updated recommendations from the Cologne Consensus Conference. Int. J. Cardiol. 2018, 272, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Godinas, L.; Guignabert, C.; Seferian, A.; Perros, F.; Bergot, E.; Sibille, Y.; Humbert, M.; Montani, D. Tyrosine kinase inhibitors in pulmonary arterial hypertension: A double-edge sword? Semin. Respir. Crit. Care Med. 2013, 34, 714–724. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef]
- Thompson, A.R.; Lawrie, A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol. Med. 2017, 23, 31–45. [Google Scholar] [CrossRef]
- Woodcock, C.-S.C.; Chan, S.Y. The Search for Disease-Modifying Therapies in Pulmonary Hypertension. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 334–354. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerabolu, D.; Weiss, A.; Kojonazarov, B.; Boehm, M.; Schlueter, B.C.; Ruppert, C.; Günther, A.; Jonigk, D.; Grimminger, F.; Ghofrani, H.-A.; et al. Targeting Jak–Stat Signaling in Experimental Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir J. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Cowan, K.N.; Rabinovitch, M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am. J. Pathol. 1997, 150, 1349–1360. [Google Scholar] [PubMed]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. TranslationalAdvances in theField ofPulmonaryHypertension.From Cancer Biology to New Pulmonary Arterial Hypertension Therapeutics. Targeting Cell Growth and Proliferation Signaling Hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Boehm, M.; Egemnazarov, B.; Grimminger, F.; Pullamsetti, S.S.; Kwapiszewska, G.; Schermuly, R.T. Kinases as potential targets for treatment of pulmonary hypertension and right ventricular dysfunction. Br. J. Pharmacol. 2021, 178, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Jasińska-Stroschein, M.; Owczarek, J.; Plichta, P.; Orszulak-Michalak, D. Concurrent Rho-Kinase and Tyrosine Kinase Platelet-Derived Growth Factor Inhibition in Experimental Pulmonary Hypertension. Pharmacology 2014, 93, 145–150. [Google Scholar] [CrossRef]
- Guignabert, C.; Phan, C.; Seferian, A.; Huertas, A.; Tu, L.; Thuillet, R.; Sattler, C.; Le Hiress, M.; Tamura, Y.; Jutant, E.-M.; et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Investig. 2016, 126, 3207–3218. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Berghausen, E.M.; Dabral, S.; Tretyn, A.; Butrous, E.; Savai, R.; Butrous, G.; Dahal, B.K.; Brandes, R.P.; Ghofrani, H.A.; et al. Role of Src Tyrosine Kinases in Experimental Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2012, 32, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahal, B.K.; Heuchel, R.; Pullamsetti, S.S.; Wilhelm, J.; Ghofrani, H.A.; Weissmann, N.; Seeger, W.; Grimminger, F.; Schermuly, R.T. Hypoxic pulmonary hypertension in mice with constitutively active platelet-derived growth factor receptor-beta. Pulm. Circ. 2011, 1, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Toba, M.; Alzoubi, A.; Koubský, K.; Ito, M.; Ota, H.; Gairhe, S.; Gerthoffer, W.; Fagan, K.A.; McMurtry, I.F.; et al. Tyrosine Kinase Inhibitors Are Potent Acute Pulmonary Vasodilators in Rats. Am. J. Respir. Cell Mol. Biol. 2011, 45, 804–808. [Google Scholar] [CrossRef] [Green Version]
- Kojonazarov, B.; Sydykov, A.; Pullamsetti, S.S.; Luitel, H.; Dahal, B.K.; Kosanovic, D.; Tian, X.; Majewski, M.; Baumann, C.; Evans, S.; et al. Effects of multikinase inhibitors on pressure overload-induced right ventricular remodeling. Int. J. Cardiol. 2013, 167, 2630–2637. [Google Scholar] [CrossRef]
- Singh, H.; Thangaraju, P.; Chakrabarti, A. Regorafenib: A novel tyrosine kinase inhibitor: A brief review of its therapeutic potential in the treatment of metastatic colorectal carcinoma and advanced gastrointestinal stromal tumors. Indian J. Cancer 2015, 52, 257. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Tak, W.Y.; Gasbarrini, A.; Santoro, A.; Colombo, M.; Lim, H.-Y.; Mazzaferro, V.; Wiest, R.; Reig, M.; Wagner, A.; et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 2013, 49, 3412–3419. [Google Scholar] [CrossRef]
- Liu, S.; Du, Y.; Ma, H.; Liang, Q.; Zhu, X.; Tian, J. Preclinical comparison of regorafenib and sorafenib efficacy for hepatocellular carcinoma using multimodality molecular imaging. Cancer Lett. 2019, 453, 74–83. [Google Scholar] [CrossRef]
- Pelosof, L.; Lemery, S.; Casak, S.; Jiang, X.; Rodríguez, L.; Pierre, V.; Bi, Y.; Liu, J.; Zirkelbach, J.F.; Patel, A.; et al. Benefit-Risk Summary of Regorafenib for the Treatment of Patients with Advanced Hepatocellular Carcinoma That Has Progressed on Sorafenib. Oncol. 2018, 23, 496–500. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.K.; Ravi, S. Regorafenib: An evidence-based review of its potential in patients with advanced liver cancer. Core Évid. 2014, 9, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Goel, G. Evolution of regorafenib from bench to bedside in colorectal cancer: Is it an attractive option or merely a “me too” drug? Cancer Manag. Res. 2018, 10, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, J.; Zhang, Y.; Cai, H.; Chen, X.-M.; Sun, D. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open Bio 2019, 9, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Zopf, D.; Fichtner, I.; Bhargava, A.; Steinke, W.; Thierauch, K.-H.; Diefenbach, K.; Wilhelm, S.; Hafner, F.-T.; Gerisch, M. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016, 5, 3176–3185. [Google Scholar] [CrossRef]
- Weiss, A.; Neubauer, M.C.; Yerabolu, D.; Kojonazarov, B.; Schlueter, B.C.; Neubert, L.; Jonigk, D.; Baal, N.; Ruppert, C.; Dorfmuller, P.; et al. Targeting cyclin-dependent kinases for the treatment of pulmonary arterial hypertension. Nat. Commun. 2019, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- Church, A.C.; Martin, D.H.; Wadsworth, R.; Bryson, G.; Fisher, A.J.; Welsh, D.J.; Peacock, A.J. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: A potential novel anti-inflammatory strategy in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L333–L347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Guo, H.; Sun, Y.; Pan, X.; Dong, J.; Gao, D.; Chen, W.; Xu, Y.; Xu, D. Valsartan attenuates pulmonary hypertension via suppression of mitogen activated protein kinase signaling and matrix metalloproteinase expression in rodents. Mol. Med. Rep. 2017, 16, 1360–1368. [Google Scholar] [CrossRef]
- Sitbon, O.; Morrell, N. Pathways in pulmonary arterial hypertension: The future is here. Eur. Respir. Rev. 2012, 21, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Schermuly, R.T.; Ellinghaus, P.; Milting, H.; Riedl, B.; Nikolova, S.; Pullamsetti, S.S.; Weissmann, N.; Dony, E.; Savai, R.; et al. Combined Tyrosine and Serine/Threonine Kinase Inhibition by Sorafenib Prevents Progression of Experimental Pulmonary Hypertension and Myocardial Remodeling. Circ 2008, 118, 2081–2090. [Google Scholar] [CrossRef]
- Moreno-Vinasco, L.; Gomberg-Maitland, M.; Maitland, M.L.; Desai, A.A.; Singleton, P.A.; Sammani, S.; Sam, L.; Liu, Y.; Husain, A.N.; Lang, R.M.; et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genom. 2008, 33, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Frost, A.E.; Barst, R.J.; Hoeper, M.M.; Chang, H.-J.; Frantz, R.P.; Fukumoto, Y.; Galie, N.; Hassoun, P.M.; Klose, H.; Matsubara, H.; et al. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J. Hear. Lung Transpl. 2015, 34, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.-A.; Morrell, N.W.; Hoeper, M.M.; Olschewski, H.; Peacock, A.J.; Barst, R.J.; Shapiro, S.; Golpon, H.; Toshner, M.; Grimminger, F.; et al. Imatinib in Pulmonary Arterial Hypertension Patients with Inadequate Response to Established Therapy. Am. J. Respir. Crit. Care Med. 2010, 182, 1171–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, H.-A.; Seeger, W.; Grimminger, F. Imatinib for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 1412–1413. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Schermuly, R.T.; Ghofrani, H.-A. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat. Rev. Drug Discov. 2010, 9, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.C.; Weissmann, A.; Ahmadi, T.; Farber, H.W. Imatinib Mesylate in the Treatment of Refractory Idiopathic Pulmonary Arterial Hypertension. Ann. Intern. Med. 2006, 145, 152–153. [Google Scholar] [CrossRef] [Green Version]
- Gomberg-Maitland, M.; Maitland, M.L.; Barst, R.J.; Sugeng, L.; Coslet, S.; Perrino, T.J.; Bond, L.; LaCouture, M.E.; Archer, S.L.; Ratain, M.J. A dosing/cross-development study of the multikinase inhibitor sorafenib in patients with pulmonary arterial hypertension. Clin. Pharmacol. Ther. 2010, 87, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Kimura, G.; Kataoka, M.; Inami, T.; Fukuda, K.; Yoshino, H.; Satoh, T. Sorafenib as a potential strategy for refractory pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 2017, 44, 46–49. [Google Scholar] [CrossRef]
- Juengpanich, S.; Topatana, W.; Lu, C.; Staiculescu, D.; Li, S.; Cao, J.; Lin, J.; Hu, J.; Chen, M.; Chen, J.; et al. Role of cellular, molecular and tumor microenvironment in hepatocellular carcinoma: Possible targets and future directions in the regorafenib era. Int. J. Cancer 2020, 147, 1778–1792. [Google Scholar] [CrossRef]
- Roman, D.; Whiteside, R. Regorafenib: Adding to the Armamentarium for Refractory Colorectal Cancer and GIST. J. Adv. Pr. Oncol. 2013, 4, 118–122. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Chen, L.; Han, L.; Li, L.; He, H.; Li, Y.; Huang, N.; Ren, H.; Pei, F.; et al. The role of MIF, cyclinD1 and ERK in the development of pulmonary hypertension in broilers. Avian Pathol. 2016, 46, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Li, H.Z.; Wang, Y.H.; Peng, X.; Shao, H.J.; Li, H.X.; Bai, S.Z.; Lu, X.X.; Wu, L.Y.; Wang, R.; et al. Exogenous spermine inhibits the proliferation of human pulmonary artery smooth muscle cells caused by chemically-induced hypoxia via the suppression of the ERK1/2- and PI3K/AKT-associated pathways. Int. J. Mol. Med. 2016, 37, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Zawada, W.M.; West, J.; Stenmark, K.R. JNK2 regulates vascular remodeling in pulmonary hypertension. Pulm. Circ. 2018, 8, 2045894018778156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Shimpo, H.; Shimamoto, A.; Chong, A.J.; Hampton, C.R.; Spring, D.J.; Yada, M.; Takao, M.; Onoda, K.; Yada, I.; et al. Specific inhibition of p38 mitogen-activated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J. Thorac. Cardiovasc. Surg. 2004, 128, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Sala, M.A.; Chen, C.; Zhang, Q.; Do-Umehara, H.C.; Wu, W.; Misharin, A.V.; Waypa, G.B.; Fang, D.; Budinger, G.R.S.; Liu, S.; et al. JNK2 up-regulates hypoxia-inducible factors and contributes to hypoxia-induced erythropoiesis and pulmonary hypertension. J. Biol. Chem. 2018, 293, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G.; et al. p38 MAPK Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef]
- Bourgeois, A.; Bonnet, S.; Breuils-Bonnet, S.; Habbout, K.; Paradis, R.; Tremblay, E.; Lampron, M.-C.; Orcholski, M.E.; Potus, F.; Bertero, T.; et al. Inhibition of CHK 1 (Checkpoint Kinase 1) Elicits Therapeutic Effects in Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 1667–1681. [Google Scholar] [CrossRef]
- Satoh, K.; Kikuchi, N.; Kurosawa, R.; Shimokawa, H. Checkpoint Kinase 1 Promotes the Development of Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 1504–1506. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Okada, M.; Hara, Y.; Yamawaki, H. Exploring calmodulin-related proteins, which mediate development of hypertension, in vascular tissues of spontaneous hypertensive rats. Biochem. Biophys. Res. Commun. 2011, 405, 47–51. [Google Scholar] [CrossRef]
- Usui, T.; Okada, M.; Hara, Y.; Yamawaki, H. Death-Associated Protein Kinase 3 Mediates Vascular Inflammation and Development of Hypertension in Spontaneously Hypertensive Rats. Hypertension 2012, 60, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Sakatsume, T.; Nijima, R.; Otani, K.; Kazama, K.; Morita, T.; Kameshima, S.; Okada, M.; Yamawaki, H. Death-associated protein kinase 3 mediates vascular structural remodelling via stimulating smooth muscle cell proliferation and migration. Clin. Sci. 2014, 127, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar]
- Dos Santos, E.O.; Carneiro-Lobo, T.C.; Aoki, M.N.; Levantini, E.; Bassères, D.S. Aurora kinase targeting in lung cancer reduces KRAS-induced transformation. Mol. Cancer 2016, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Bowers, N.L.; Betticher, D.C.; Gautschi, O.; Ratschiller, D.; Hoban, P.R.; Booton, R.; Santibáñez-Koref, M.F.; Heighway, J. Overexpression of aurora B kinase (AURKB) in primary non-small cell lung carcinoma is frequent, generally driven from one allele, and correlates with the level of genetic instability. Br. J. Cancer 2005, 93, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, M.; Koga, T.; Takayama, K.; Ijichi, K.; Yano, T.; Maehara, Y.; Nakanishi, Y.; Sueishi, K. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer 2013, 80, 85–90. [Google Scholar] [CrossRef]
- Vischioni, B.; Oudejans, J.J.; Vos, W.; Rodriguez, J.A.; Giaccone, G. Frequent overexpression of aurora B kinase, a novel drug target, in non–small cell lung carcinoma patients. Mol. Cancer Ther. 2006, 5, 2905–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.L.; Wang, L.; Zhang, Z.; Hill, N.S.; Polgar, P. Participation of PLK1 and FOXM1 in the hyperplastic proliferation of pulmonary artery smooth muscle cells in pulmonary arterial hypertension. PLoS ONE 2019, 14, e0221728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.K.; Kundumani-Sridharan, V.; Kumar, S.; Verma, S.K.; Kotla, S.; Mukai, H.; Heckle, M.R.; Rao, G.N. Protein Kinase N1 Is a Novel Substrate of NFATc1-mediated Cyclin D1-CDK6 Activity and Modulates Vascular Smooth Muscle Cell Division and Migration Leading to Inward Blood Vessel Wall Remodeling. J. Biol. Chem. 2012, 287, 36291–36304. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-H.; Kim, S.W.; Lee, C.Y.; Lim, K.H.; Lee, J.; Choi, E.; Lim, S.; Lee, S.; Hwang, K.-C. A spleen tyrosine kinase inhibitor attenuates the proliferation and migration of vascular smooth muscle cells. Biol. Res. 2017, 50, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, D.L.; Krejsgaard, T.; Berthelsen, J.; Fredholm, S.; Willerslev-Olsen, A.; Sibbesen, N.; Bonefeld, C.M.; Andersen, M.H.; Francavilla, C.; Olsen, J.V.; et al. B-lymphoid tyrosine kinase (Blk) is an oncogene and a potential target for therapy with dasatinib in cutaneous T-cell lymphoma (CTCL). Leukemia 2014, 28, 2109–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuder, R. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wu, L.-W.; Zhang, Z.-Y.; Chen, M.-L.; Li, Y.-L.; Zhang, C. The anti-tumor effect of regorafenib in lung squamous cell carcinoma in vitro. Biochem. Biophys. Res. Commun. 2018, 503, 1123–1129. [Google Scholar] [CrossRef]
- Boran, T.; Akyildiz, A.G.; Jannuzzi, A.T.; Alpertunga, B. Extended regorafenib treatment can be linked with mitochondrial damage leading to cardiotoxicity. Toxicol Lett. 2021, 336, 39–49. [Google Scholar] [CrossRef]
- Hsiao, F.C.; Yeh, C.N.; Chu, P.H. Regorafenib-Related Myocardial Injury during Atrial Fibrillation. Acta Cardiol. Sin. 2016, 32, 243–246. [Google Scholar]
- Saito, Y.; Atsushi, M.; Masaru, M.; Yuki, Y. Cardiovascular toxicity of regorafenib for heavily-treated osteosarcoma. Pediatr. Int. 2020. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J. Risk of regorafenib-induced cardiovascular events in patients with solid tumors: A systematic review and meta-analysis. Medicine 2018, 97, e12705. [Google Scholar] [CrossRef] [PubMed]
- Elseud, Y.A.; Shaaban, A.; Mohanty, A.; Albarrak, J. Safety and tolerability of regorafenib: A real-life experience. J. Gastrointest Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Arnold, N.; Braithwaite, A.T.; Pickworth, J.A.; Lu, C.; Novoyatleva, T.; Kiely, D.G.; Grimminger, F.; Ghofrani, H.-A.; Weissmann, N.; et al. Eplerenone attenuates pathological pulmonary vascular rather than right ventricular remodeling in pulmonary arterial hypertension. BMC Pulm. Med. 2018, 18, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146. [Google Scholar] [CrossRef] [PubMed]
- Novoyatleva, T.; Kojonazarov, B.; Owczarek, A.; Veeroju, S.; Rai, N.; Henneke, I.; Böhm, M.; Grimminger, F.; Ghofrani, H.-A.; Seeger, W.; et al. Evidence for the Fucoidan/P-Selectin Axis as a Therapeutic Target in Hypoxia-induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Budas, G.R.; Boehm, M.; Kojonazarov, B.; Viswanathan, G.; Tian, X.; Veeroju, S.; Novoyatleva, T.; Grimminger, F.; Hinojosa-Kirschenbaum, F.; Ghofrani, H.-A.; et al. ASK1 Inhibition Halts Disease Progression in Preclinical Models of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 373–385. [Google Scholar] [CrossRef]
- Alack, K.; Weiss, A.; Krüger, K.; Höret, M.; Schermuly, R.; Frech, T.; Eggert, M.; Mooren, F.-C. Profiling of human lymphocytes reveals a specific network of protein kinases modulated by endurance training status. Sci. Rep. 2020, 10, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.-S.; Kwapiszewska, G.; et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veeroju, S.; Kojonazarov, B.; Weiss, A.; Ghofrani, H.A.; Weissmann, N.; Grimminger, F.; Seeger, W.; Novoyatleva, T.; Schermuly, R.T. Therapeutic Potential of Regorafenib—A Multikinase Inhibitor in Pulmonary Hypertension. Int. J. Mol. Sci. 2021, 22, 1502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031502
Veeroju S, Kojonazarov B, Weiss A, Ghofrani HA, Weissmann N, Grimminger F, Seeger W, Novoyatleva T, Schermuly RT. Therapeutic Potential of Regorafenib—A Multikinase Inhibitor in Pulmonary Hypertension. International Journal of Molecular Sciences. 2021; 22(3):1502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031502
Chicago/Turabian StyleVeeroju, Swathi, Baktybek Kojonazarov, Astrid Weiss, Hossein Ardeschir Ghofrani, Norbert Weissmann, Friedrich Grimminger, Werner Seeger, Tatyana Novoyatleva, and Ralph Theo Schermuly. 2021. "Therapeutic Potential of Regorafenib—A Multikinase Inhibitor in Pulmonary Hypertension" International Journal of Molecular Sciences 22, no. 3: 1502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031502