
Interaction of TLR4 and TLR8 in the Innate Immune Response against Mycobacterium Tuberculosis

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Cohort Characteristics and Genotyping
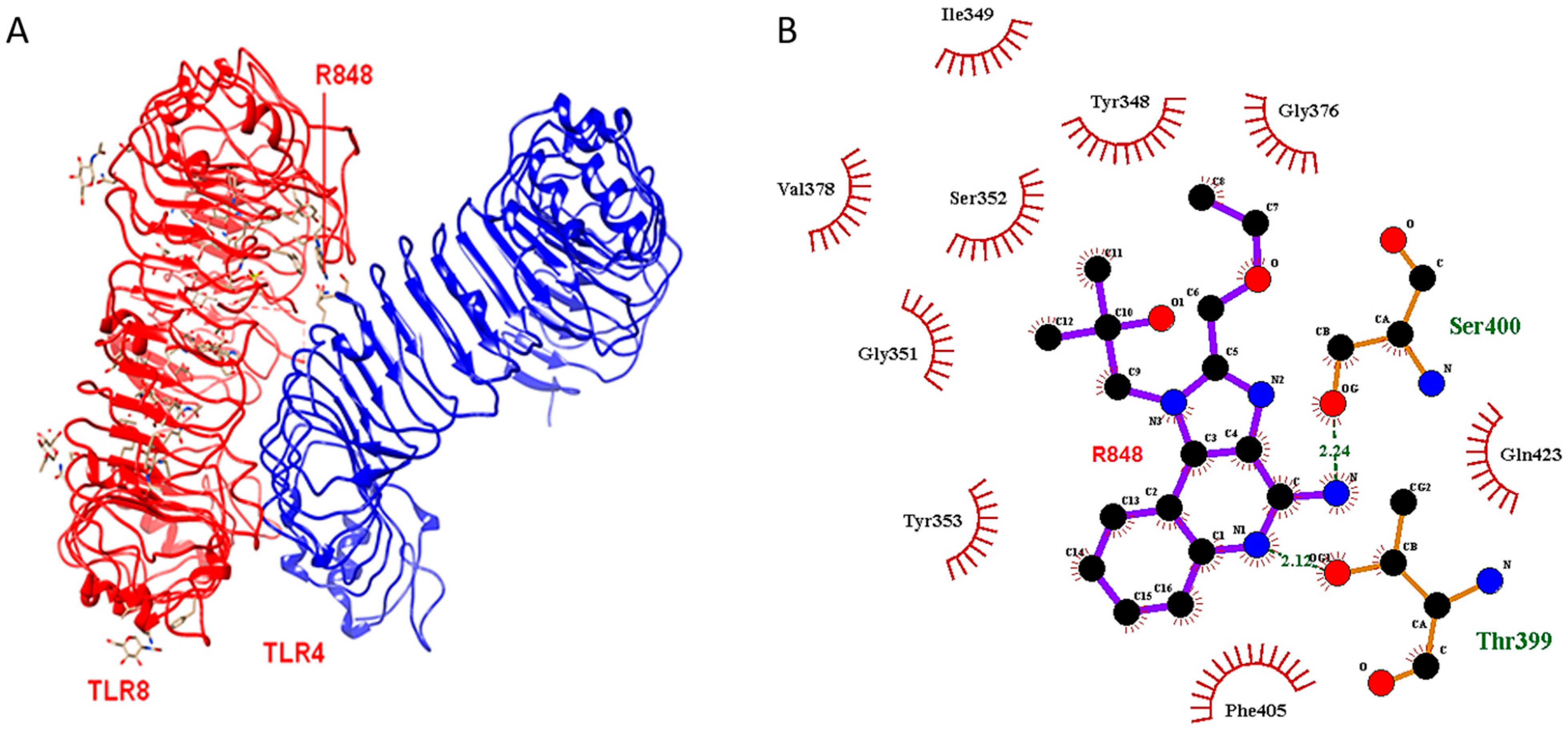
2.2. Docking Outcome
2.3. Mass Spectrometry
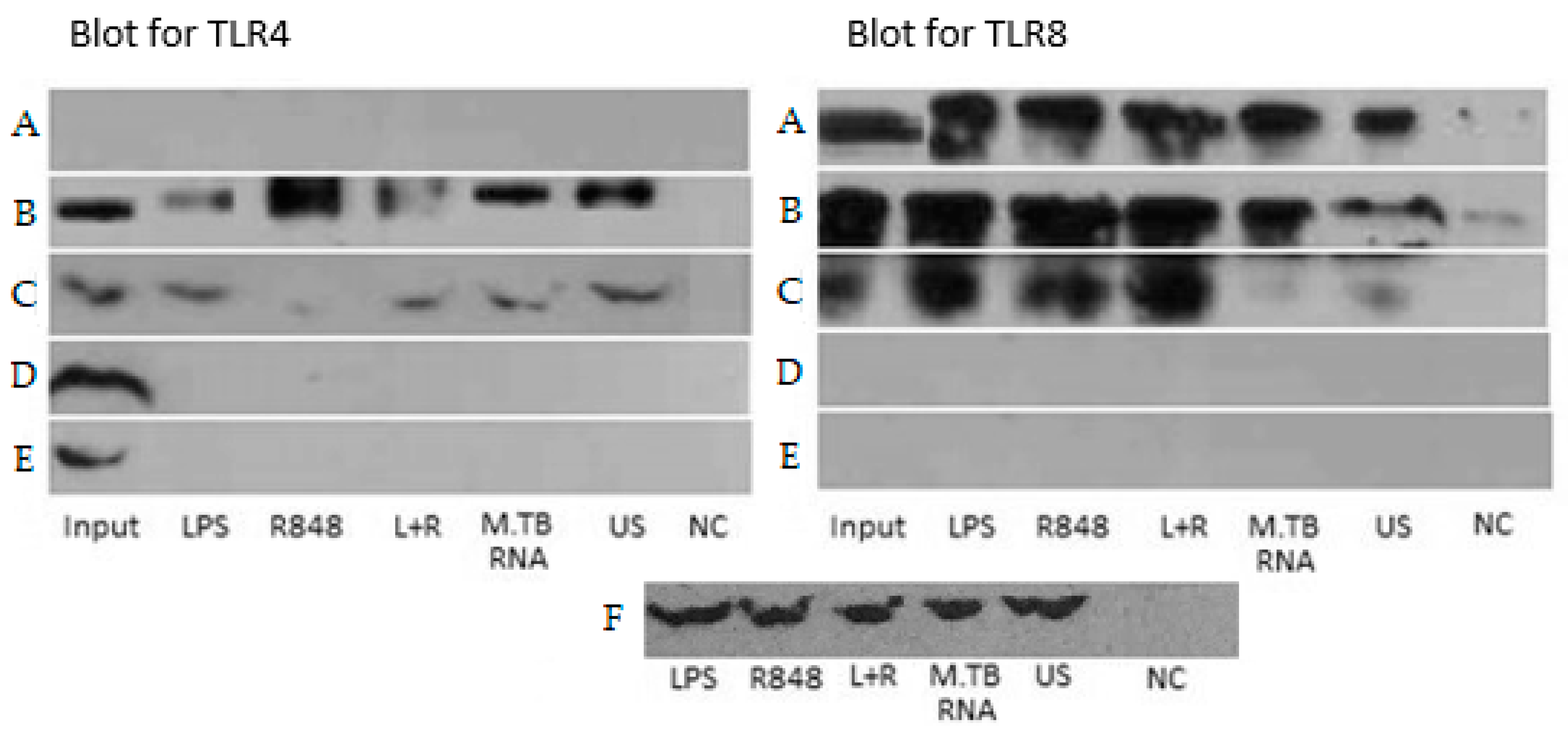
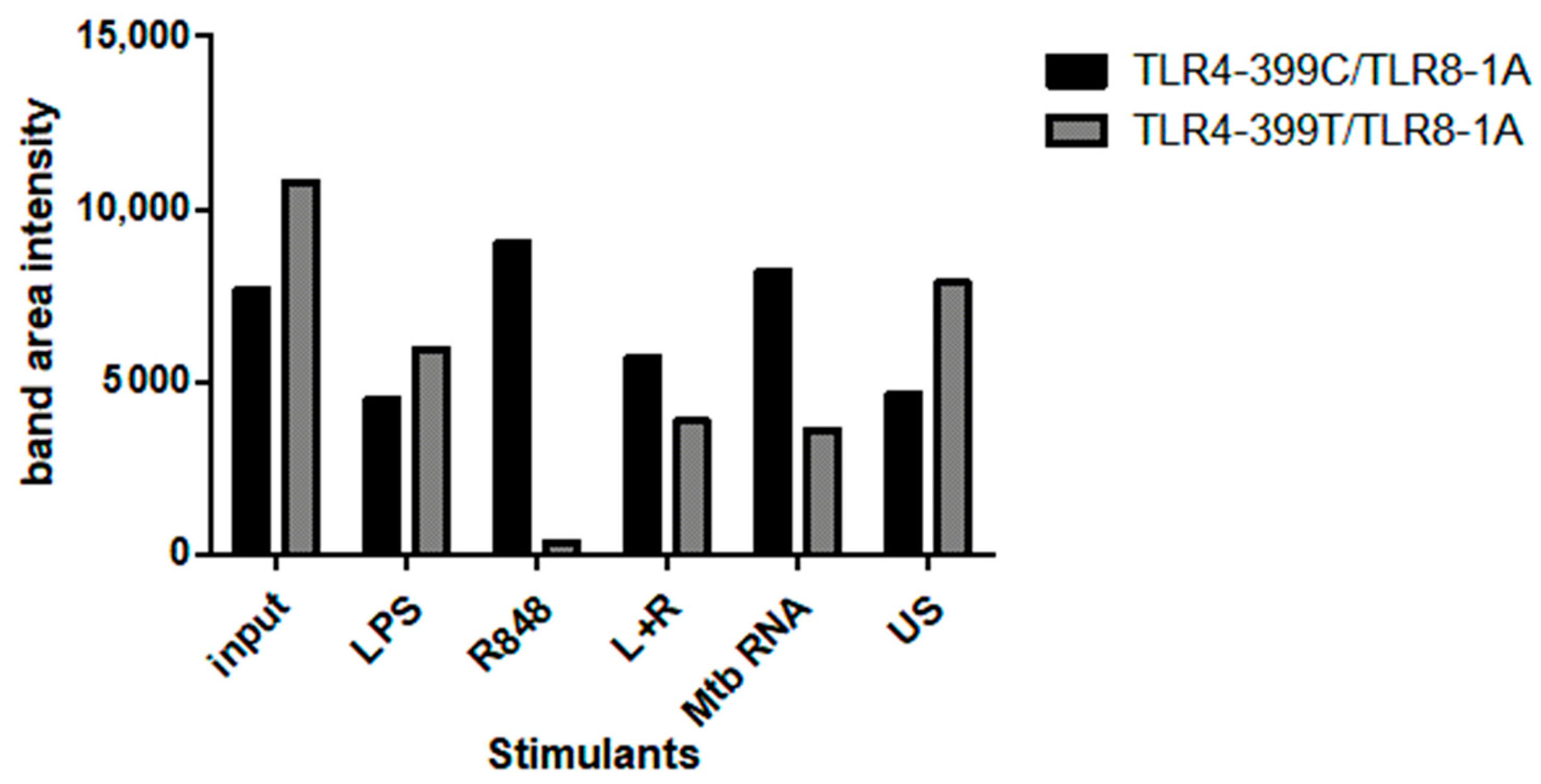
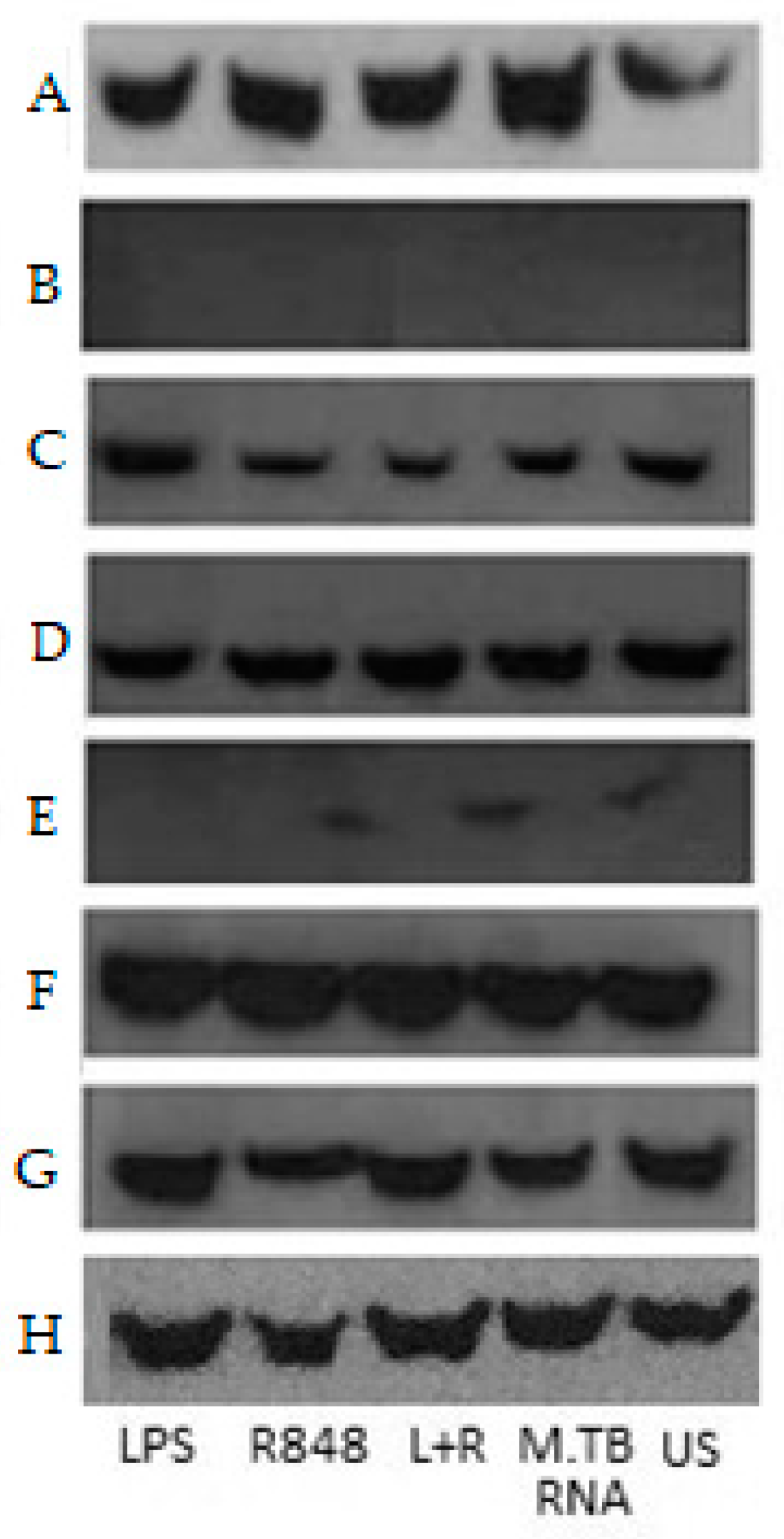
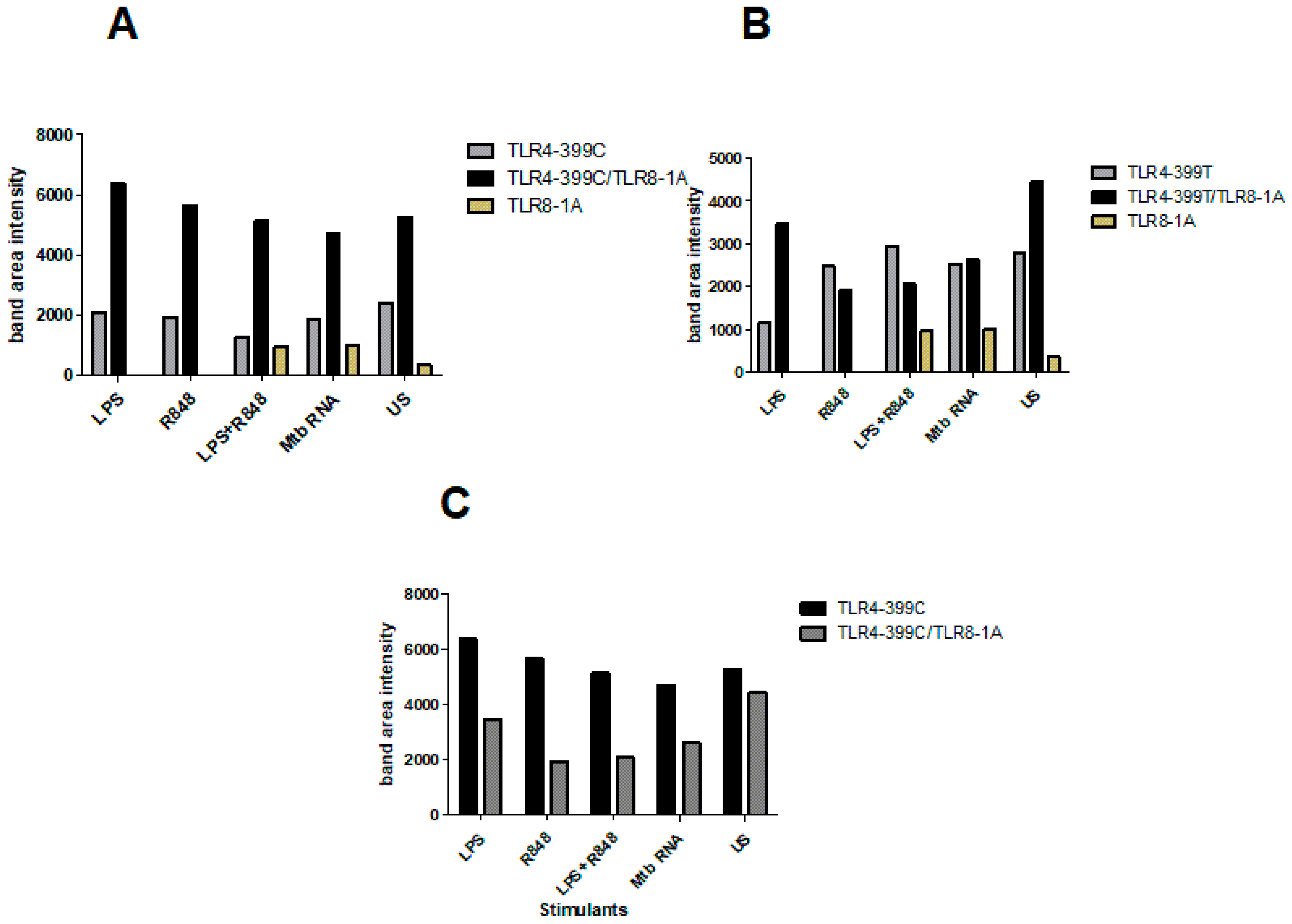
2.4. Co-Immunoprecipitation
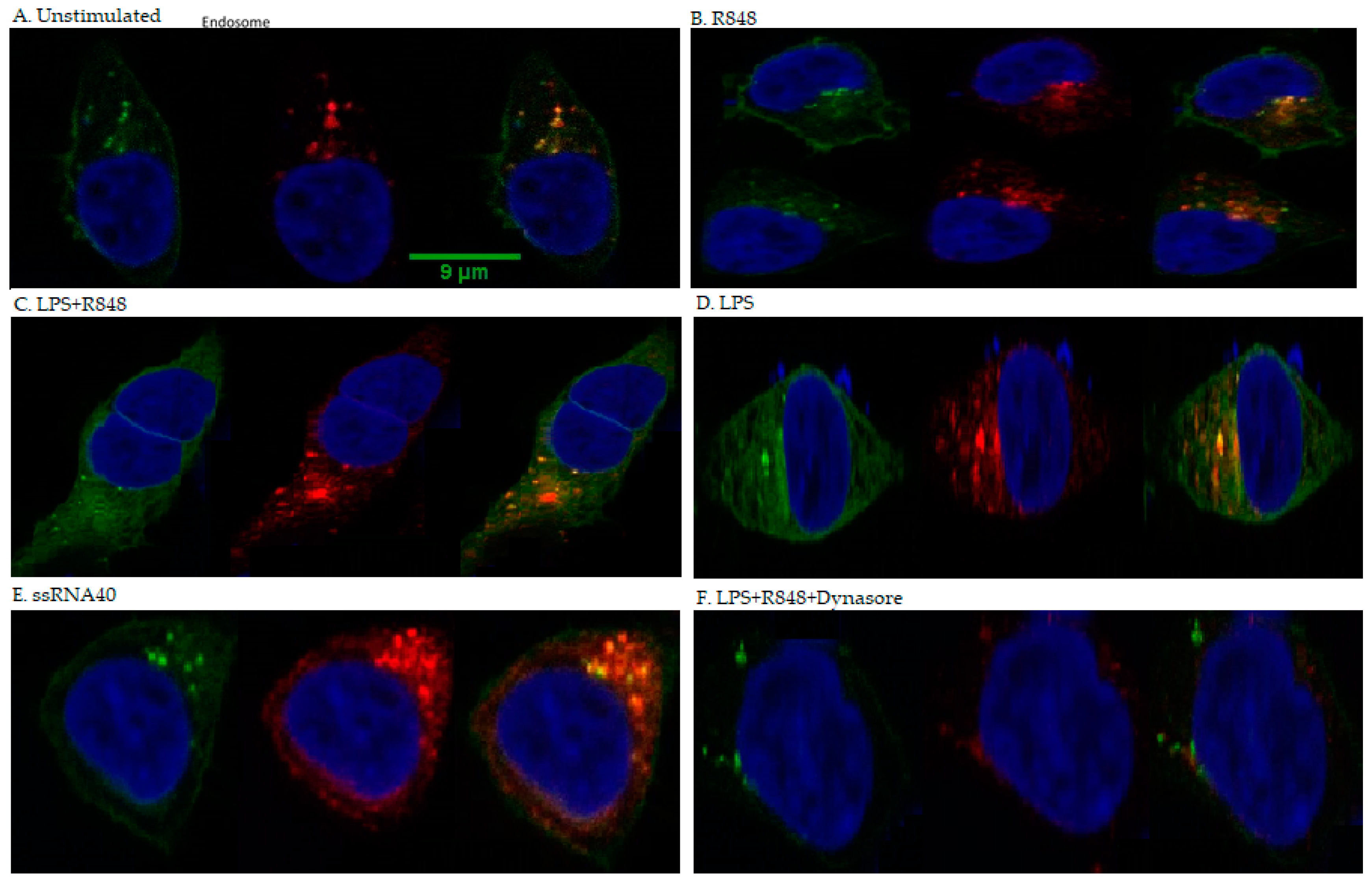
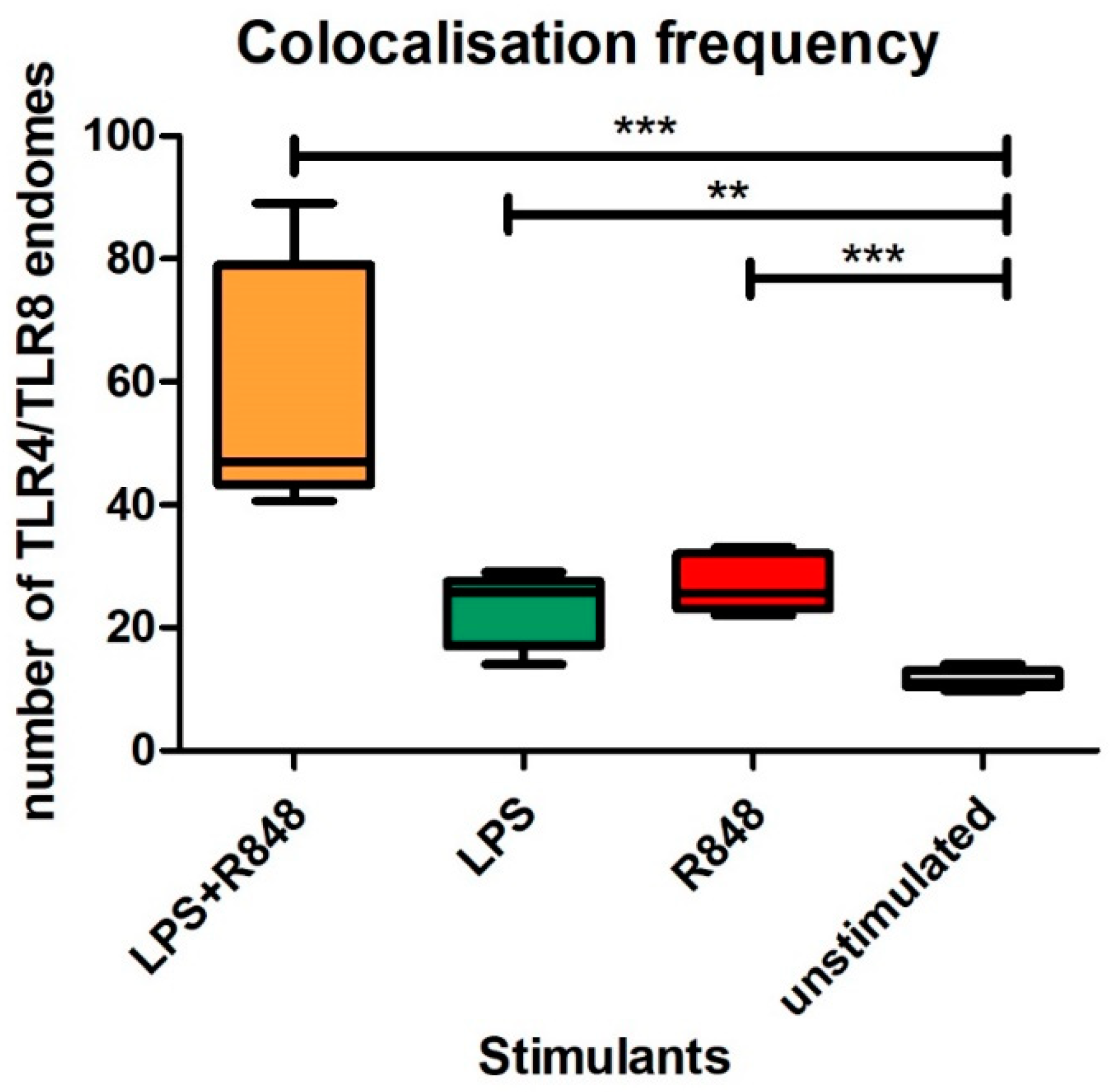
2.5. Co-Localisation
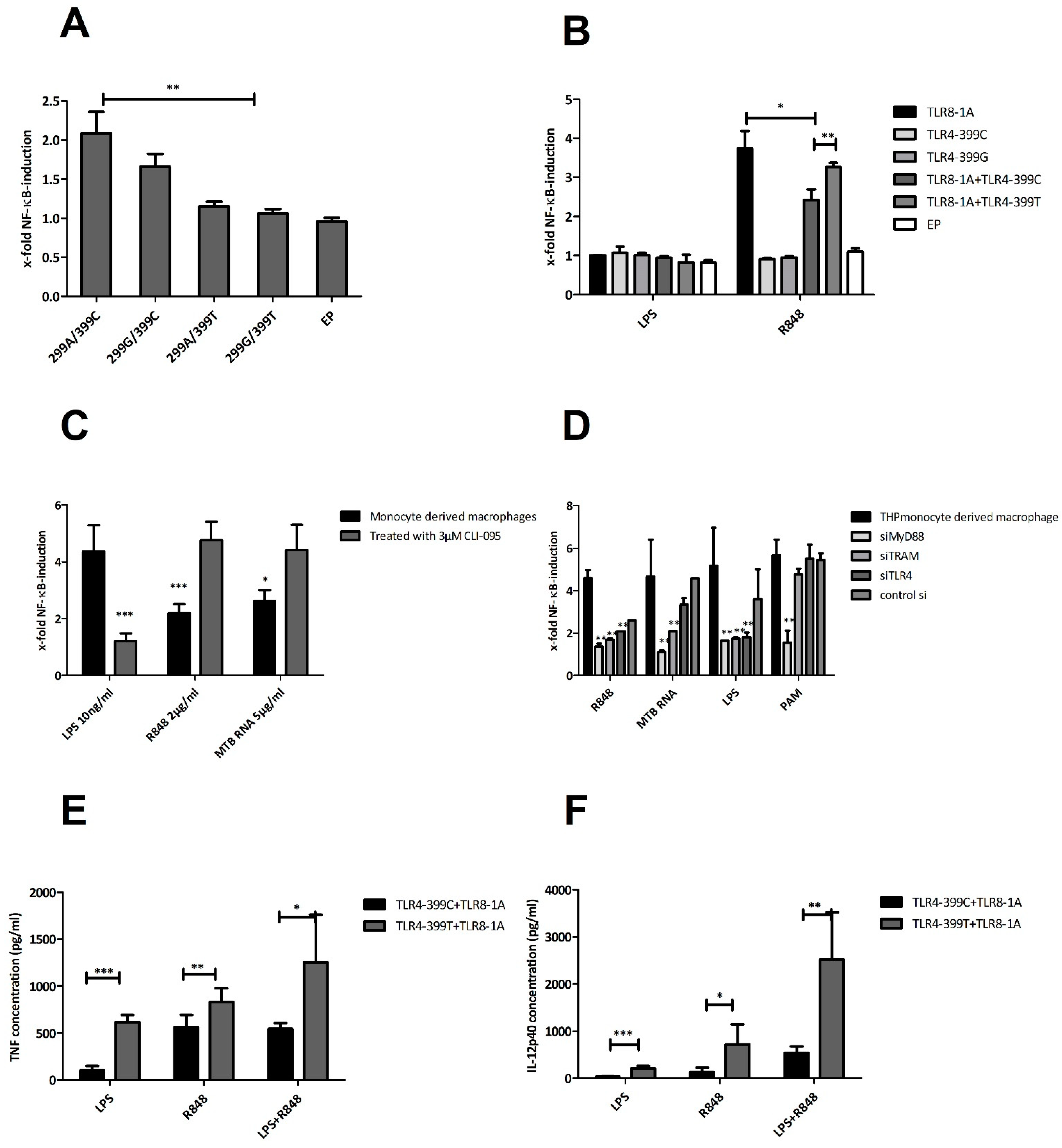
2.6. Functional Studies
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. SNP Analysis
4.3. Modelling and Molecular Docking
4.4. In Vitro Experiments
4.4.1. Stimulants and Reagents
4.4.2. Mutagenesis
4.4.3. Cell Line Experiments
4.4.4. Transient Transfection
4.4.5. Immunofluorescence Staining of TLR8HA/Confocal Microscopy
4.4.6. Co-Immunoprecipitation
4.4.7. Western Blot Procedure
4.5. Mass Spectrometry (MS)
4.5.1. Sample Preparation
4.5.2. Mass Spectrometric and Statistical Analysis
4.6. PBMC Experiments
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Adenine |
| APC | Antigen-presenting cell |
| ARDS | acute respiratory distress syndrome |
| BCG | Bacillus Calmette–Guérin |
| BMI | Body mass index |
| BSA | Bovine serum albumin |
| C | Cytosine |
| DC | Dendritic cell |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| ELISA | Enzyme-linked immunosorbent assay |
| EPTB | Extrapulmonary TB |
| FCS | Fetal calf serum |
| G | Guanine |
| HA | Human influenza hemagglutinin |
| HC | Healthy control |
| HEK | Human embryonic kidney |
| HHC | Healthy household contact |
| IFN | Interferon |
| IL | Interleukin |
| IP | Immunoprecipitation |
| IRF | Interferon response factor |
| LAM | Lipoarabinomannan |
| LM | Lipomannan |
| LPS | Lipopolysaccharide |
| LRT | Likelihood ratio test |
| MHC | Major histocompatibility complex |
| Mtb | Mycobacerium tuberculosis |
| MyD88 | Myeloid differentiation primary response 88 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PAMP | Pathogen-associated molecular pattern |
| PMB | Polymyxin B |
| PRR | Pattern recognition receptor |
| PTB | Pulmonary TB |
| SEAP | Secreted embryonic alkaline phosphatase |
| T | Thymine |
| TB | Tuberculosis |
| Th | T helper |
| TLR | Toll-like receptor |
| TNF | Tumour necrosis factor |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| WB | Western blotting |
References
- Medzhitov, R.; Janeway, C., Jr. Advances in immunology: Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Reis e Sousa, C. Activation of dendritic cells: Translating innate into adaptive immunity. Curr. Opin. Immunol. 2004, 16, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- Kirschning, C.J.; Schumann, R.R. TLR2: Cellular sensor for microbial and endogenous molecular patterns. Curr. Top. Microbiol. Immunol. 2002, 270, 121–144. [Google Scholar]
- Faridgohar, M.; Nikoueinejad, H. New findings of Toll-like receptors involved in Mycobacterium tuberculosis infection. Pathog. Glob. Health 2017, 111, 256–264. [Google Scholar] [CrossRef]
- Chang, J.-S.; Huggett, J.F.; Dheda, K.; Kim, L.U.; Zumla, A.; Rook, G.A.W. Myobacterium tuberculosis Induces Selective Up-Regulation of TLRs in the Mononuclear Leukocytes of Patients with Active Pulmonary Tuberculosis. J. Immunol. 2006, 176, 3010–3018. [Google Scholar] [CrossRef] [Green Version]
- Sepehri, Z.; Kiani, Z.; Kohan, F.; Ghavami, S. Toll-Like Receptor 4 as an Immune Receptor against Mycobacterium tuberculosis: A Systematic Review. Lab. Med. 2019, 50, 117–129. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll- like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Da Silva Correia, J.; Soldau, K.; Christen, U.; Tobias, P.S.; Ulevitch, R.J. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. Transfer from CD14 to TLR4 and MD-2. J. Biol. Chem. 2001, 276, 21129–21135. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Zanoni, I.; Cullen, T.W.; Goodman, A.L.; Kagan, J.C. Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity 2015, 43, 909–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; He, X.; Wang, H.; Wang, Z.; Kelly, G.T.; Wang, X.; Chen, Y.; Wang, T.; Qian, Z. TLR4-NOX2 axis regulates the phagocytosis and killing of Mycobacterium tuberculosis by macrophages. BMC Pulm. Med. 2017, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Ramírez, L.M.; Estrada-García, I.; López-Marín, L.M.; Segura-Salinas, E.; Méndez-Aragón, P.; Van Soolingen, D.; Torres-González, R.; Chacón-Salinas, R.; Estrada-Parra, S.; Maldonado-Bernal, C.; et al. Mycobacterium tuberculosis lipids regulate cytokines, TLR-2/4 and MHC class II expression in human macrophages. Tuberculosis 2008, 88, 212–220. [Google Scholar] [CrossRef]
- Jang, A.-R.; Kim, G.; Hong, J.J.; Kang, S.M.; Shin, S.J.; Park, J.-H. Mycobacterium tuberculosis ESAT6 Drives the Activation and Maturation of Bone Marrow-Derived Dendritic Cells via TLR4-Mediated Signaling. Immune Netw. 2019, 19, e13. [Google Scholar] [CrossRef]
- Kim, W.S.; Jung, I.D.; Kim, J.-S.; Kim, H.M.; Kwon, K.W.; Park, Y.-M.; Shin, S.J. Mycobacterium tuberculosis GrpE, A Heat-Shock Stress Responsive Chaperone, Promotes Th1-Biased T Cell Immune Response via TLR4-Mediated Activation of Dendritic Cells. Front. Cell. Infect. Microbiol. 2018, 8, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, N.; Varman, R.; Nair, S.; Das, G.; Ghosh, S.; Mukhopadhyay, S. Endocytosis of Mycobacterium tuberculosis Heat Shock Protein 60 Is Required to Induce Interleukin-10 Production in Macrophages. J. Biol. Chem. 2013, 288, 24956–24971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doz, E.; Rose, S.; Court, N.; Front, S.; Vasseur, V.; Charron, S.; Gilleron, M.; Puzo, G.; Fremaux, I.; Delneste, Y.; et al. Mycobacterial Phosphatidylinositol Mannosides Negatively Regulate Host Toll-like Receptor 4, MyD88-dependent Proinflammatory Cytokines, and TRIF-dependent Co-stimulatory Molecule Expression. J. Biol. Chem. 2009, 284, 23187–23196. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, J.; Ignatowicz, L.; Kallenius, G.; Svenson, S.B.; Pawlowski, A.; Hamasur, B. Divergent Effects of Mycobacterial Cell Wall Glycolipids on Maturation and Function of Human Monocyte-Derived Dendritic Cells. PLoS ONE 2012, 7, e42515. [Google Scholar] [CrossRef]
- Ugolini, M.; Gerhard, J.; Burkert, S.; Jensen, K.J.; Georg, P.; Ebner, F.; Volkers, S.M.; Thada, S.; Dietert, K.; Bauer, L.; et al. Recognition of microbial viability via TLR8 drives TFHcell differentiation and vaccine responses. Nat. Immunol. 2018, 19, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Thada, S.; Burkert, S.; Sivangala, R.; Hussain, A.; Sur, S.; Dittrich, N.; Conrad, M.L.; Slevogt, H.; Latha Gaddam, S.; Schumann, R.R. A SNP upstream of the cyclic GMP-AMP synthase (cGAS) gene protects from relapse and extra-pulmonary TB and relates to vaccination status in an Indian cohort. Genes Immun. 2020, 21, 13–26. [Google Scholar] [CrossRef]
- Burkert, S.; Schumann, R.R. RNA Sensing of Mycobacterium tuberculosis and Its Impact on TB Vaccination Strategies. Vaccines 2020, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Funami, K.; Tatematsu, M.; Seya, T.; Matsumoto, M. Endosomal Localization of TLR8 Confers Distinctive Proteolytic Processing on Human Myeloid Cells. J. Immunol. 2014, 193, 5118–5128. [Google Scholar] [CrossRef] [Green Version]
- Keegan, C.; Krutzik, S.; Schenk, M.; Scumpia, P.O.; Lu, J.; Pang, Y.L.J.; Russell, B.S.; Lim, K.S.; Shell, S.; Prestwich, E.; et al. Mycobacterium tuberculosis Transfer RNA Induces IL-12p70 via Synergistic Activation of Pattern Recognition Receptors within a Cell Network. J. Immunol. 2018, 200, 3244–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergne, I.; Fratti, R.A.; Hill, P.J.; Chua, J.; Belisle, J.; Deretic, V. Mycobacterium tuberculosis Phagosome Maturation Arrest: Mycobacterial Phosphatidylinositol Analog Phosphatidylinositol Mannoside Stimulates Early Endosomal Fusion. Mol. Biol. Cell 2004, 15, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.R.; Patel, J.; Robertson, B.D.; Rae, A.; Young, D.B. Mycobacterial Mutants with Defective Control of Phagosomal Acidification. PLoS Pathog. 2005, 1, e33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.; Nasiri, M.R.; Sanaei, R.; Anoosheh, S.; Farnia, P.; Sepanjnia, A.; Tajik, N. The NRAMP1, VDR, TNF-α, ICAM1, TLR2 and TLR4 gene polymorphisms in Iranian patients with pulmonary tuberculosis: A case–control study. Infect. Genet. Evol. 2016, 39, 92–98. [Google Scholar] [CrossRef]
- Davila, S.; Hibberd, M.L.; Hari Dass, R.; Wong, H.E.E.; Sahiratmadja, E.; Bonnard, C.; Alisjahbana, B.; Szeszko, J.S.; Balabanova, Y.; Drobniewski, F.; et al. Genetic Association and Expression Studies Indicate a Role of Toll-Like Receptor 8 in Pulmonary Tuberculosis. PLoS Genet. 2008, 4, e1000218. [Google Scholar] [CrossRef] [Green Version]
- Berrocal-Almanza, L.C.; Goyal, S.; Hussain, A.; Klassert, T.E.; Driesch, D.; Grozdanovic, Z.; Sumanlatha, G.; Ahmed, N.; Valluri, V.; Conrad, M.L.; et al. S100A12 is up-regulated in pulmonary tuberculosis and predicts the extent of alveolar infiltration on chest radiography: An observational study. Sci. Rep. 2016, 6, 31798. [Google Scholar] [CrossRef] [Green Version]
- Dittrich, N.; Berrocal-Almanza, L.C.; Thada, S.; Goyal, S.; Slevogt, H.; Sumanlatha, G.; Hussain, A.; Sur, S.; Burkert, S.; Oh, D.-Y.; et al. Toll-like receptor 1 variations influence susceptibility and immune response to Mycobacterium tuberculosis. Tuberculosis 2015, 95, 328–335. [Google Scholar] [CrossRef]
- Pelka, K.; Bertheloot, D.; Reimer, E.; Phulphagar, K.; Schmidt, S.V.; Christ, A.; Stahl, R.; Watson, N.; Miyake, K.; Hacohen, N.; et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity 2018, 48, 911–922.e7. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Matsunaga, N.; Hazeki, K.; Nakamura, K.; Takashima, K.; Seya, T.; Hazeki, O.; Kitazaki, T.; Iizawa, Y. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 2006. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Zhao, G.W.; Gao, C.H.; Chi, X.W.; Zeng, T.; Hu, Y.W.; Zheng, L.; Wang, Q. Mannose-capped Lipoarabinomannan from Mycobacterium tuberculosis induces IL-37 production via upregulating ERK1/2 and p38 in human type II alveolar epithelial cells. Int. J. Clin. Exp. Med. 2015, 8, 7279–7287. [Google Scholar] [PubMed]
- Mpofu, C.M.; Campbell, B.J.; Subramanian, S.; Marshall–Clarke, S.; Hart, C.A.; Cross, A.; Roberts, C.L.; McGoldrick, A.; Edwards, S.W.; Rhodes, J.M. Microbial Mannan Inhibits Bacterial Killing by Macrophages: A Possible Pathogenic Mechanism for Crohn’s Disease. Gastroenterology 2007, 133, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Mockenhaupt, F.P.; Hamann, L.; von Gaertner, C.; Bedu-Addo, G.; von Kleinsorgen, C.; Schumann, R.R.; Bienzle, U. Common Polymorphisms of Toll-Like Receptors 4 and 9 Are Associated with the Clinical Manifestation of Malaria during Pregnancy. J. Infect. Dis. 2006, 194, 184–188. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Ioana, M.; Alonso, S.; Izagirre, N.; Hervella, M.; Joosten, L.A.B.; van der Meer, J.W.M.; de la Rúa, C.; Netea, M.G. The Evolutionary History of TLR4 Polymorphisms in Europe. J. Innate Immun. 2012, 4, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Schurz, H.; Daya, M.; Möller, M.; Hoal, E.G.; Salie, M. TLR1, 2, 4, 6 and 9 Variants Associated with Tuberculosis Susceptibility: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0139711. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, K.; Kong, X.; Tao, Z.; Wang, Y.; Liu, Y. Association of polymorphisms in toll-like receptors 4 and 9 with risk of pulmonary Tuberculosis: A meta-analysis. Med. Sci. Monit. 2015, 21, 1097–1106. [Google Scholar]
- Richard, K.; Piepenbrink, K.H.; Shirey, K.A.; Gopalakrishnan, A.; Nallar, S.; Prantner, D.J.; Perkins, D.J.; Lai, W.; Vlk, A.; Toshchakov, V.Y.; et al. A mouse model of human TLR4 D299G/T399I SNPs reveals mechanisms of altered LPS and pathogen responses. J. Exp. Med. 2021, 218, e20200675. [Google Scholar] [CrossRef]
- Bösl, K.; Giambelluca, M.; Haug, M.; Bugge, M.; Espevik, T.; Kandasamy, R.K.; Bergstrøm, B. Coactivation of TLR2 and TLR8 in Primary Human Monocytes Triggers a Distinct Inflammatory Signaling Response. Front. Physiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, D.J.; Richard, K.; Hansen, A.M.; Lai, W.; Nallar, S.; Koller, B.; Vogel, S.N. Autocrine–paracrine prostaglandin E 2 signaling restricts TLR4 internalization and TRIF signaling. Nat. Immunol. 2018, 19, 1309–1318. [Google Scholar] [CrossRef]
- World Health Organization. WHO | WHO End TB Strategy; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Moreno-Eutimio, M.A.; López-Macías, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Brandão, S.C.S.; Ramos, J.d.O.X.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2020. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Taube, S.; Hamouda, O.; Kücherer, C.; Poggensee, G.; Jessen, H.; Eckert, J.K.; Neumann, K.; Storek, A.; Pouliot, M.; et al. A functional toll-like receptor 8 variant is associated with HIV disease restriction. J. Infect. Dis. 2008, 198, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for cellprofiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TLR SNPs (Nucleotide Change) | Alleles | N | Allele Frequency [N(%)] Controls Primary TB | OR [95% CI] * | p-Value | |
|---|---|---|---|---|---|---|
| TLR4-Asp299Gly (A > G) | G | 533 | 72 (27.48) | 100 (33.22) | 0.72 [0.49–1.07] | 0.101 |
| TLR4-Thr399Ile (C > T) | T | 552 | 68 (23.37) | 105 (31.44) | 1.57 [1.04–2.36] | 0.027 |
| TLR8-Met1Val (A > G) | A | 556 | 139 (47.60) | 199 (58.70) | 1.68 [1.08–2.63] | 0.022 |
| TLR8-1, when TLR4-399CT/T | A | 395 | 34 (50.00) | 59 (56.19) | 1.97 [1.15–3.37] | 0.013 |
| TLR8-1, when TLR4-399CC | A | 155 | 105 (47.09) | 137 (59.83) | 1.19 [0.52–2.72] | 0.681 |
| TLR SNPs | Alleles | N | Allele Frequency [N(%)] Primary TB Relapses | OR [95% CI] * | p-Value | |
|---|---|---|---|---|---|---|
| TLR4-Asp299Gly | G | 383 | 100 (33.22) | 36 (39.13) | 0.80 [0.49–1.32] | 0.381 |
| TLR4-Thr399Ile | T | 376 | 105 (31.44) | 33 (38.82) | 1.36 [0.81–2.28] | 0.242 |
| TLR8-Met1Val | A | 355 | 140 (58.70) | 68 (72.34) | 1.99 [1.03–3.82] | 0.035 |
| TLR8-1, when TLR4-399CT/T | A | 111 | 59 (56.19) | 25 (75.76) | 2.90 [0.87–9.59] | 0.069 |
| TLR8-1, when TLR4-399CC | A | 231 | 137 (59.82) | 37 (71.15) | 1.62 [0.69–3.81] | 0.265 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thada, S.; Horvath, G.L.; Müller, M.M.; Dittrich, N.; Conrad, M.L.; Sur, S.; Hussain, A.; Pelka, K.; Gaddam, S.L.; Latz, E.; et al. Interaction of TLR4 and TLR8 in the Innate Immune Response against Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2021, 22, 1560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041560
Thada S, Horvath GL, Müller MM, Dittrich N, Conrad ML, Sur S, Hussain A, Pelka K, Gaddam SL, Latz E, et al. Interaction of TLR4 and TLR8 in the Innate Immune Response against Mycobacterium Tuberculosis. International Journal of Molecular Sciences. 2021; 22(4):1560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041560
Chicago/Turabian StyleThada, Shruthi, Gabor L. Horvath, Mario M. Müller, Nickel Dittrich, Melanie L. Conrad, Saubashya Sur, Abid Hussain, Karin Pelka, Suman Latha Gaddam, Eicke Latz, and et al. 2021. "Interaction of TLR4 and TLR8 in the Innate Immune Response against Mycobacterium Tuberculosis" International Journal of Molecular Sciences 22, no. 4: 1560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041560