
Chronic Treatment of Ascorbic Acid Leads to Age-Dependent Neuroprotection against Oxidative Injury in Hippocampal Slice Cultures


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
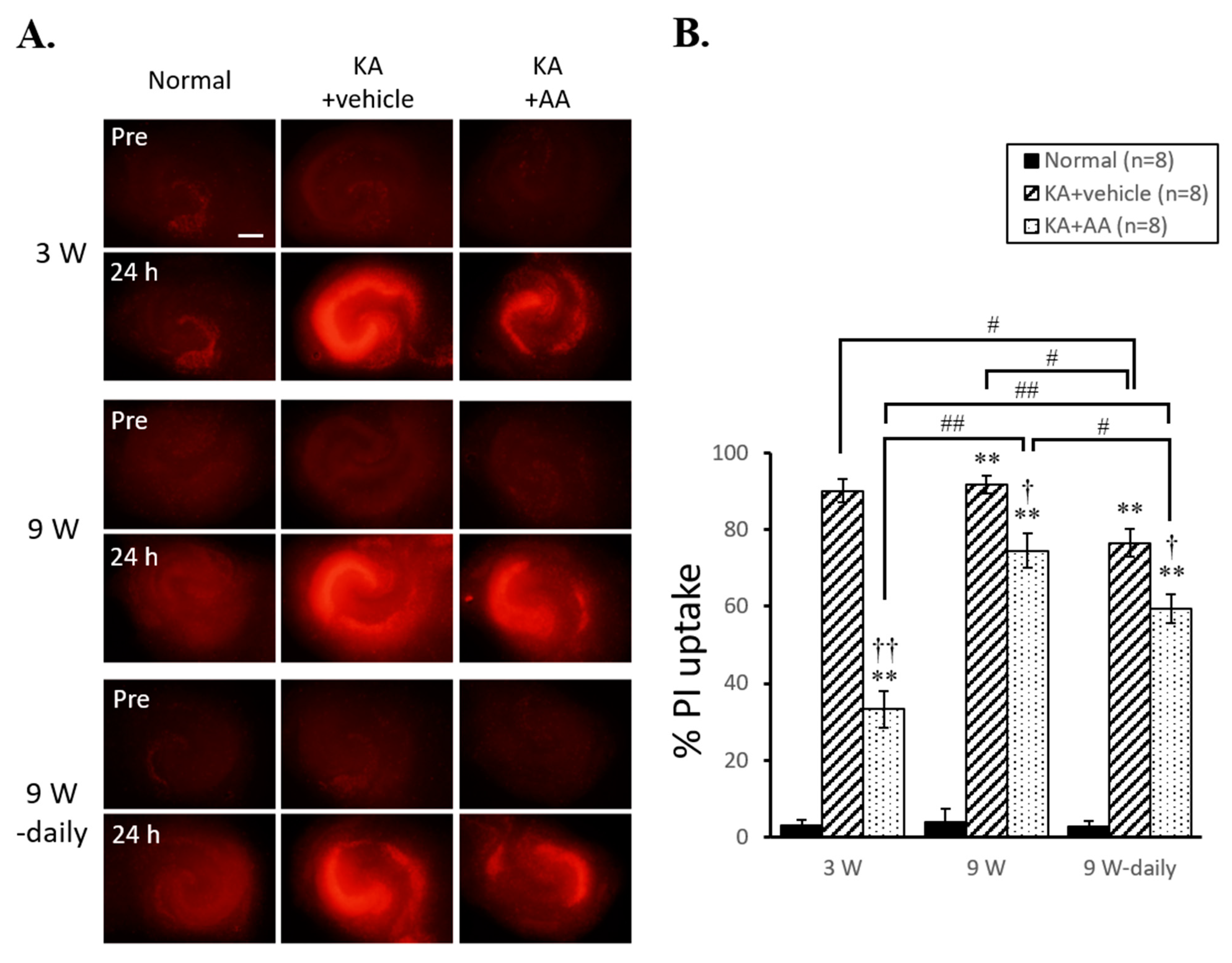
2.1. Different Neuroprotection Effects of AA
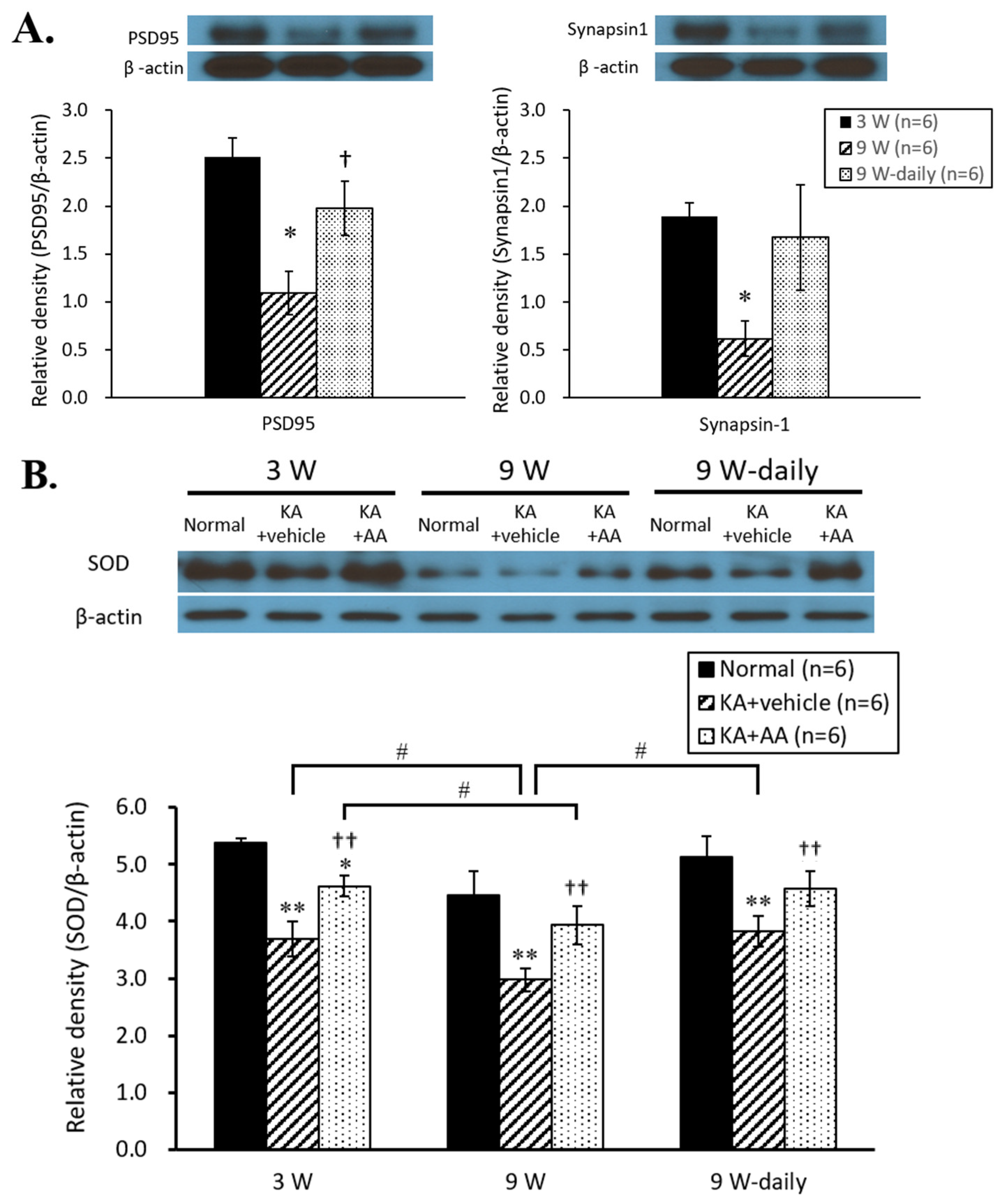
2.2. Activation of Antioxidant Signals by AA Treatment in Aging
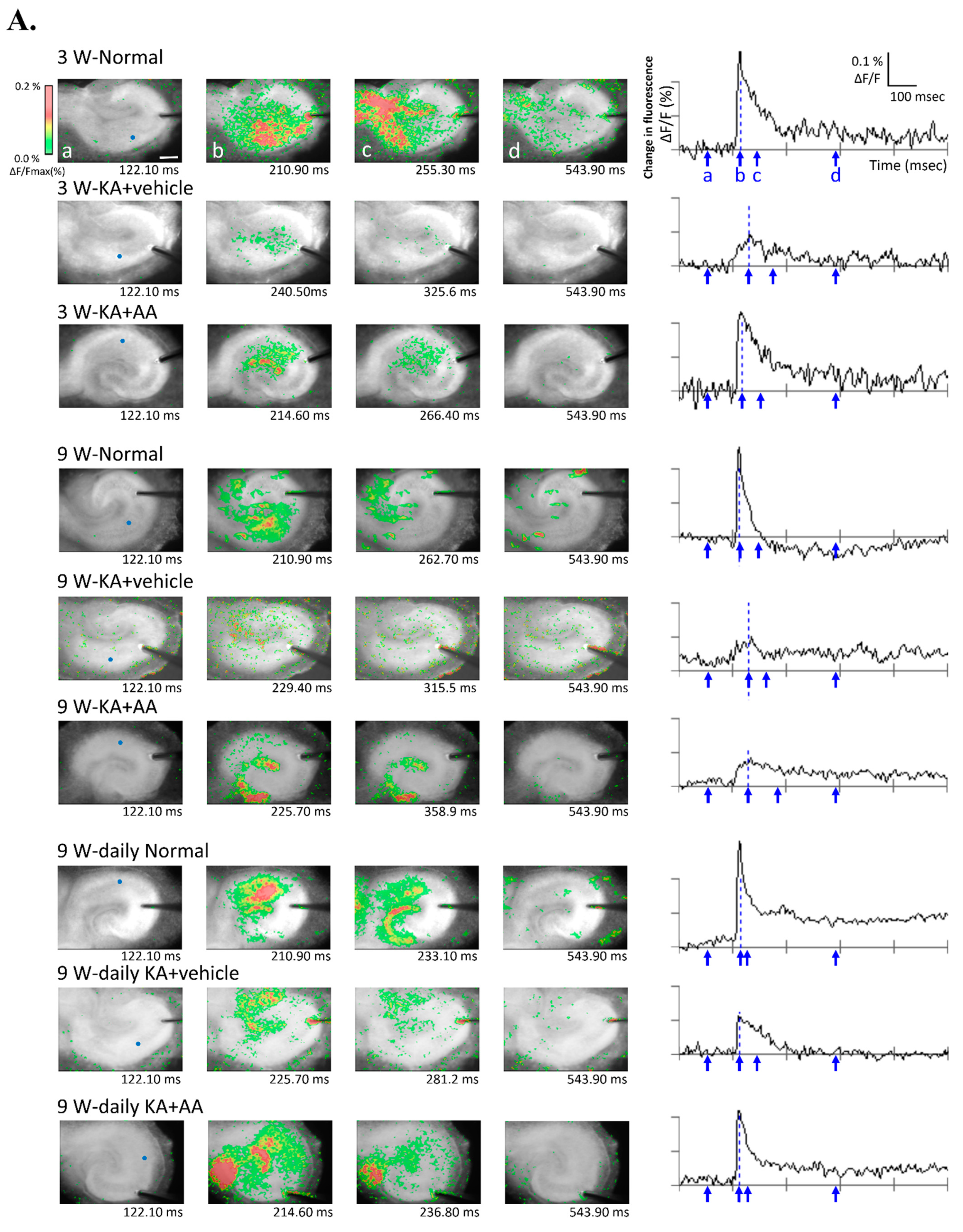
2.3. Effect of AA on Neuronal Activity
3. Discussion
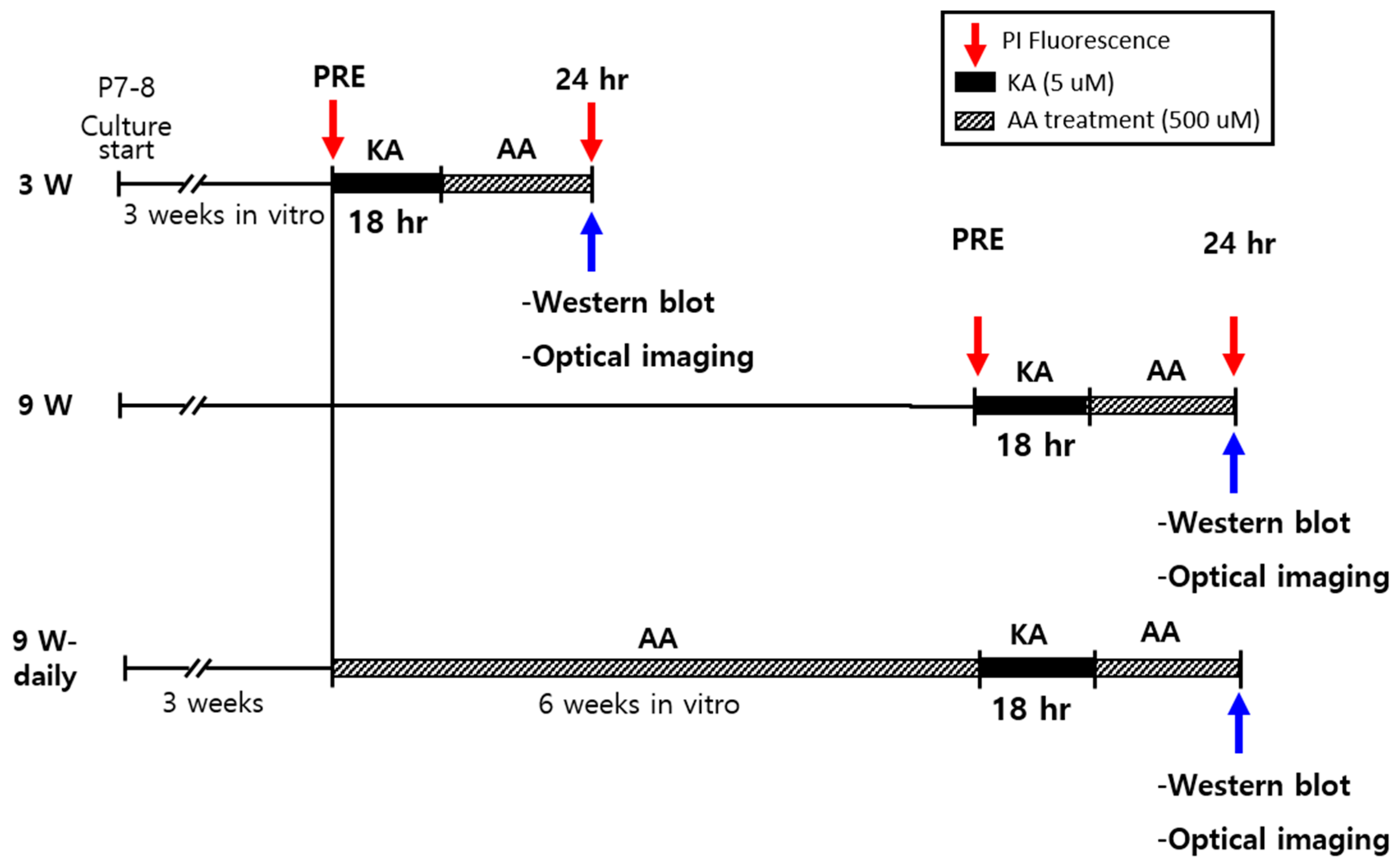
4. Materials and Methods
4.1. Organotypic Hippocampal Slices Cultures (OHSCs)
4.2. Propidium Iodide (PI) Staining
4.3. Western Blot Analysis
4.4. Optical Recording
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.M.; Austad, S.N.; Johnson, T.E. Genetic analysis of ageing: Role of oxidative damage and environmental stresses. Nat. Genet. 1996, 13, 25–34. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Heikkila, R.E. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J. Biol. Chem. 1974, 249, 2447–2452. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction—A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Xiong, Y.; Zhou, S.; Sun, Y.; Zhao, Y.; Ren, X.; Zhang, Y.; Zhang, N. Vitamin C and E Supplements Enhance the Antioxidant Capacity of Erythrocytes Obtained from Aged Rats. Rejuvenation Res. 2016, 20, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.-Q.; Shen, T.-T.; Wang, F.; Wu, P.-F.; Chen, J.-G. Preventive and Therapeutic Potential of Vitamin C in Mental Disorders. Curr. Med. Sci 2018, 38, 1–10. [Google Scholar] [CrossRef]
- Kocot, J.; Luchowska-Kocot, D.; Kielczykowska, M.; Musik, I.; Kurzepa, J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients 2017, 9, 659. [Google Scholar] [CrossRef] [Green Version]
- May, J.M.; Qu, Z.-C. Ascorbic acid prevents oxidant-induced increases in endothelial permeability. Biofactors 2011, 37, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional distribution of ascorbate in human brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1997, 82, 1213–1223. [Google Scholar] [CrossRef]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Preventive and therapeutic potential of ascorbic acid in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 921–929. [Google Scholar] [CrossRef]
- Siqueira, I.R.; Elsner, V.R.; Leite, M.C.; Vanzella, C.; Moysés, F.d.S.; Spindler, C.; Godinho, G.; Battú, C.; Wofchuk, S.; Souza, D.O.; et al. Ascorbate uptake is decreased in the hippocampus of ageing rats. Neurochem. Int. 2011, 58, 527–532. [Google Scholar] [CrossRef]
- Michels, A.J.; Joisher, N.; Hagen, T.M. Age-related decline of sodium-dependent ascorbic acid transport in isolated rat hepatocytes. Arch. Biochem. Biophys. 2003, 410, 112–120. [Google Scholar] [CrossRef]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharm. Res. 2019, 146, 104321. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [Green Version]
- Buchs, P.A.; Stoppini, L.; Muller, D. Structural modifications associated with synaptic development in area CA1 of rat hippocampal organotypic cultures. Brain Res. Dev. Brain Res. 1993, 71, 81–91. [Google Scholar] [CrossRef]
- De Simoni, A.; Griesinger, C.B.; Edwards, F.A. Development of rat CA1 neurones in acute versus organotypic slices: Role of experience in synaptic morphology and activity. J. Physiol. 2003, 550, 135–147. [Google Scholar] [CrossRef]
- Mielke, J.G.; Comas, T.; Woulfe, J.; Monette, R.; Chakravarthy, B.; Mealing, G.A.R. Cytoskeletal, synaptic, and nuclear protein changes associated with rat interface organotypic hippocampal slice culture development. Brain Res. Dev. Brain Res. 2005, 160, 275–286. [Google Scholar] [CrossRef]
- López-Alarcón, C.; Denicola, A. Evaluating the antioxidant capacity of natural products: A review on chemical and cellular-based assays. Anal. Chim. Acta 2013, 763, 1–10. [Google Scholar] [CrossRef]
- Friedman, L.K.; Goldstein, B.; Rafiuddin, A.; Roblejo, P.; Friedman, S. Lack of resveratrol neuroprotection in developing rats treated with kainic acid. Neuroscience 2013, 230, 39–49. [Google Scholar] [CrossRef]
- Kuruba, R.; Hattiangady, B.; Parihar, V.K.; Shuai, B.; Shetty, A.K. Differential Susceptibility of Interneurons Expressing Neuropeptide Y or Parvalbumin in the Aged Hippocampus to Acute Seizure Activity. PLoS ONE 2011, 6, e24493. [Google Scholar] [CrossRef] [Green Version]
- Birringer, M. Hormetics: Dietary triggers of an adaptive stress response. Pharm. Res. 2011, 28, 2680–2694. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Iavicoli, I.; Di Paola, R.; Koverech, A.; Cuzzocrea, S.; Rizzarelli, E.; Calabrese, E.J. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta 2012, 1822, 753–783. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-Induced Oxidative Stress in Temporal Lobe Epilepsy. Biomed. Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Acuna, A.I.; Beltran, F.A.; Torres-Diaz, L.; Castro, M.A. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef] [PubMed]
- Weisenburger, S.; Vaziri, A. A Guide to Emerging Technologies for Large-Scale and Whole-Brain Optical Imaging of Neuronal Activity. Annu. Rev. Neurosci. 2018, 41, 431–452. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Bruce, A.J.; Malfroy, B.; Baudry, M. beta-Amyloid toxicity in organotypic hippocampal cultures: Protection by EUK-8, a synthetic catalytic free radical scavenger. Proc. Natl. Acad. Sci. USA 1996, 93, 2312–2316. [Google Scholar] [CrossRef] [Green Version]
- Borsello, T.; Croquelois, K.; Hornung, J.P.; Clarke, P.G.H. N-methyl-D-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 2003, 18, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Park, J.H.; Won, R.; Lee, H.; Nam, T.S.; Lee, B.H. Inhibition of hexokinase leads to neuroprotection against excitotoxicity in organotypic hippocampal slice culture. J. Neurosci. Res. 2011, 89, 96–107. [Google Scholar] [CrossRef]
- Tominaga, T.; Tominaga, Y.; Yamada, H.; Matsumoto, G.; Ichikawa, M. Quantification of optical signals with electrophysiological signals in neural activities of Di-4-ANEPPS stained rat hippocampal slices. J. Neurosci. Methods 2000, 102, 11–23. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.H.; Kim, U.J.; Cha, M.; Lee, B.H. Chronic Treatment of Ascorbic Acid Leads to Age-Dependent Neuroprotection against Oxidative Injury in Hippocampal Slice Cultures. Int. J. Mol. Sci. 2021, 22, 1608. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041608
Lee KH, Kim UJ, Cha M, Lee BH. Chronic Treatment of Ascorbic Acid Leads to Age-Dependent Neuroprotection against Oxidative Injury in Hippocampal Slice Cultures. International Journal of Molecular Sciences. 2021; 22(4):1608. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041608
Chicago/Turabian StyleLee, Kyung Hee, Un Jeng Kim, Myeounghoon Cha, and Bae Hwan Lee. 2021. "Chronic Treatment of Ascorbic Acid Leads to Age-Dependent Neuroprotection against Oxidative Injury in Hippocampal Slice Cultures" International Journal of Molecular Sciences 22, no. 4: 1608. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041608