
Construction and Validation of a Prognostic Gene-Based Model for Overall Survival Prediction in Hepatocellular Carcinoma Using an Integrated Statistical and Bioinformatic Approach

 , and
, and
Abstract
:1. Introduction
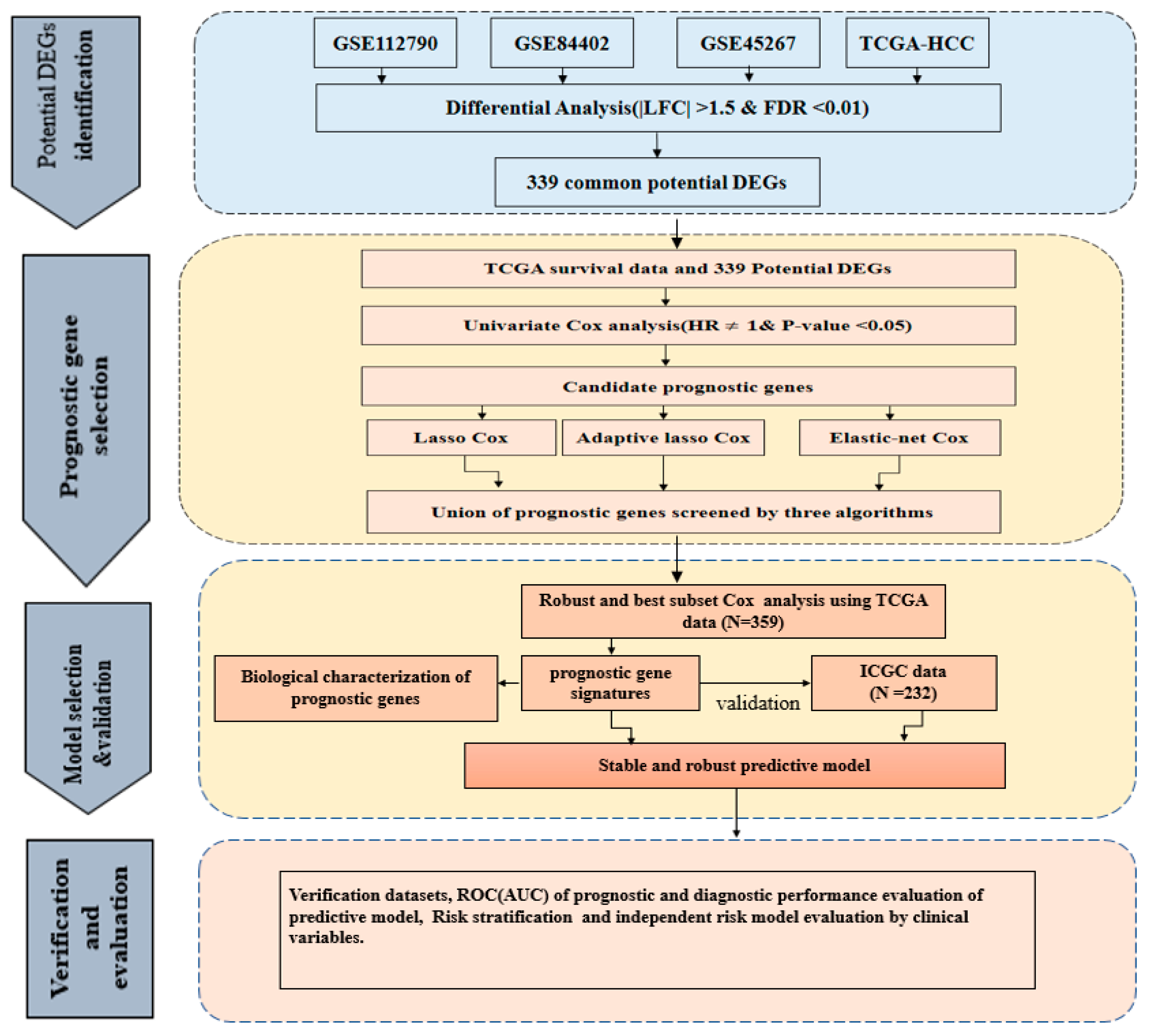
2. Results
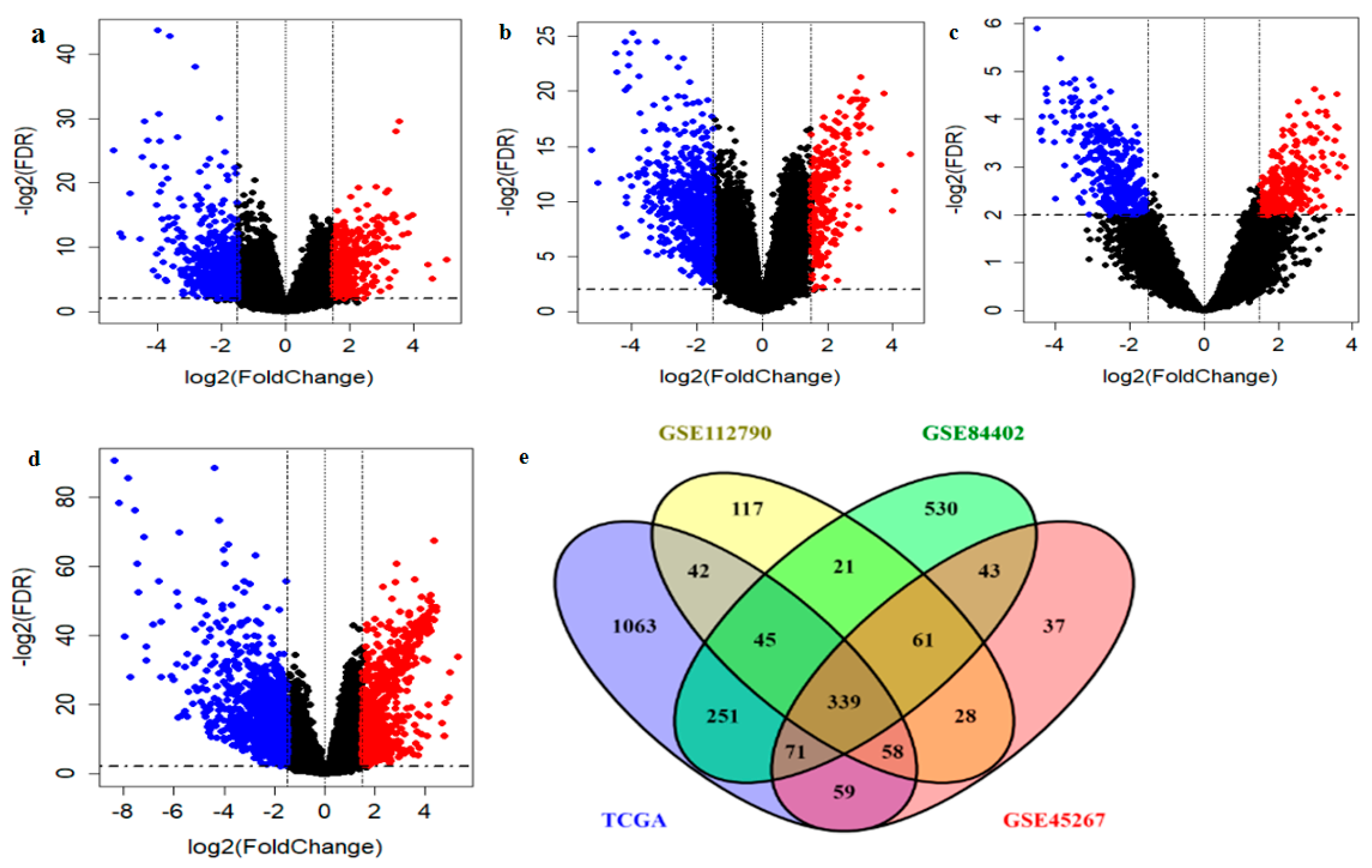
2.1. Identification and Validation of Potential DEGs
2.2. Identification of Prognosis-Related DEGs
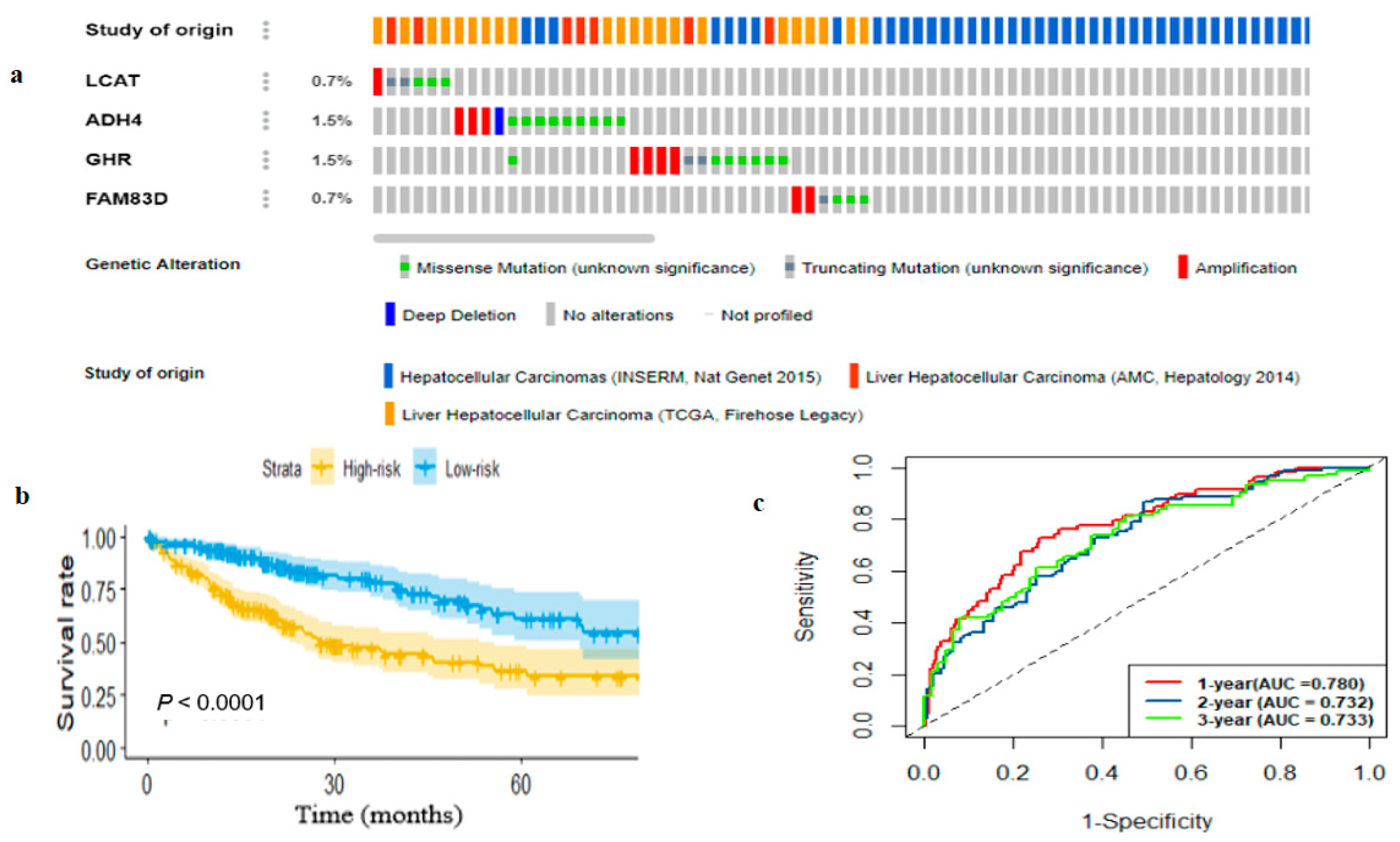
2.3. Genetic Alterations and Survival-Related Gene Expression Profiles
2.4. Development and Estimation of the Four-Gene Signature
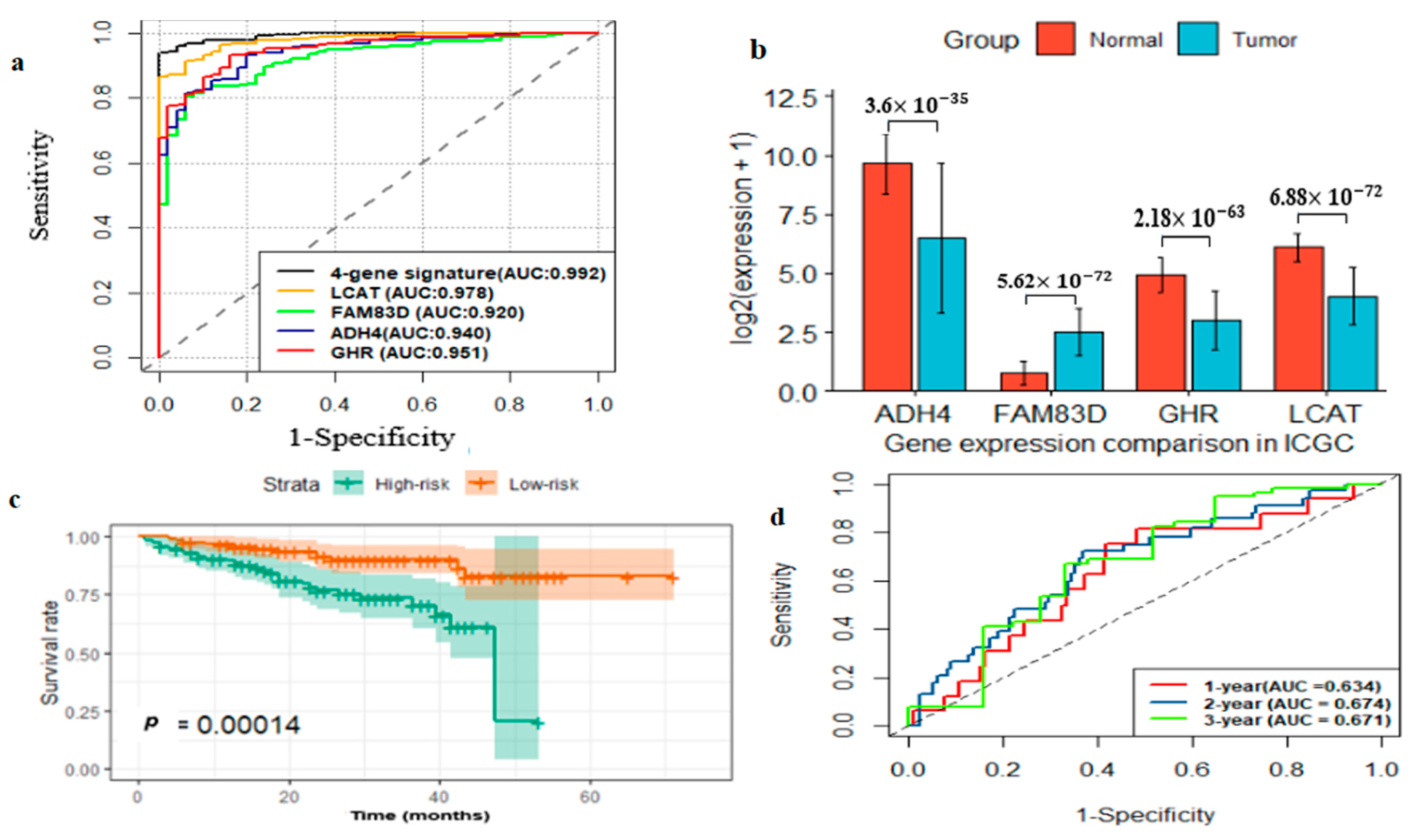
2.5. External Prognostic and Diagnostic Validation of Four-Gene Signature
2.6. External Prognostic and Diagnostic Verification of the Four-Gene Signature
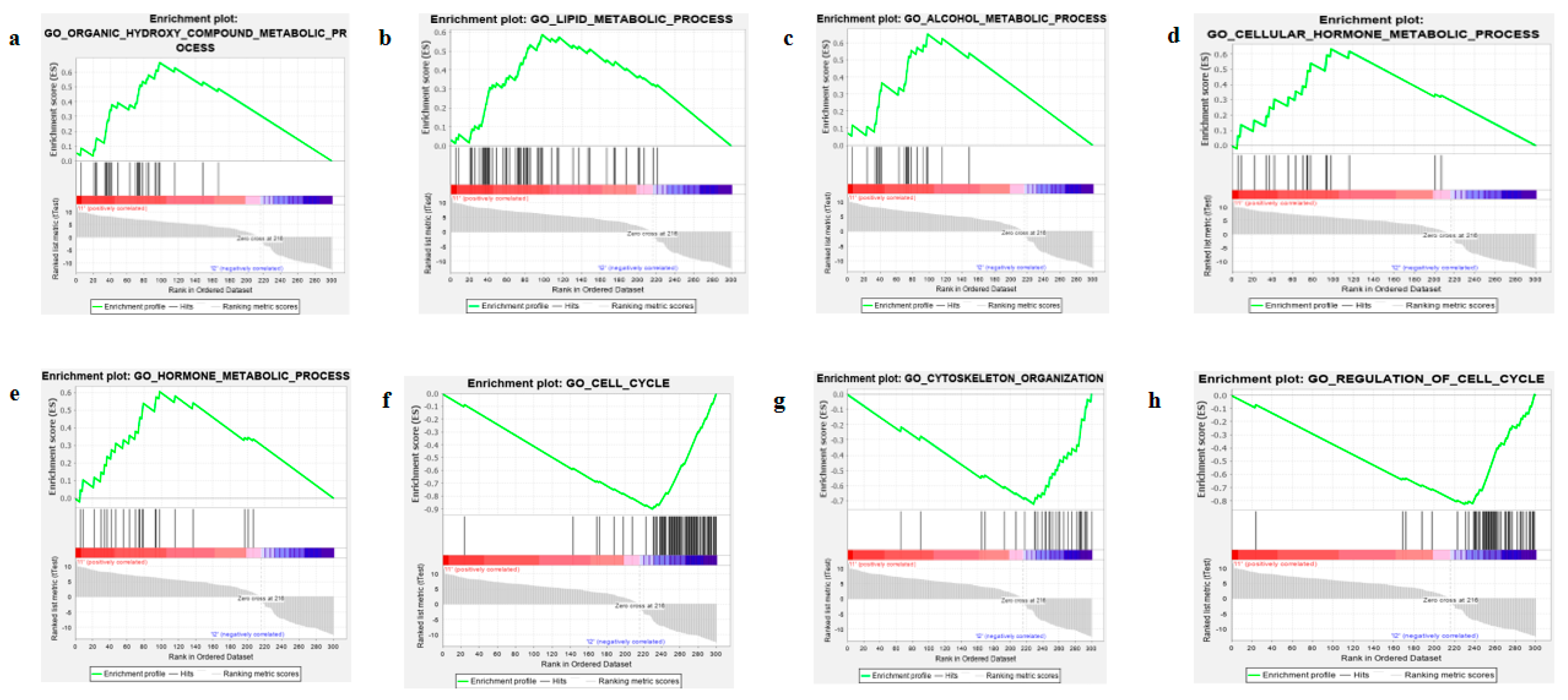
2.7. Identification of Biological Pathways of Four-Gene Signture and Thier Coe-Expressed Degs
3. Discussion
4. Materials and Methods
4.1. HCC Sample Source
4.2. Data Preprocessing and Identfication of DEGs
4.3. Prognosis-Related Gene Selection and Development of Prognostic Model
- Construct all possible models C (K, τ) using τ subset of identified genes.
- Calculate Akaike information criterion (AIC) for each constructed model in a.
- Choose an optimal prognostic model, whose AIC is the smallest τ prognostic genes using the “glmulti” package (in R platform) [36].
4.4. Construction and Estimation of Prognostic Gene Signature
4.5. Evaluation of the Diagnostic Performance of the Multi-Gene Signature in HCC
4.6. Clinical HCC Patient Samples
4.7. RNA Sequencing (RNA-Seq) and Data Analysis
4.8. Biological Characterization of the Identified Prognostic Genes
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene expression omnibus |
| DEG | Differentially expressed gene |
| KM | Kaplan–Meier |
| ICGC | International Cancer Genome Consortium |
| RS | Risk score |
| AUC | Area under the curve |
| ROC | Receiver operating characteristic |
| FDR | False discovery rate |
| HR | Hazard ratio |
| RSEM | RNA-Seq by Expectation–Maximization |
| FAM83D | Family with sequence similarity 83-member D |
| ADH4 | Alcohol dehydrogenase 4 |
| GHR | Growth hormone receptor |
| LCAT | Lecithin-cholesterol acyltransferase |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.M.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Tian, G.; Yang, S.; Yuan, J.; Threapleton, D.; Zhao, Q.; Chen, F.; Cao, H.; Jiang, T.; Li, L. Comparative efficacy of treatment strategies for hepatocellular carcinoma: Systematic review and network meta-analysis. BMJ Open 2018, 8, e021269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.-M.; Zeng, H.-D.; Zhang, C.-Y.; Xu, J.-W. Identification of a six-gene signature predicting overall survival for hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, A.; Kumada, T.; Tsuji, K.; Takaguchi, K.; Itobayashi, E.; Kariyama, K.; Ochi, H.; Tajiri, K.; Hirooka, M.; Shimada, N.; et al. Validation of Modified ALBI Grade for More Detailed Assessment of Hepatic Function in Hepatocellular Carcinoma Patients: A Multicenter Analysis. Liver Cancer 2019, 8, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, D.; Gu, J.; Xu, F.; Zhu, Q.; Lu, C. A large cohort study identifying a novel prognosis prediction model for lung adenocarcinoma through machine learning strategies. BMC Cancer 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiang, Z.; Chen, X.; Zhu, X.; Peng, X. A seven-gene signature model predicts overall survival in kidney renal clear cell carcinoma. Hereditas 2020, 157, 1–8. [Google Scholar] [CrossRef]
- Chen, W.-B.; Ou, M.; Tang, D.; Dai, Y.; Du, W. Identification and Validation of Immune-Related Gene Prognostic Signature for Hepatocellular Carcinoma. J. Immunol. Res. 2020, 2020, 1–14. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Ning, G.; Wang, Y.-S.; Lv, W.-B. 14-CpG-Based Signature Improves the Prognosis Prediction of Hepatocellular Carcinoma Patients. Biomed. Res. Int. 2020, 2020, 9762067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Zou, H. The Adaptive Lasso and Its Oracle Properties. J. Am. Stat. Assoc. 2006, 101, 1418–1429. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Walian, P.J.; Hang, B.; Mao, J. Prognostic significance of FAM83D gene expression across human cancer types. Oncotarget 2015, 7, 3332–3340. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Liu, W.; Liu, X.; Yuan, Q.; Ou, Y.; Qi, Y.; Huang, W.; Wang, Y.; Huang, J. Upregulation of FAM83D affects the proliferation and invasion of hepatocellular carcinoma. Oncotarget 2015, 6, 24132–24147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Gao, H.; Zhang, J.; Xue, D. FAM83D is associated with gender, AJCC stage, overall survival and disease-free survival in hepatocellular carcinoma. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Yu, S.; Lok, S.I.S.; Wong, A.S.T.; Jiao, Y.; Lee, L.T.O. FAM83D promotes ovarian cancer progression and its potential application in diagnosis of invasive ovarian cancer. J. Cell. Mol. Med. 2019, 23, 4569–4581. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, G.; Yi, B.; Pan, G.; Chen, X. A robust twelve-gene signature for prognosis prediction of hepatocellular carcinoma. Cancer Cell Int. 2020, 20, 1–18. [Google Scholar] [CrossRef]
- Wei, R.-R.; Zhang, M.-Y.; Rao, H.-L.; Pu, H.-Y.; Zhang, H.-Z.; Wang, H.-Y. Identification of ADH4 as a novel and potential prognostic marker in hepatocellular carcinoma. Med. Oncol. 2012, 29, 2737–2743. [Google Scholar] [CrossRef]
- Wang, X.; Liao, X.; Yang, C.; Huang, K.; Yu, T.; Yu, L.; Han, C.; Zhu, G.; Zeng, X.; Liu, Z.; et al. Identification of prognostic biomarkers for patients with hepatocellular carcinoma after hepatectomy. Oncol. Rep. 2019, 41, 1586–1602. [Google Scholar] [CrossRef]
- Gebre-Medhin, M.; Kindblom, L.-G.; Wennbo, H.; Törnell, J.; Meis-Kindblom, J.M. Growth Hormone Receptor Is Expressed in Human Breast Cancer. Am. J. Pathol. 2001, 158, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, A.; Subramani, R.; Nandy, S.B.; Terreros, D.; Dwivedi, A.K.; Saltzstein, E.; Lakshmanaswamy, R. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, Y.; Wong, H.Y.; Nikolajsen, L.F.; Steinocher, H.; Papadopulos, A.; Tunny, K.A.; Meunier, F.A.; Smith, A.G.; Kragelund, B.B.; Brooks, A.J.; et al. A growth hormone receptor SNP promotes lung cancer by impairment of SOCS2-mediated degradation. Oncogene 2017, 37, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, F.; Yang, L.; Dai, B.; Xue, H.; Zhang, L.; Ge, R.; Sun, Y. Bioinformatics profiling identifies seven immune-related risk signatures for hepatocellular carcinoma. PeerJ 2020, 8, e8301. [Google Scholar] [CrossRef]
- Zhang, B.-H.; Yang, J.; Jiang, L.; Lyu, T.; Kong, L.-X.; Tan, Y.-F.; Li, B.; Zhu, Y.-F.; Xi, A.-Y.; Xu, X.; et al. Development and validation of a 14-gene signature for prognosis prediction in hepatocellular carcinoma. Genomics 2020, 112, 2763–2771. [Google Scholar] [CrossRef]
- Lee, S.; Lim, H. Review of statistical methods for survival analysis using genomic data. Genom. Inf. 2019, 17, e41. [Google Scholar] [CrossRef]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huo, X.; Yang, X.-R.; He, J.; Cheng, L.; Wang, N.; Deng, X.; Jingyuan, F.; Wang, N.; Wang, C.; et al. STAT3-mediated upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer metastasis by regulating SOX4. Mol. Cancer 2017, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Tsai, Y.S.; Huang, Y.-H.; Liang, Y.-J.; Sun, Y.-Y. Lymphoid Enhancer Factor 1 Contributes to Hepatocellular Carcinoma Progression through Transcriptional Regu-lation of Epithelial-Mesenchymal Transition Regulators and Stemness Genes. Hepatol. Commun. 2018, 2, 1392–1407. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-W.; Hsieh, T.-H.; Huang, S.-Y.; Chau, G.Y.; Tung, C.-Y.; Su, C.-W.; Wu, J.-C. Forfeited hepatogenesis program and increased embryonic stem cell traits in young hepatocellular carcinoma (HCC) comparing to elderly HCC. BMC Genom. 2013, 14, 736. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, M.M.; Hu, C.; Roe, D.; Chen, Z.; Halonen, M.; Guerra, S. Least absolute shrinkage and selection operator type methods for the identification of serum biomarkers of overweight and obesity: Simulation and application. BMC Med Res. Methodol. 2016, 16, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, V.; de Mazancourt, C. glmulti: An R Package for Easy Automated Model Selection with (Generalized) Linear Models. J. Stat. Softw. 2010, 34, 29. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Elastic Net | Lasso | Adaptive Lasso |
|---|---|---|---|
| FAM83D | 0.091 | 0.104 | 0.113 |
| CDC20 | 0.048 | 0.058 | 0.004 |
| TPX2 | 0.016 | - | 0.084 |
| UBE2S | - | - | 0.024 |
| LECT2 | −0.001 | - | - |
| ANXA10 | −0.002 | - | - |
| DNASE1L3 | −0.004 | - | - |
| PON1 | −0.009 | −0.008 | - |
| CD5L | −0.009 | −0.009 | - |
| CYP2C9 | −0.016 | −0.016 | - |
| ADH4 | −0.019 | −0.021 | - |
| CFHR3 | −0.021 | −0.024 | −0.008 |
| GHR | −0.035 | −0.037 | - |
| LCAT | −0.080 | −0.091 | −0.135 |
| Gene Symbol | Gene Full Name | GSE112790 | GSE84402 | GSE45267 | TCGA |
|---|---|---|---|---|---|
| LFC | |||||
| GHR | Growth Hormone Receptor | −2.371 | −2.300 | −2.632 | −3.031 |
| LCAT | lecithin-cholesterol acyltransferase | −3.359 | −3.761 | −2.935 | −3.478 |
| FAM83D | family with sequence similarity 83 member D | 2.808 | 2.114 | 2.255 | 2.348 |
| ADH4 | Alcohol Dehydrogenase 4 | −2.910 | −2.328 | −4.360 | −4.360 |
| Factors | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| HR | p-Value | HR | p-Value | |
| TCGA dataset | ||||
| Age(>60 vs. ) | 1.27(0.88–1.85) | 0.205 | ||
| Gender (Male vs. Female) | 0.78(0.54–1.15) | 0.209 | ||
| Stage (III+IV vs. I+II) | 2.54(1.74–3.69) | <0.001 | 1.05(0.15–7.66) | 0.960 |
| T-stage (T3+T4 vs. T1+T2) | 2.56(1.76–3.74) | <0.001 | 2.35(0.32–17.12 | 0.400 |
| Grade (G3+G4 vs. G1+G2) | 1.14(0.78–1.66) | 0.506 | ||
| RS (High vs. Low risk) | 2.28(1.54–3.36) | <0.001 | 2.20(1.48–3.25) | <0.001 |
| ICGC dataset | ||||
| Age(>60 vs. ≤60) | 1.28(0.70–2.35) | 0.427 | ||
| Gender (Male vs. Female) | 0.48(0.26–0.90) | 0.022 | 0.38(0.20–0.73) | 0.004 |
| Stage (III+IV vs. I+II) | 2.18(1.19–4.01) | 0.012 | 2.66(1.42–5.01) | 0.002 |
| RS (High vs. Low risk) | 3.52(1.77–6.99) | <0.001 | 3.57(1.78–7.18) | <0.001 |
| KEGG Terms | Count | FDR | Enriched Genes |
|---|---|---|---|
| hsa00830:Retinol metabolism | 14 | 1.02 × | CYP3A4, CYP2B6, CYP2C9, CYP2C8, ADH1C, CYP26A1, ADH1B, ADH1A, CYP1A2, RDH5, ADH4, AOX1, CYP2A6, RDH16 |
| hsa01100:Metabolic pathways | 64 | 2.85 × | CYP3A4, NAMPT, CNDP1, OGDHL, ADH1C, ADH1B, ADH1A, ASPA, TDO2, MAT1A, ST3GAL6, GSTZ1, HPD, AADAT, ALDH6A1, FBP1, CYP26A1, CYP2E1, CYP1A2, ACADL, TAT, CTH, FOLH1, SDS, SQLE, RRM2, HAO2, BHMT, HSD11B1, CYP2A6, AKR1D1, SLC27A5, XDH, ASS1, CYP2B6, ALDOB, KMO, GLS2, TYMS, ACSL1, ADH4, DHODH, ENO3, DMGDH, ACSL4, CYP2C9, CYP2C8, NAT2, ACMSD, EPHX2, FTCD, IDO2, MAN1C1, PCK1, ACSM3, GBA3, AOX1, AGXT2, CYP4F3, CYP4F2, RDH16, CYP8B1, ACSM5, NNMT |
| hsa04610:Complement and coagulation cascades | 13 | 2.42 × | F11, MBL2, C7, C9, MASP2, C6, F9, C1R, PLG, C8A, C8B, KLKB1, SERPINE1 |
| hsa05204:Chemical carcinogenesis | 13 | 0.001 | CYP3A4, CYP2C9, CYP2C8, NAT2, ADH1C, ADH1B, ADH1A, CYP1A2, CYP2E1, CYP3A43, ADH4, HSD11B1, CYP2A6 |
| hsa00982:Drug metabolism—cytochrome P450 | 12 | 0.002 | CYP3A4, CYP2B6, CYP2C9, CYP2C8, ADH4, AOX1, ADH1C, ADH1B, CYP2A6, ADH1A, CYP2E1, CYP1A2 |
| hsa04110:Cell cycle | 14 | 0.026 | CDC6, CDK1, TTK, CDC20, PTTG1, MCM2, MCM6, CCNB1, CCNE2, CCNB2,MAD2L1, BUB1, BUB1B, CCNA2 |
| hsa00980:Metabolism of xenobiotics by cytochrome P450 | 11 | 0.027 | CYP3A4, CYP2B6, CYP2C9, ADH4, HSD11B1, ADH1C, ADH1B, CYP2A6, ADH1A, CYP2E1, CYP1A2 |
| hsa00350:Tyrosine metabolism | 8 | 0.036 | ADH4, AOX1, ADH1C, ADH1B, GSTZ1, ADH1A, TAT, HPD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dessie, E.Y.; Tu, S.-J.; Chiang, H.-S.; Tsai, J.J.P.; Chang, Y.-S.; Chang, J.-G.; Ng, K.-L. Construction and Validation of a Prognostic Gene-Based Model for Overall Survival Prediction in Hepatocellular Carcinoma Using an Integrated Statistical and Bioinformatic Approach. Int. J. Mol. Sci. 2021, 22, 1632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041632
Dessie EY, Tu S-J, Chiang H-S, Tsai JJP, Chang Y-S, Chang J-G, Ng K-L. Construction and Validation of a Prognostic Gene-Based Model for Overall Survival Prediction in Hepatocellular Carcinoma Using an Integrated Statistical and Bioinformatic Approach. International Journal of Molecular Sciences. 2021; 22(4):1632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041632
Chicago/Turabian StyleDessie, Eskezeia Yihunie, Siang-Jyun Tu, Hui-Shan Chiang, Jeffrey J.P. Tsai, Ya-Sian Chang, Jan-Gowth Chang, and Ka-Lok Ng. 2021. "Construction and Validation of a Prognostic Gene-Based Model for Overall Survival Prediction in Hepatocellular Carcinoma Using an Integrated Statistical and Bioinformatic Approach" International Journal of Molecular Sciences 22, no. 4: 1632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041632