
Discovery of a Small Molecule Inhibitor of Human Adenovirus Capable of Preventing Escape from the Endosome

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
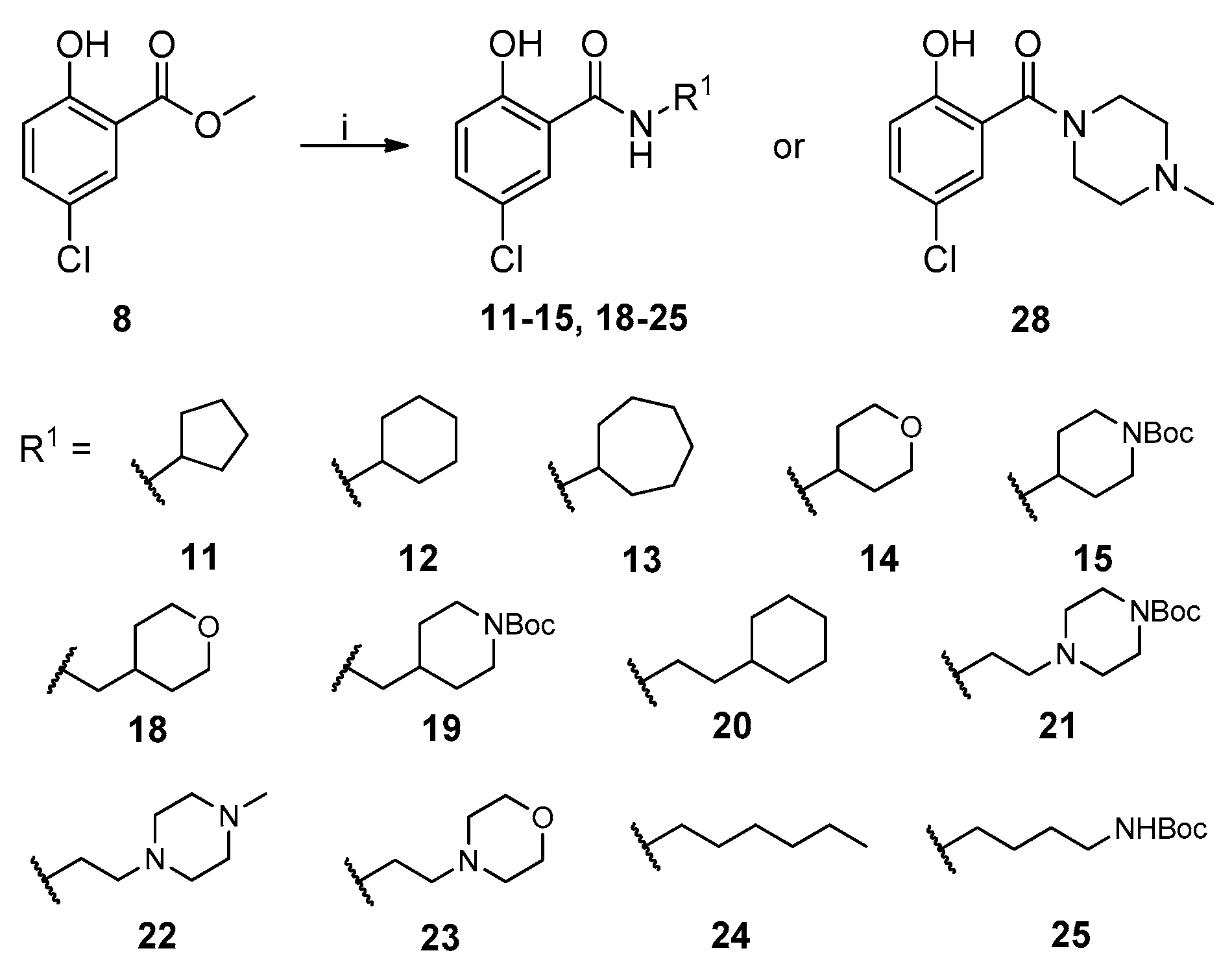
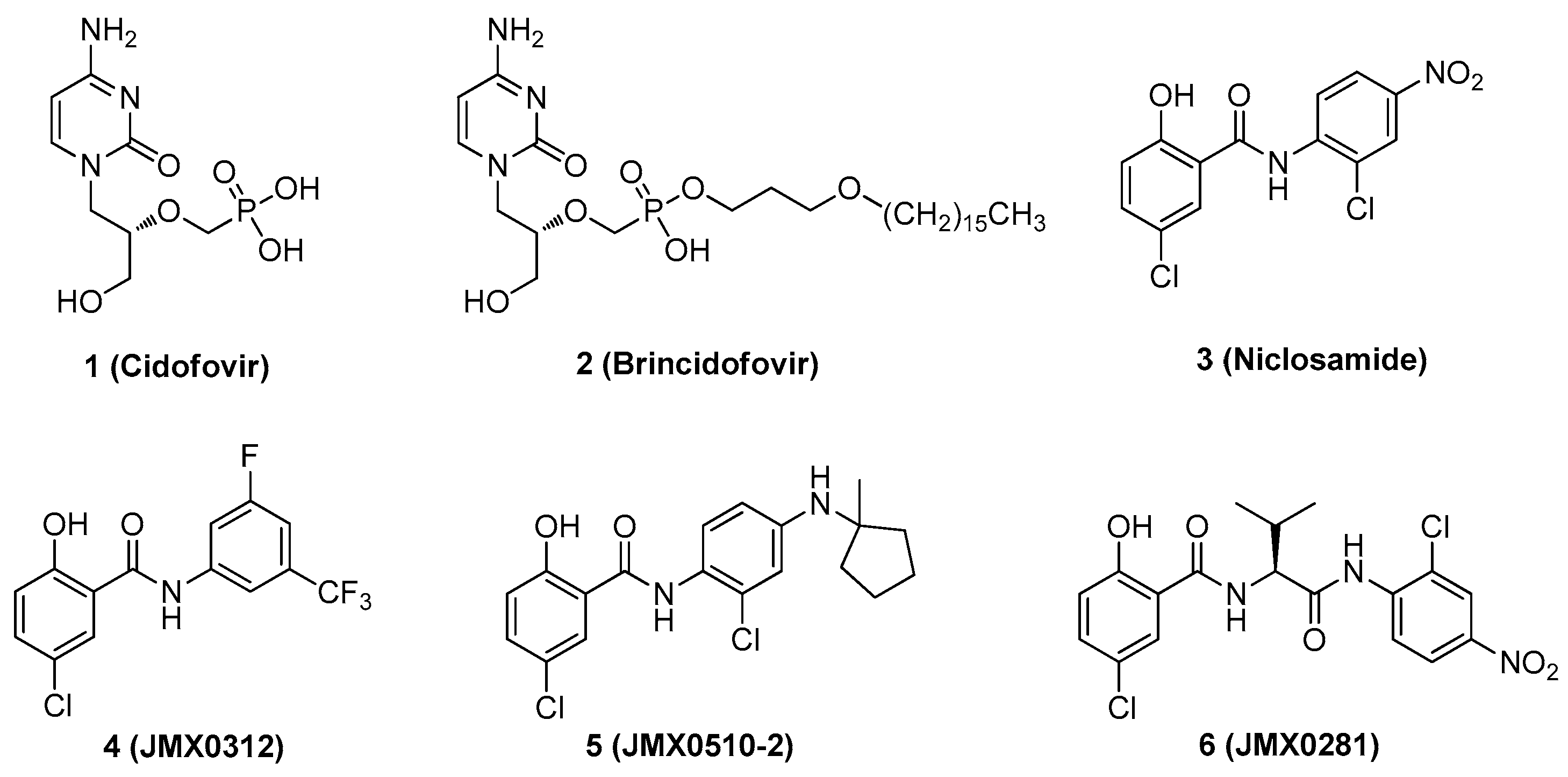
2.1. Chemistry
2.2. In Vitro Evaluation of Human Adenovirus Inhibition
2.3. Antiviral Mechanism of Action Studies
2.3.1. Impact of Compound 16 on HAdV Replication
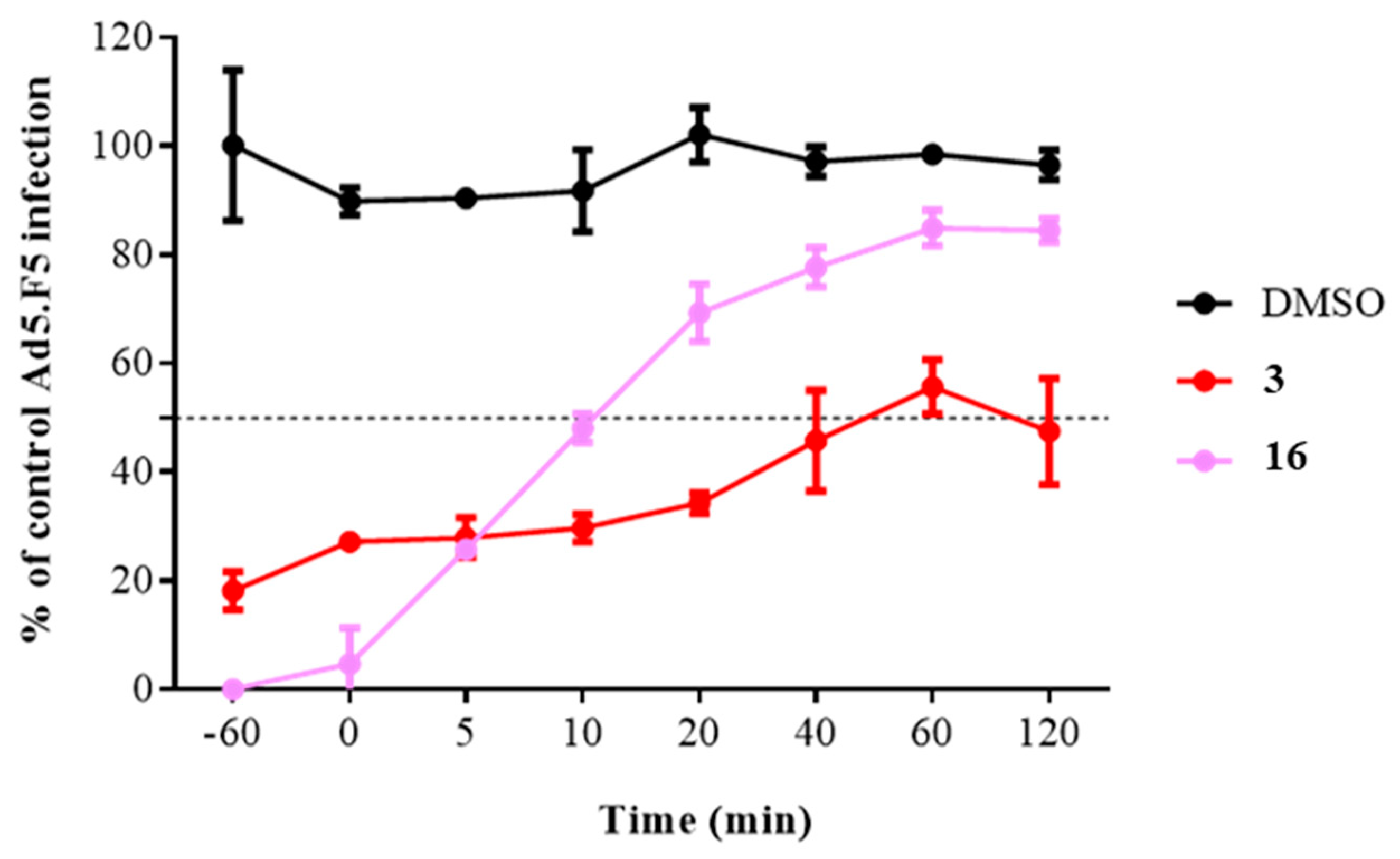
2.3.2. Time of Addition Assay
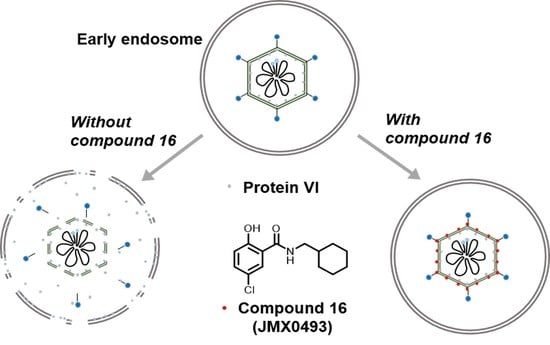
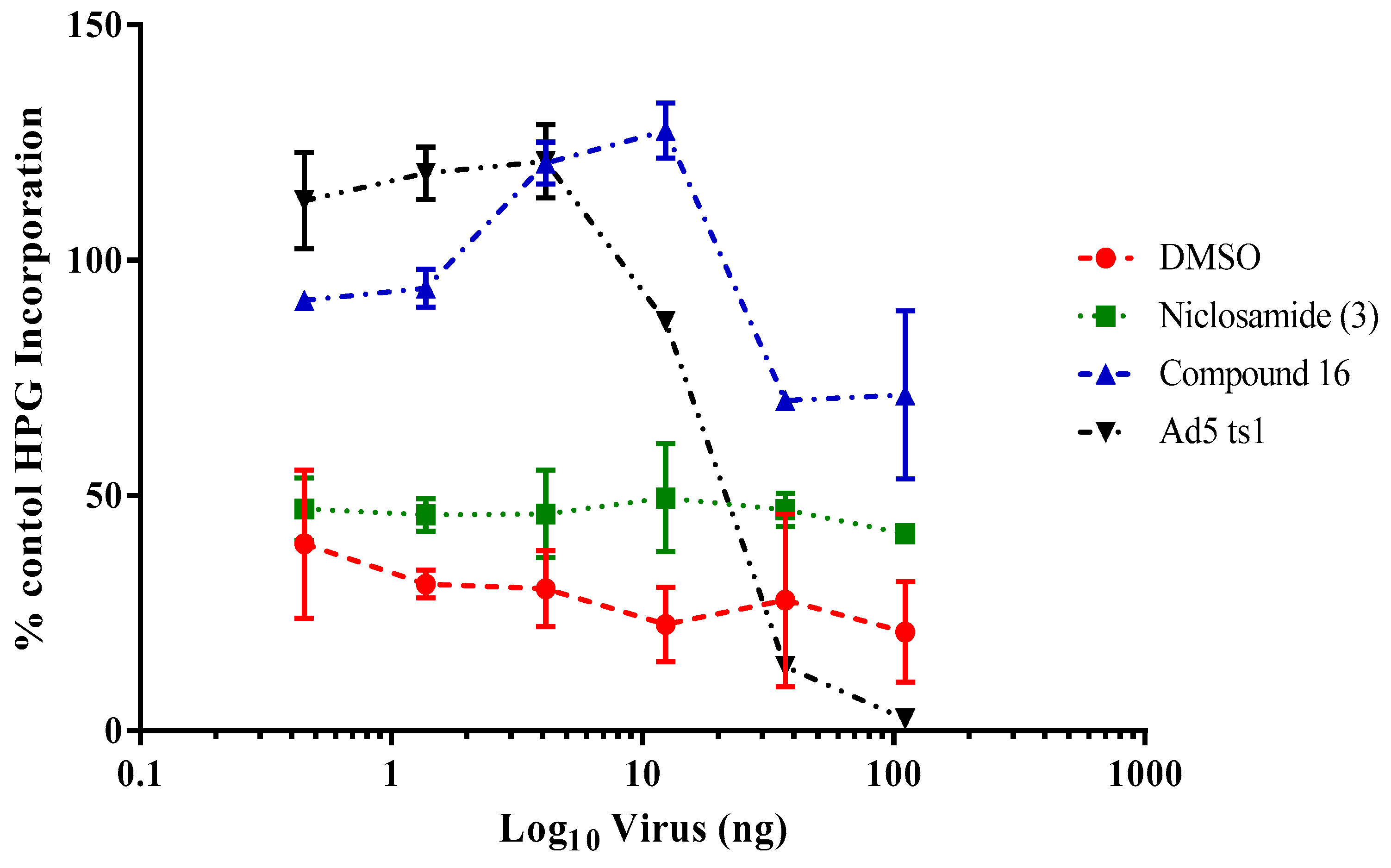
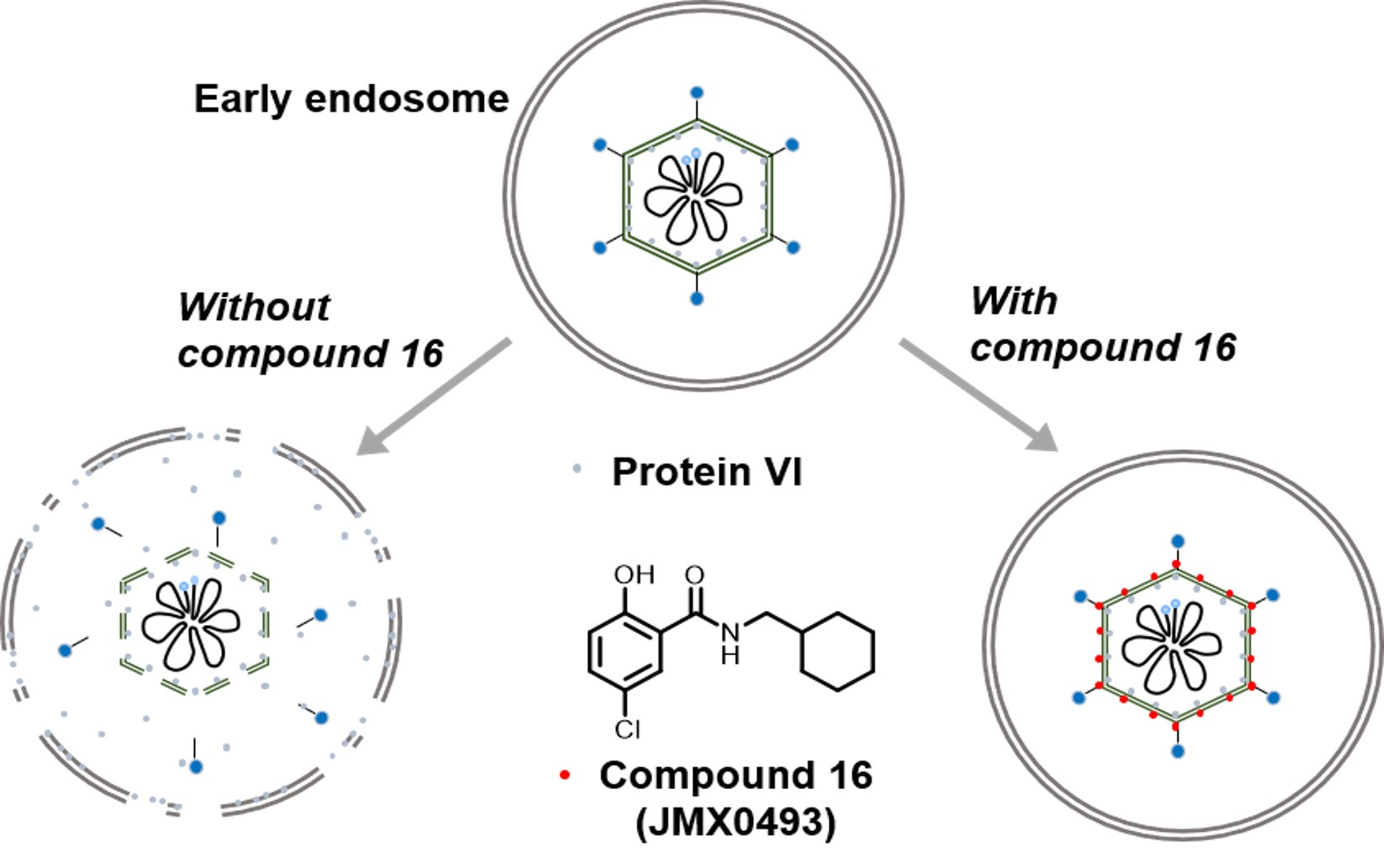
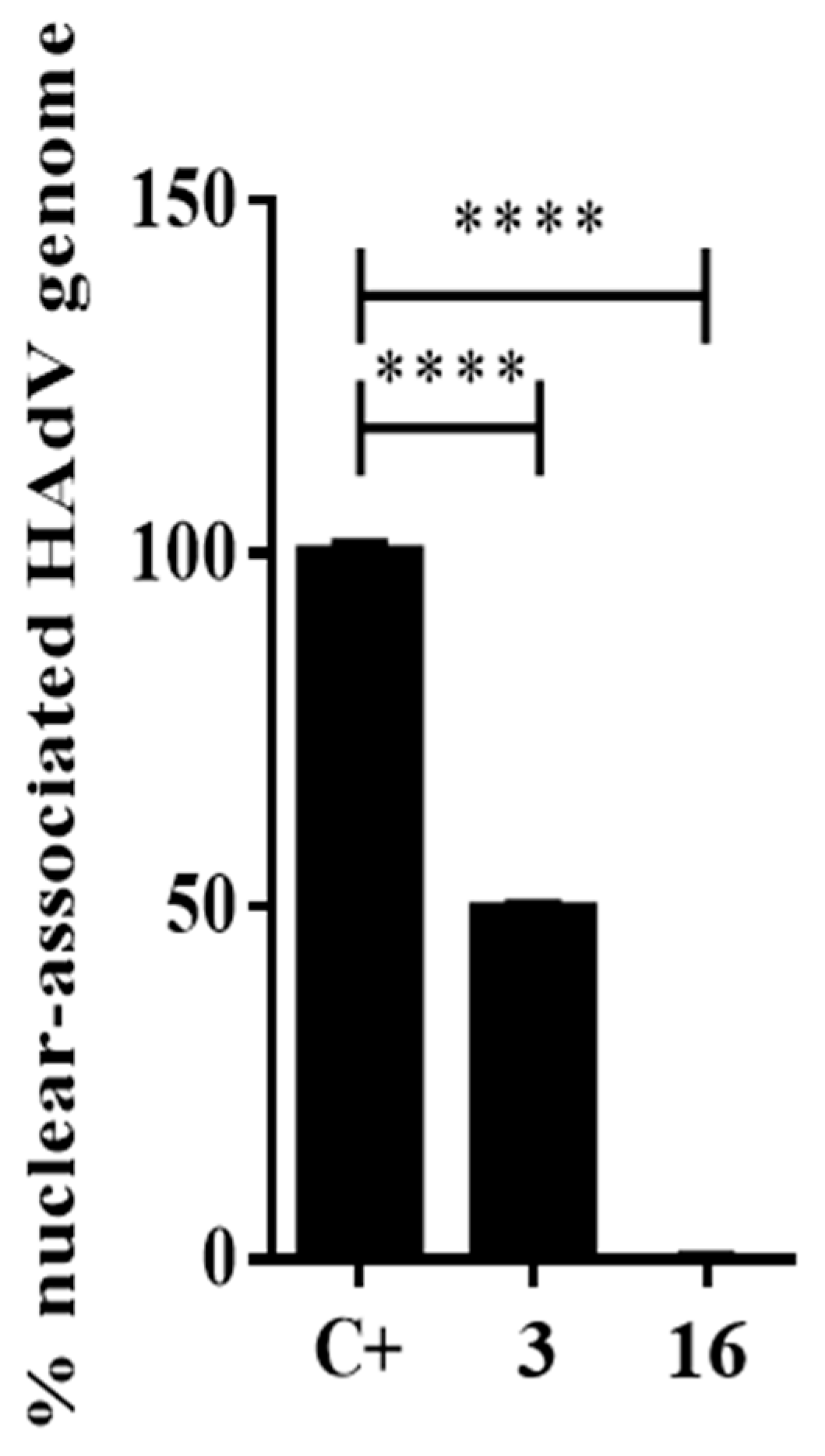
2.3.3. Impacts of Compound 16 on HAdV Escape from the Endosome
2.4. Synergistic Activity of the Selected Compounds
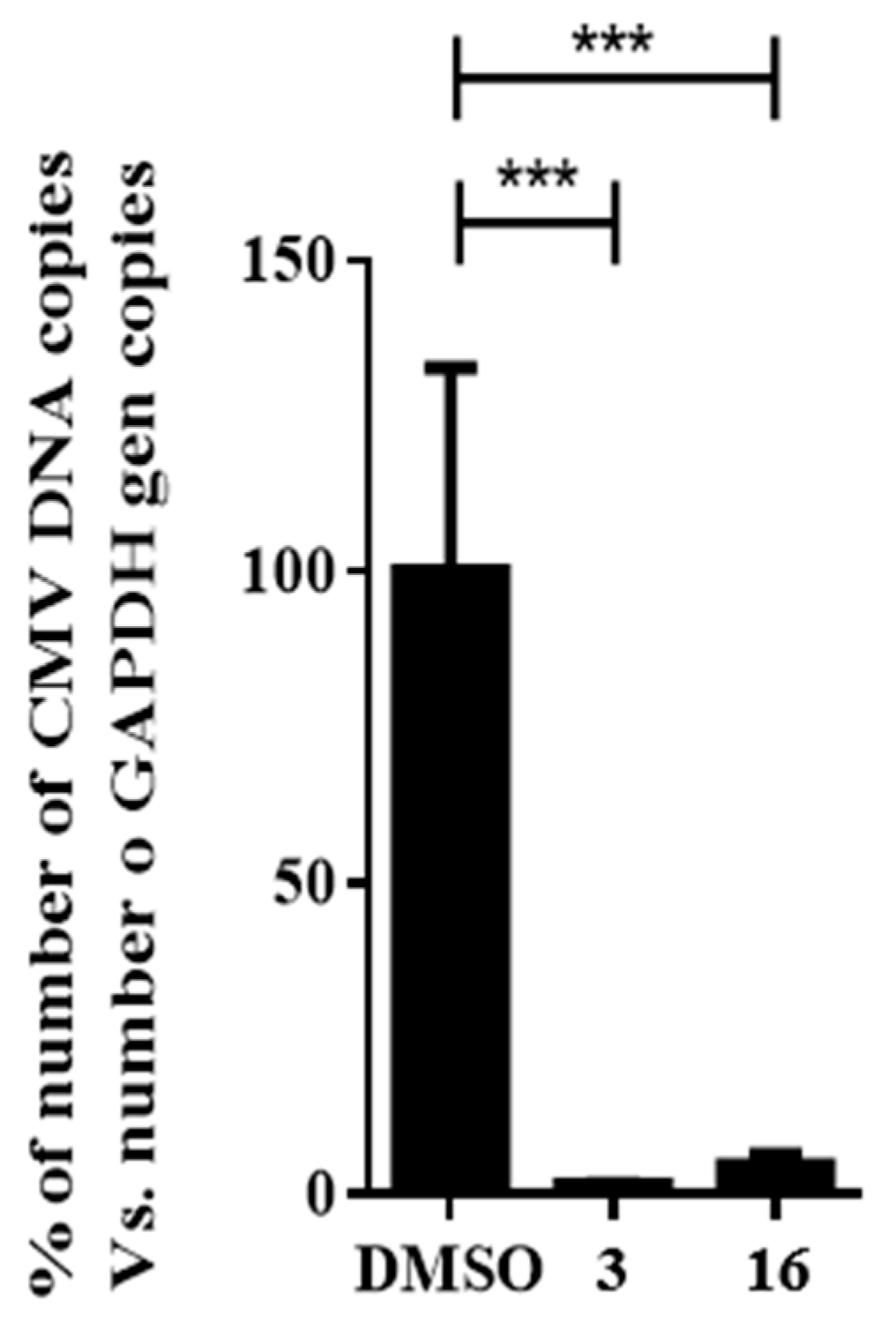
2.5. Impact of Compound 16 on HCMV Replication
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.2. Biology
4.2.1. Cell Lines and Virus Strain
4.2.2. Plaque Assay
4.2.3. Cytotoxicity Assay
4.2.4. Virus Yield Reduction
4.2.5. DNA Quantification by Real-Time PCR
4.2.6. Nuclear-Associated HAdV Genomes
4.2.7. Time of Addition Assay
4.2.8. HAdV-Mediated Endosome Disruption
4.2.9. Thermostability Assay
4.2.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| allo-HSCT | allogenic hematopoietic stem cell transplant |
| BCV | brincidofovir |
| CAP | community-acquired pneumonia |
| CC50 | half-maximal cytotoxic concentration |
| DCM | dichloromethane |
| DMAP | 4-(dimethylamino)pyridine |
| DMSO | dimethyl sulfoxide |
| EDCI | 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| EtOAc | ethyl acetate |
| HAdVs | human adenoviruses |
| HCMV | human cytomegalovirus |
| HPG | L-homopropargylglycine |
| HPLC | high-performance liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| IC50 | half-maximal inhibitory concentration |
| p.i. | postinfection |
| SAR | structure–activity relationship |
| SOT | solid-organ transplant |
| TLC | thin layer chromatography |
| TMS | tetramethylsilane |
| UV | ultraviolet |
References
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef] [Green Version]
- HAdV Working Group Human Adenovirus Working Group. Available online: http://hadvwg.gmu.edu/ (accessed on 22 January 2020).
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, S.; Raybould, J.E.; Sastry, S.; De la Cruz, O. Respiratory viruses in transplant recipients: More than just a cold. Clinical syndromes and infection prevention principles. Int. J. Infect. Dis. 2017, 62, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandkovsky, U.; Vargas, L.; Florescu, D.F. Adenovirus: Current epidemiology and emerging approaches to prevention and treatment. Curr. Infect. Dis. Rep. 2014, 16, 416. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, P.; Petterson, T.; Robin, M.; Sivaprakasam, P.; Vainorius, E.; Brundage, T.; Chandak, A.; Mozaffari, E.; Nichols, G.; Voigt, S. Incidence of adenovirus infection in hematopoietic stem cell transplantation recipients: Findings from the advance study. Biol. Blood Marrow Transplant. 2019, 25, 810–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonnalagadda, S.; Rodríguez, O.; Estrella, B.; Sabin, L.L.; Sempértegui, F.; Hamer, D.H. Etiology of severe pneumonia in Ecuadorian children. PLoS ONE 2017, 12, e0171687. [Google Scholar] [CrossRef]
- Kajon, A.E.; Ison, M.G. Severe infections with human adenovirus 7d in 2 adults in family, Illinois, USA, 2014. Emerg. Infect. Dis. 2016, 22, 730–733. [Google Scholar] [CrossRef]
- Tan, D.; Fu, Y.; Xu, J.; Wang, Z.; Cao, J.; Walline, J.; Zhu, H.; Yu, X. Severe adenovirus community-acquired pneumonia in immunocompetent adults: Chest radiographic and CT findings. J. Thorac. Dis. 2016, 8, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Zhu, H.; Fu, Y.; Tong, F.; Yao, D.; Walline, J.; Xu, J.; Yu, X. Severe community-acquired pneumonia caused by human adenovirus in immunocompetent adults: A multicenter case series. PLoS ONE 2016, 11, e0151199. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.W.; Song, Y.G.; Lee, S.H. Severe community-acquired adenovirus pneumonia treated with oral ribavirin: A case report. BMC Res. Notes 2017, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.X.; Zhao, M.M.; Pu, Z.H.; Wang, Y.Q.; Liu, Y. Clinical data analysis of 19 cases of community-acquired adenovirus pneumonia in immunocompetent adults. Int. J. Clin. Exp. Med. 2015, 8, 19051–19057. [Google Scholar]
- Lenaerts, L.; De Clercq, E.; Naesens, L. Clinical features and treatment of adenovirus infections. Rev. Med. Virol. 2008, 18, 357–374. [Google Scholar] [CrossRef]
- Martínez-Aguado, P.; Serna-Gallego, A.; Marrugal-Lorenzo, J.A.; Gómez-Marín, I.; Sánchez-Céspedes, J. Antiadenovirus drug discovery: Potential targets and evaluation methodologies. Drug Discov. Today 2015, 20, 1235–1242. [Google Scholar] [CrossRef]
- Wold, W.S.M.; Tollefson, A.E.; Ying, B.; Spencer, J.F.; Toth, K. Drug development against human adenoviruses and its advancement by Syrian hamster models. FEMS Microbiol. Rev. 2019, 43, 380–388. [Google Scholar] [CrossRef]
- Andersson, E.K.; Strand, M.; Edlund, K.; Lindman, K.; Enquist, P.-A.; Spjut, S.; Allard, A.; Elofsson, M.; Mei, Y.-F.; Wadell, G. Small-molecule screening using a whole-cell viral replication reporter gene assay identifies 2-{[2-(benzoylamino)benzoyl]amino}-benzoic acid as a novel antiadenoviral compound. Antimicrob. Agents Chemother. 2010, 54, 3871–3877. [Google Scholar] [CrossRef] [Green Version]
- Oberg, C.T.; Strand, M.; Andersson, E.K.; Edlund, K.; Tran, N.P.; Mei, Y.F.; Wadell, G.; Elofsson, M. Synthesis, biological evaluation, and structure-activity relationships of 2-[2-(benzoylamino)benzoylamino]benzoic acid analogues as inhibitors of adenovirus replication. J. Med. Chem. 2012, 55, 3170–3181. [Google Scholar] [CrossRef]
- Sanchez-Cespedes, J.; Moyer, C.L.; Whitby, L.R.; Boger, D.L.; Nemerow, G.R. Inhibition of adenovirus replication by a trisubstituted piperazin-2-one derivative. Antivir. Res. 2014, 108, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Cespedes, J.; Martinez-Aguado, P.; Vega-Holm, M.; Serna-Gallego, A.; Candela, J.I.; Marrugal-Lorenzo, J.A.; Pachon, J.; Iglesias-Guerra, F.; Vega-Perez, J.M. New 4-acyl-1-phenylaminocarbonyl-2-phenylpiperazine derivatives as potential inhibitors of adenovirus infection. Synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2016, 59, 5432–5448. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, S.; Marrugal-Lorenzo, J.A.; Vega-Holm, M.; Serna-Gallego, A.; Álvarez-Vidal, J.; Berastegui-Cabrera, J.; Pérez del Palacio, J.; Díaz, C.; Aiello, F.; Pachón, J.; et al. Optimization of piperazine-derived ureas privileged structures for effective antiadenovirus agents. Eur. J. Med. Chem. 2020, 185, 111840. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.M.; Sortino, K.; Sethna, P.; Bae, A.; Lanier, R.; Bambara, R.A.; Dewhurst, S. Cidofovir diphosphate inhibits adenovirus 5 DNA polymerase via both nonobligate chain termination and direct inhibition, and polymerase mutations confer cidofovir resistance on intact virus. Antimicrob. Agents Chemother. 2019, 63, e01925-18. [Google Scholar] [CrossRef] [Green Version]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antivir. Res. 2009, 82, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Tippin, T.K.; Morrison, M.E.; Brundage, T.M.; Mommeja-Marin, H. Brincidofovir is not a substrate for the human organic anion transporter 1: A mechanistic explanation for the lack of nephrotoxicity observed in clinical studies. Ther. Drug Monit. 2016, 38, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, K.; Spencer, J.F.; Dhar, D.; Sagartz, J.E.; Buller, R.M.; Painter, G.R.; Wold, W.S. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc. Natl. Acad. Sci. USA 2008, 105, 7293–7297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimley, M.S.; Marón, G.M.; Prasad, V.K.; Jacobsohn, D.A.; Young, J.-A.H.; Chittick, G.; Brundage, T.M.; Momméja-Marin, H. Preliminary results from the advise study evaluating brincidofovir (CMX001, BCV) for the treatment of disseminated and high-risk adenovirus (AdV) infection. Biol. Blood Marrow Transplant. 2015, 21, 108–109. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.K.; Papanicolaou, G.A.; Marón, G.M.; Vainorius, E.; Brundage, T.M.; Chittick, G.; Nichols, G.; Grimley, M.S. 46-Treatment of adenovirus (AdV) infection in allogeneic hematopoietic cell transplant (allo HCT) patients (pts) with brincidofovir: Final 36 week results from the advise trial. Biol. Blood Marrow Transplant. 2017, 23, 57–58. [Google Scholar] [CrossRef]
- Chittick, G.; Morrison, M.; Brundage, T.; Nichols, W.G. Short-term clinical safety profile of brincidofovir: A favorable benefit–risk proposition in the treatment of smallpox. Antivir. Res. 2017, 143, 269–277. [Google Scholar] [CrossRef]
- Grimley, M.S.; Chemaly, R.F.; Englund, J.A.; Kurtzberg, J.; Chittick, G.; Brundage, T.M.; Bae, A.; Morrison, M.E.; Prasad, V.K. Brincidofovir for asymptomatic adenovirus viremia in pediatric and adult allogeneic hematopoietic cell transplant recipients: A randomized placebo-controlled phase II trial. Biol. Blood Marrow Transplant. 2017, 23, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Xu, J.; Files, M.; Cirillo, J.D.; Endsley, J.J.; Zhou, J.; Endsley, M.A. Dual activity of niclosamide to suppress replication of integrated HIV-1 and Mycobacterium tuberculosis (Beijing). Tuberculosis 2019, 116, 28–33. [Google Scholar] [CrossRef]
- Xu, J.; Xie, X.; Ye, N.; Zou, J.; Chen, H.; White, M.A.; Shi, P.Y.; Zhou, J. Design, synthesis, and biological evaluation of substituted 4,6-dihydrospiro[[1,2,3]triazolo[4,5-b]pyridine-7,3’-indoline]-2’,5(3H)-dione analogues as potent NS4B inhibitors for the treatment of dengue virus infection. J. Med. Chem. 2019, 62, 7941–7960. [Google Scholar] [CrossRef]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.Y.; Zhou, J. Therapeutic potential of spirooxindoles as antiviral agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, J.; Lang, Y.; Fan, X.; Kuo, L.; D’Brant, L.; Hu, S.; Samrat, S.K.; Trudeau, N.; Tharappel, A.M.; et al. JMX0207, a niclosamide derivative with improved pharmacokinetics, suppresses ZIKA virus infection both in vitro and in vivo. ACS Infect. Dis. 2020, 6, 2616–2628. [Google Scholar] [CrossRef]
- Xu, J.; Xie, X.; Chen, H.; Zou, J.; Xue, Y.; Ye, N.; Shi, P.-Y.; Zhou, J. Design, synthesis and biological evaluation of spiropyrazolopyridone derivatives as potent dengue virus inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127162. [Google Scholar] [CrossRef]
- Marrugal-Lorenzo, J.A.; Serna-Gallego, A.; Berastegui-Cabrera, J.; Pachón, J.; Sánchez-Céspedes, J. Repositioning salicylanilide anthelmintic drugs to treat adenovirus infections. Sci. Rep. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Berastegui-Cabrera, J.; Chen, H.; Pachón, J.; Zhou, J.; Sánchez-Céspedes, J. Structure–activity relationship studies on diversified salicylamide derivatives as potent inhibitors of human adenovirus infection. J. Med. Chem. 2020, 63, 3142–3160. [Google Scholar] [CrossRef]
- Xu, J.; Berastegui-Cabrera, J.; Ye, N.; Carretero-Ledesma, M.; Pachón-Díaz, J.; Chen, H.; Pachón-Ibáñez, M.E.; Sánchez-Céspedes, J.; Zhou, J. Discovery of novel substituted N-(4-Amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as potent human adenovirus inhibitors. J. Med. Chem. 2020, 63, 12830–12852. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [Green Version]
- Rancourt, C.; Keyvani-Amineh, H.; Sircar, S.; Labrecque, P.; Weber, J.M. Proline 137 is critical for adenovirus protease encapsidation and activation but not enzyme activity. Virology 1995, 209, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.G.; Nemerow, G.R. Mechanism of adenovirus neutralization by Human α-defensins. Cell Host Microbe 2008, 3, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner, L.B.; Wenk, M.R.; Guan, X.L.; Grzybek, M.; Coskun, Ü.; Greber, U.F. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe 2015, 18, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Matthews, H.; Deakin, J.; Rajab, M.; Idris-Usman, M.; Nirmalan, N.J. Investigating antimalarial drug interactions of emetine dihydrochloride hydrate using CalcuSyn-based interactivity calculations. PLoS ONE 2017, 12, e0173303. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, E.K.; Nemerow, G.R.; Smith, J.G. Direct evidence from single-cell analysis that human α-defensins block adenovirus uncoating to neutralize infection. J. Virol. 2010, 84, 4041–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepomuceno, R.R.; Pache, L.; Nemerow, G.R. Enhancement of gene transfer to human myeloid cells by adenovirus-fiber complexes. Mol. Ther. 2007, 15, 571–578. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Rivera, A.A.; Wang, M.; Suzuki, K.; Uil, T.G.; Krasnykh, V.; Curiel, D.T.; Nettelbeck, D.M. Mode of transgene expression after fusion to early or late viral genes of a conditionally replicating adenovirus via an optimized internal ribosome entry site in vitro and in vivo. Virology 2004, 320, 121–134. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
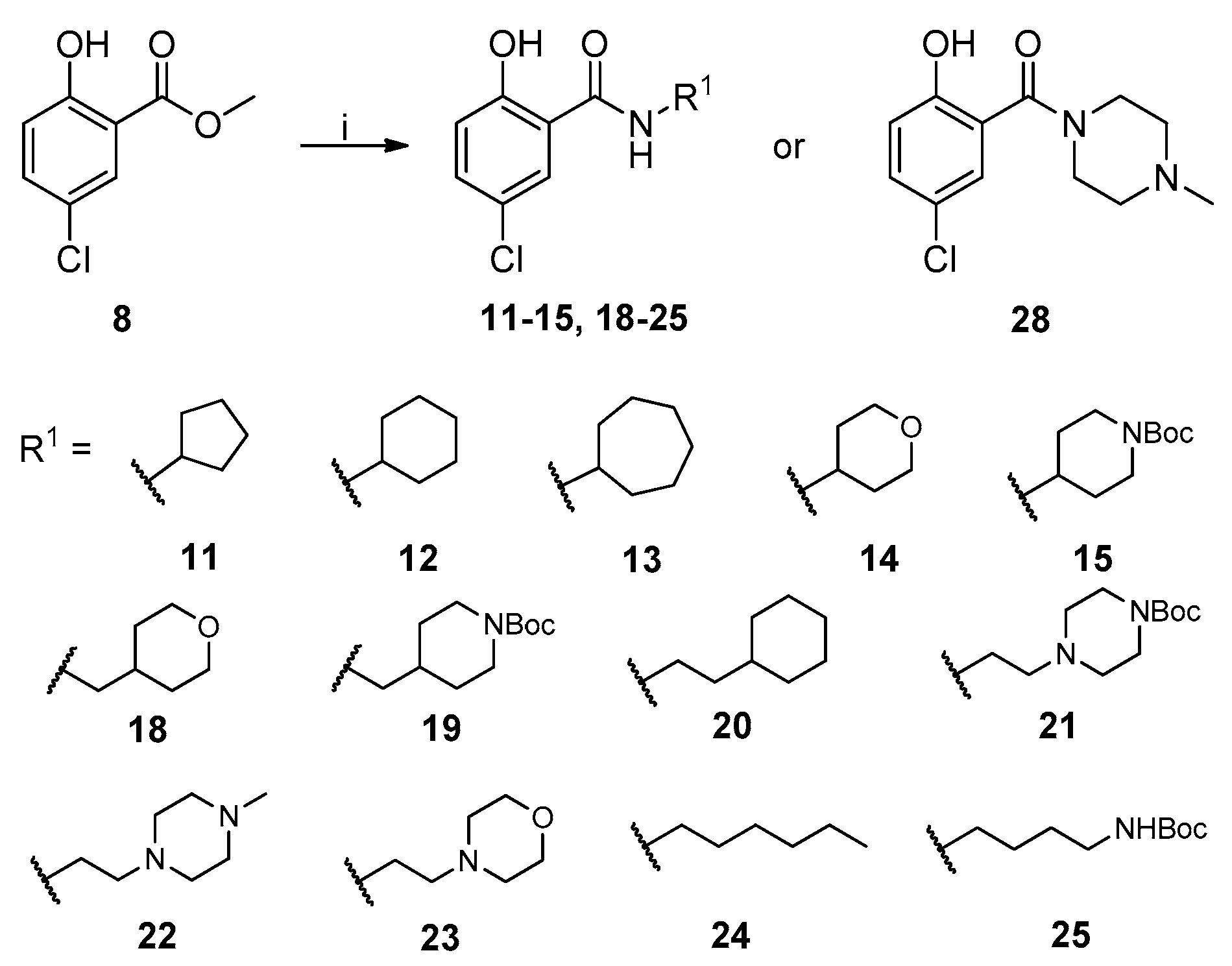{kind=link}
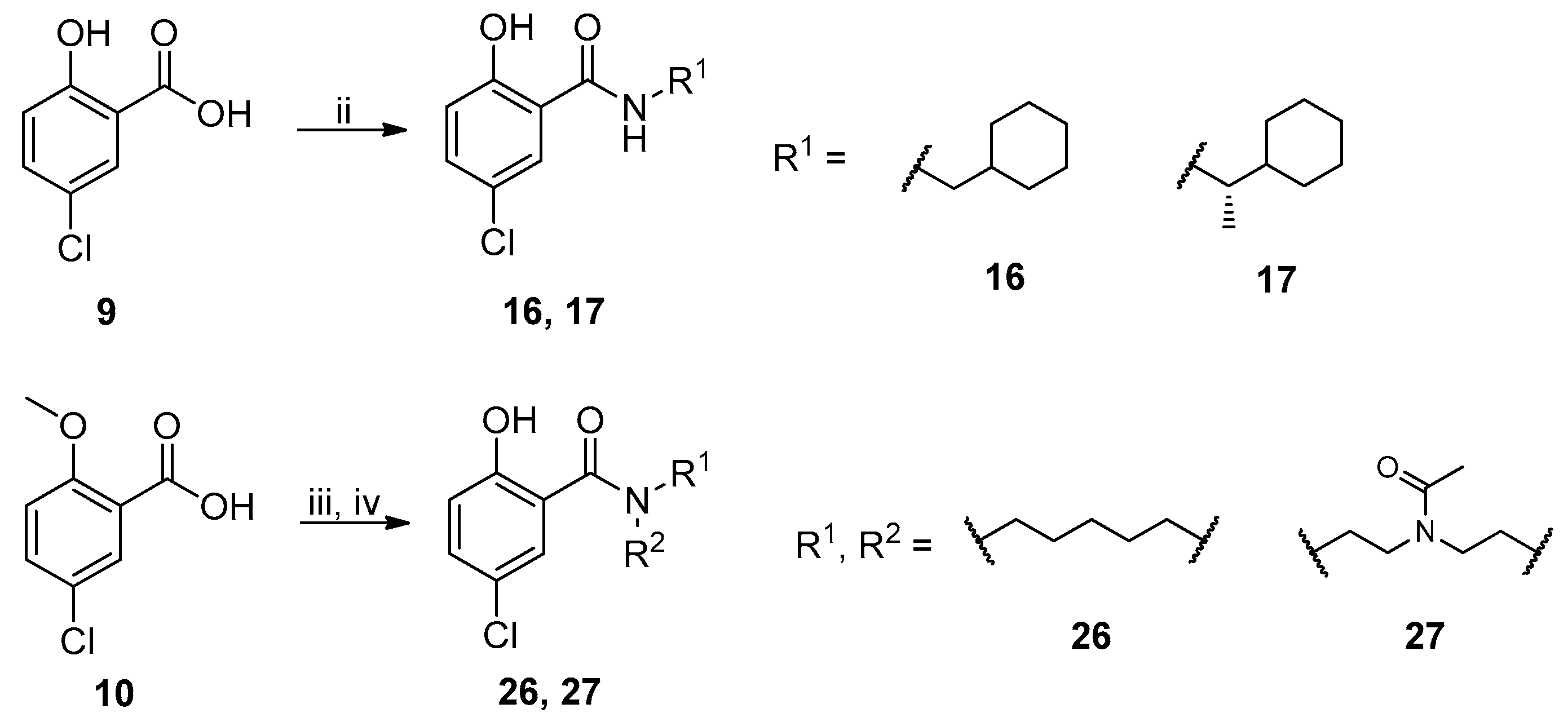{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Plaque Assay (10 μM) a | CC50 (μM) b | Selectivity Index (SI) c | |
| (%) Inhibition | IC50 (μM) | |||||
| 3 | NA | 100 ± 0 | 0.6 ± 0.05 | 22.9 ± 9.8 | 38.2 | |
| 11 | H |  | 22.6 ± 7.4 | NT d | NT | NT |
| 12 | H |  | 57.6 ± 0.2 | NT | NT | NT |
| 13 | H |  | 99.6 ± 0.2 | 4.7 ± 0.1 | 30.9 ± 6.3 | 6.6 |
| 14 | H |  | 0.0 ± 0.0 | NT | NT | NT |
| 15 | H |  | 52.2 ± 7.1 | NT | NT | NT |
| 16 | H |  | 100 ± 0 | 0.78 ± 0.01 | 91.2 ± 18.4 | 116.9 |
| 17 | H |  | 100 ± 0 | 0.51 ± 0.14 | 23.0 ± 1.3 | 45.1 |
| 18 | H |  | 0.0 ± 0.0 | NT | NT | NT |
| 19 | H |  | 88.5 ± 1.9 | NT | NT | NT |
| 20 | H |  | 97.8 ± 1.7 | 1.0 ± 0.1 | 66.3 ± 7.9 | 64.3 |
| 21 | H |  | 22.2 ± 13.2 | NT | NT | NT |
| 22 | H |  | 0.00 ± 0.00 | NT | NT | NT |
| 23 | H |  | 0.00 ± 0.00 | NT | NT | NT |
| 24 | H |  | 98.5 ± 2.1 | 3.73 ± 0.64 | 23.8 ± 3.1 | 6.4 |
| 25 | H |  | 74.0 ± 5.7 | NT | NT | NT |
| 26 |  | 0.00 ± 0.00 | NT | NT | NT | |
| 27 |  | 0.00 ± 0.00 | NT | NT | NT | |
| 28 |  | 0.00 ± 0.00 | NT | NT | NT | |
| Compound | Virus Yield Reduction (fold) a |
|---|---|
| niclosamide | 82 ± 35 |
| 16 | 208 ± 108 |
| Combinatory Index (CI) Values in Plaque Assay | ||||
|---|---|---|---|---|
| Combination (Ratio) | ED50 | ED75 | ED90 | r |
| 4 and 16 (1:4.3) | 0.26 | 0.38 | 0.56 | 0.87 |
| 5 and 16 (1:2.9) | 1.09 | 1.08 | 1.14 | 0.92 |
| 6 and 16 (1:1.73) | 0.40 | 0.27 | 0.19 | 0.94 |
| 4, 5, and 16 (1:1.5:4.3) | 0.33 | 0.26 | 0.22 | 0.94 |
| 5, 6, and 16 (1:1.7:2.9) | 2.01 | 1.17 | 0.72 | 0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Berastegui-Cabrera, J.; Carretero-Ledesma, M.; Chen, H.; Xue, Y.; Wold, E.A.; Pachón, J.; Zhou, J.; Sánchez-Céspedes, J. Discovery of a Small Molecule Inhibitor of Human Adenovirus Capable of Preventing Escape from the Endosome. Int. J. Mol. Sci. 2021, 22, 1617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041617
Xu J, Berastegui-Cabrera J, Carretero-Ledesma M, Chen H, Xue Y, Wold EA, Pachón J, Zhou J, Sánchez-Céspedes J. Discovery of a Small Molecule Inhibitor of Human Adenovirus Capable of Preventing Escape from the Endosome. International Journal of Molecular Sciences. 2021; 22(4):1617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041617
Chicago/Turabian StyleXu, Jimin, Judith Berastegui-Cabrera, Marta Carretero-Ledesma, Haiying Chen, Yu Xue, Eric A. Wold, Jerónimo Pachón, Jia Zhou, and Javier Sánchez-Céspedes. 2021. "Discovery of a Small Molecule Inhibitor of Human Adenovirus Capable of Preventing Escape from the Endosome" International Journal of Molecular Sciences 22, no. 4: 1617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041617