
Structural Characterization of Daunomycin-Peptide Conjugates by Various Tandem Mass Spectrometric Techniques

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
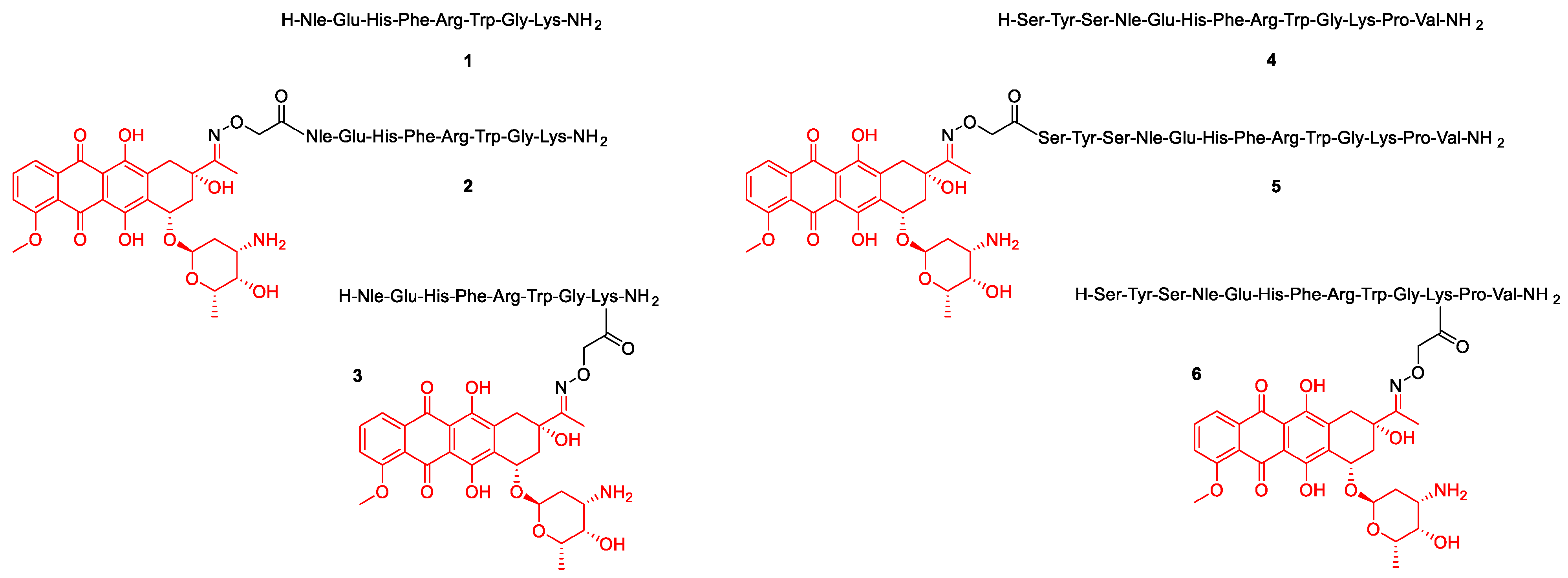
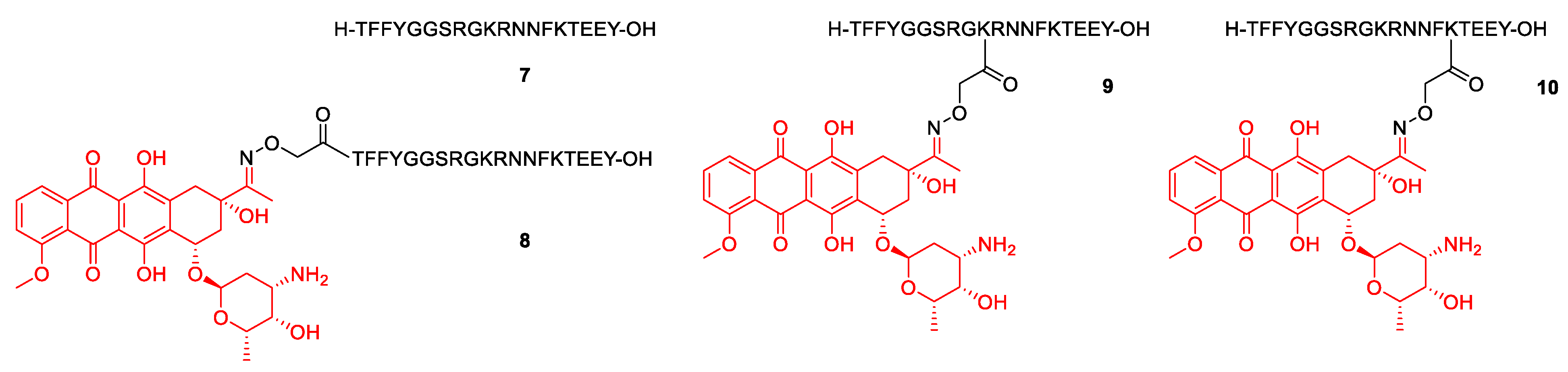
2.1. Synthesis
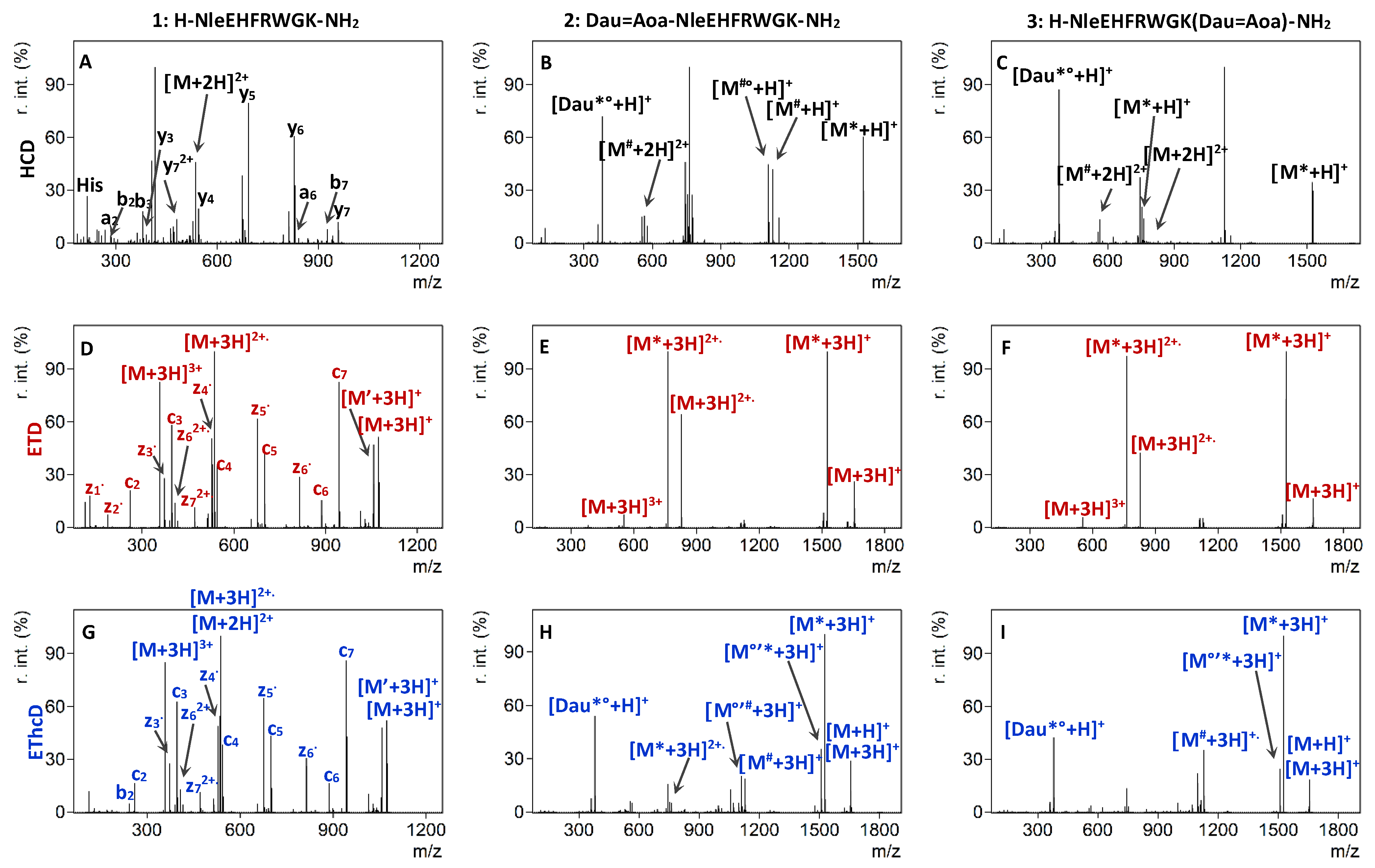
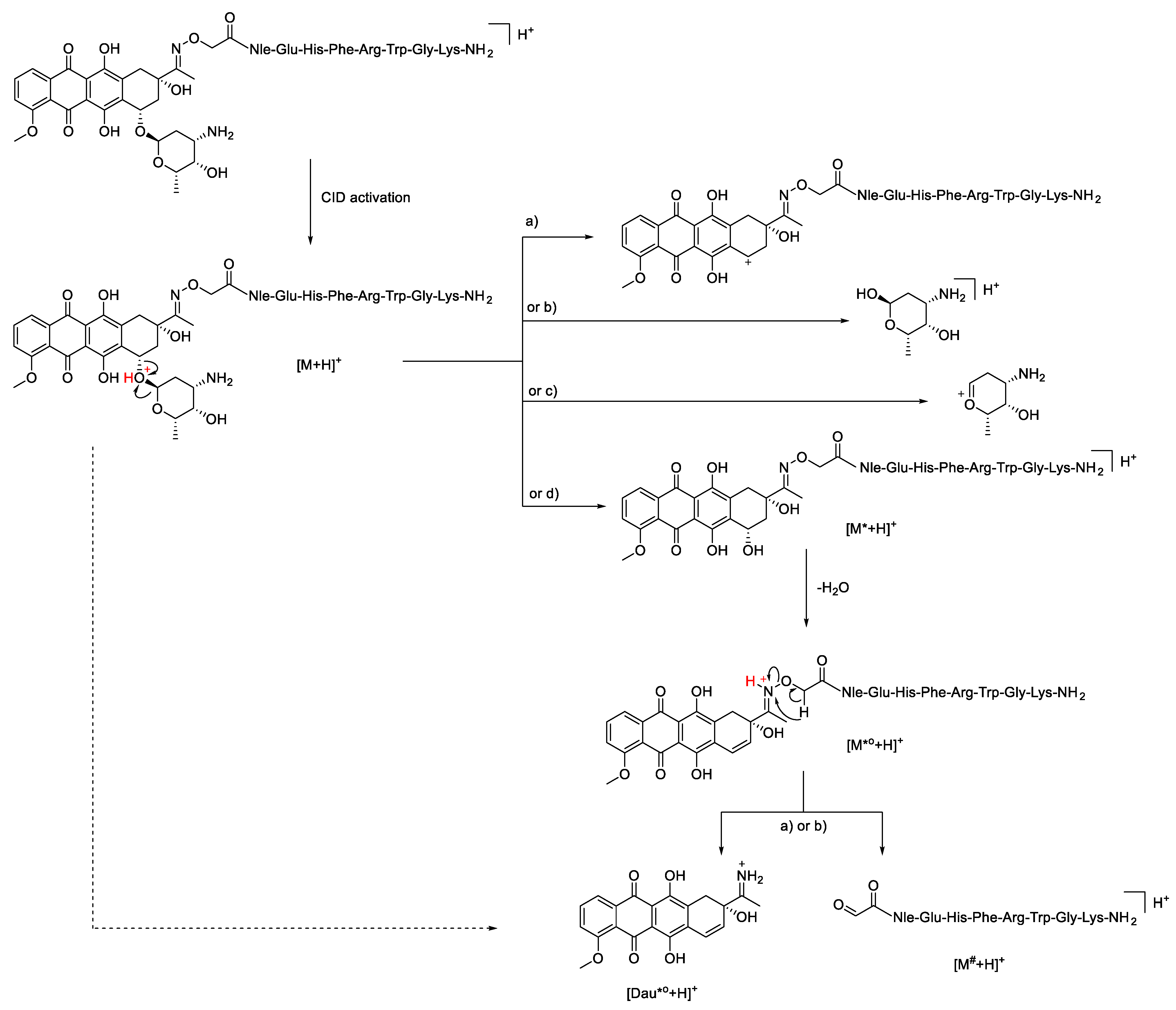
2.2. HCD Experiments
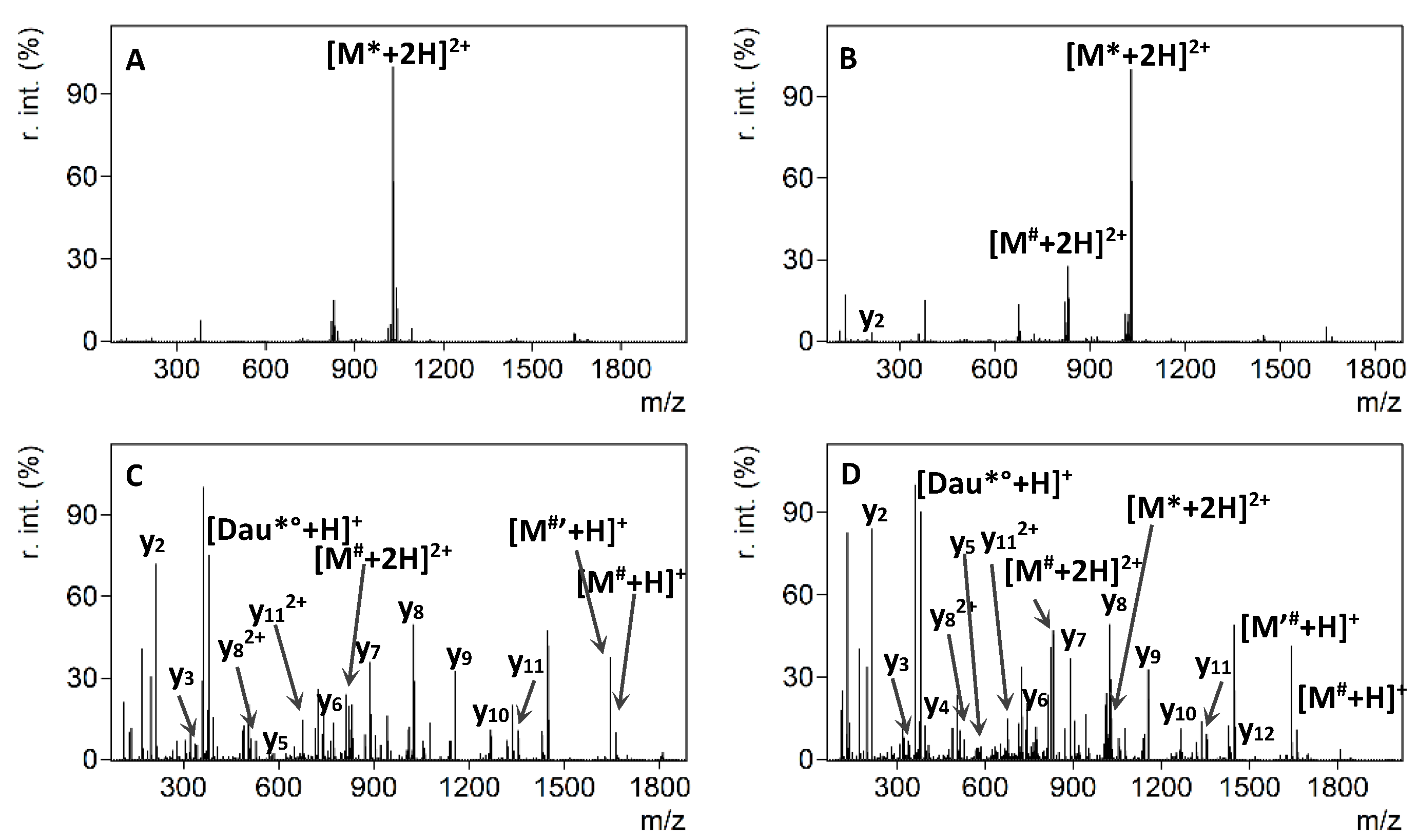
2.3. ETD and EThcD Experiments
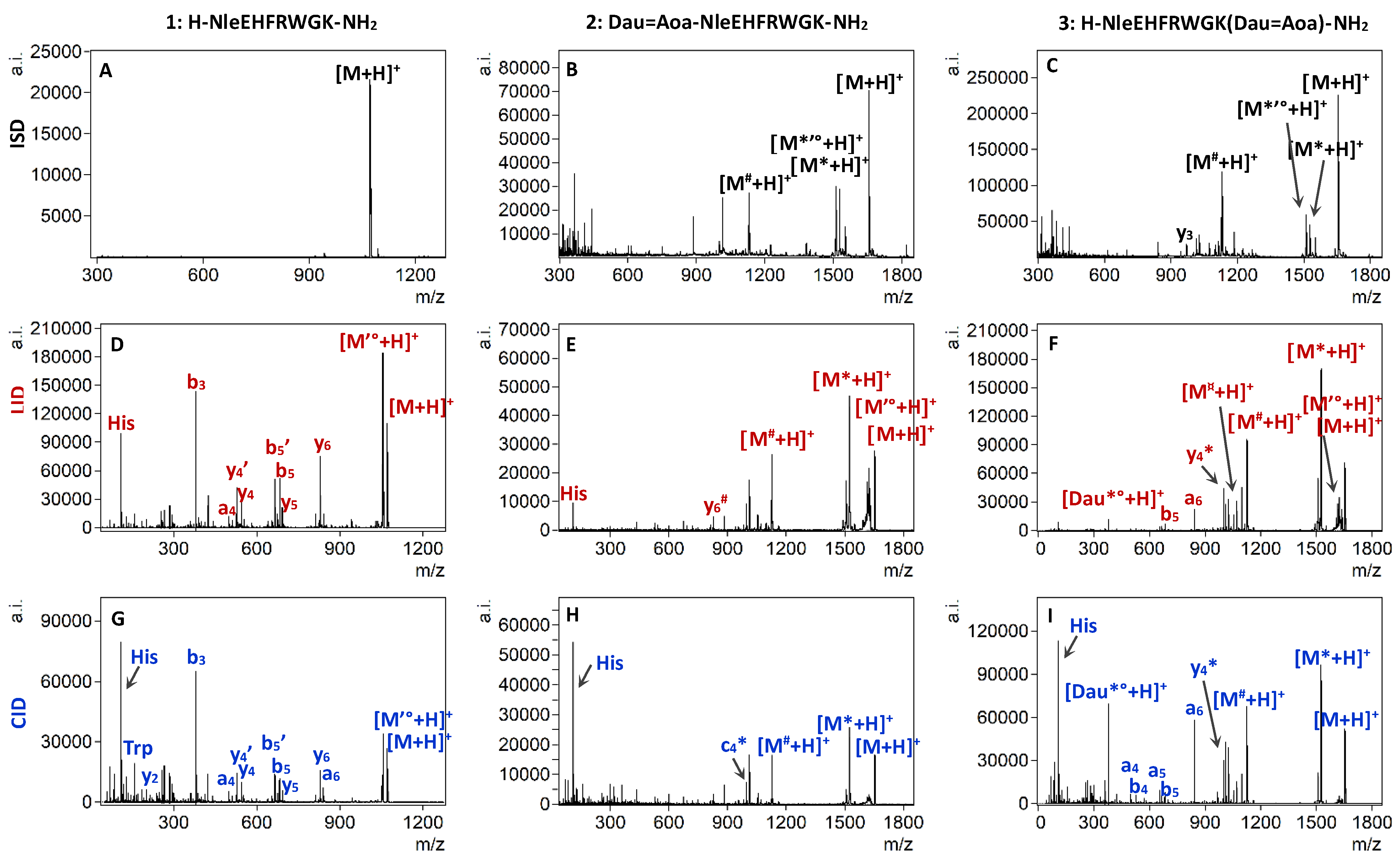
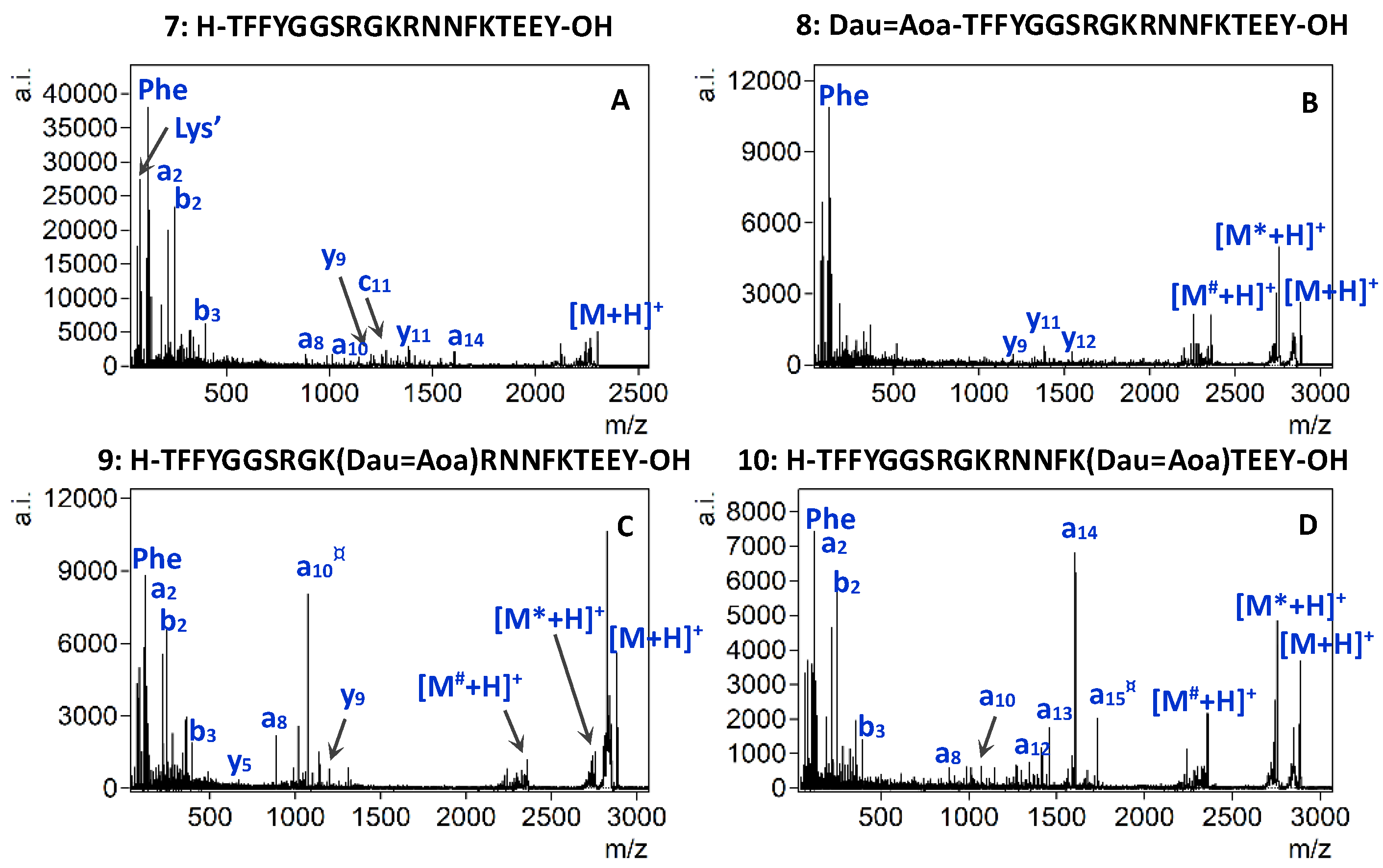
2.4. MALDI-TOF/TOF Experiments
3. Materials and Methods
3.1. Synthesis of the Daunomycin-Peptide Conjugates
3.2. Instruments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the design principles of peptide–drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, V.; Laakkonen, P. Seek & Destroy, use of targeting peptides for cancer detection and drug delivery. Bioorg. Med. Chem. 2018, 26, 2797–2806. [Google Scholar] [PubMed]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-targeted delivery systems based on peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandini, R.; Suli-Vargha, H.; Libert, A.; Loir, B.; Botyánszki, J.; Medzihradszky, K.; Ghanem, G. Receptor-mediated cyotoxicity of α-MSH fragments containing melphalan in a human melanoma cell line. Int. J. Cancer 1994, 56, 129–133. [Google Scholar] [CrossRef]
- Castrucci, A.M.L.; Hadley, M.E.; Sawyer, T.K.; Wilkes, B.C.; Al-Obeidi, F.; Staples, D.J.; De Vaux, A.E.D.E.; Dym, O.; Hintz, M.F.; Riehm, J.P.; et al. α-Melanotropin: The Minimal Active Sequence in the Lizard Skin Bioassay. Gen. Comp. Endocrinol. 1989, 163, 157–163. [Google Scholar] [CrossRef]
- Orbán, E.; Mezo, G.; Schlage, P.; Csík, G.; Kulić, Ž.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin-GnRH-III bioconjugates and DNA-binding properties of daunorubicin-amino acid metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef]
- Miklán, Z.; Orbán, E.; Csík, G.; Schlosser, G.; Magyar, A.; Hudecz, F. New daunomycin-oligoarginine conjugates: Synthesis, characterization, and effect on human leukemia and human hepatoma cells. Biopolymers 2009, 92, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Orbán, E.; Manea, M.; Marquadt, A.; Bánóczi, Z.; Csik, G.; Fellinger, E.; Bõsze, S.; Hudecz, F. A new daunomycin-peptide conjugate: Synthesis, characterization and the effect on the protein expression profile of HL-60 cells in vitro. Bioconjug. Chem. 2011, 22, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Kiss, K.; Biri-Kovács, B.; Szabó, R.; Ranđelović, I.; Enyedi, K.N.; Schlosser, G.; Orosz, Á.; Kapuvári, B.; Tóvári, J.; Mező, G. Sequence modification of heptapeptide selected by phage display as homing device for HT-29 colon cancer cells to improve the anti-tumour activity of drug delivery systems. Eur. J. Med. Chem. 2019, 176, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randelovic, I.; Schuster, S.; Kapuvári, B.; Fossati, G.; Steinkühler, C.; Mezo, G.; Tóvári, J. Improved in vivo anti-tumor and anti-metastatic effect of GnRH-III-daunorubicin analogs on colorectal and breast carcinoma bearing mice. Int. J. Mol. Sci. 2019, 20, 4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripodi, A.A.P.; Ranđelović, I.; Biri-Kovács, B.; Szeder, B.; Mező, G.; Tóvári, J. In Vivo Tumor Growth Inhibition and Antiangiogenic Effect of Cyclic NGR Peptide-Daunorubicin Conjugates Developed for Targeted Drug Delivery. Pathol. Oncol. Res. 2019, 26, 1879–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkowitz, S.A.; Engen, J.R.; Mazzeo, J.R.; Jones, G.B. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat. Rev. Drug Discov. 2012, 11, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Pethő, L.; Mező, G.; Schlosser, G. Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules 2019, 24, 2981. [Google Scholar] [CrossRef] [Green Version]
- Al-Majidi, M.; Szabó, D.; Dókus, L.; Steckel, A.; Mező, G.; Schlosser, G. Energy-resolved HCD fragmentation of daunorubicin-peptide conjugates. J. Mass Spectrom. 2020, 55, e4641. [Google Scholar] [CrossRef] [PubMed]
- Sleno, L.; Campagna-Slater, V.; Volmer, D.A. Dissociation reactions of protonated anthracycline antibiotics following electrospray ionization-tandem mass spectrometry. Int. J. Mass Spectrom. 2006, 255–256, 130–138. [Google Scholar] [CrossRef]
- Tripodi, A.A.P.; Tóth, S.; Enyedi, K.N.; Schlosser, G.; Szakács, G.; Mező, G. Development of novel cyclic NGR peptide—Daunomycin conjugates with dual targeting property. Beilstein J. Org. Chem. 2018, 14, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dókus, L.E.; Lajkó, E.; Ranđelović, I.; Mező, D.; Schlosser, G.; Kőhidai, L.; Tóvári, J.; Mező, G. Phage Display-Based Homing Peptide-Daunomycin Conjugates for Selective Drug Targeting to PANC-1 Pancreatic Cancer. Pharmaceutics 2020, 12, 576. [Google Scholar] [CrossRef] [PubMed]
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frese, C.K.; Altelaar, A.F.M.; Van Den Toorn, H.; Nolting, D.; Griep-Raming, J.; Heck, A.J.R.; Mohammed, S. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 2012, 84, 9668–9673. [Google Scholar] [CrossRef] [PubMed]
- Medzihradszky, K.F.; Campbell, J.M.; Baldwin, M.A.; Falick, A.M.; Juhasz, P.; Vestal, M.L.; Burlingame, A.L. The Characteristics of Peptide Collision-Induced Dissociation Using a High-Performance MALDI-TOF/TOF Tandem Mass Spectrometer. Anal. Chem. 2000, 72, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Manea, M.; Orbán, E.; Csámpai, A.; Bősze, S.; Szabó, R.; Tejeda, M.; Gaál, D.; Kapuvári, B.; Przybylski, M.; et al. Development of an oxime bond containing daunorubicin-gonadotropin-releasing hormone-III conjugate as a potential anticancer drug. Bioconjug. Chem. 2009, 20, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Steckel, A.; Schlosser, G. An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation. Molecules 2019, 24, 611. [Google Scholar] [CrossRef] [Green Version]
- Monneret, C.; Sellier, N. Desorption chemical ionization mass spectrometry of anthracyclines and of trisaccharides related to aclacinomycin A and marcellomycin. Biol. Mass Spectrom. 1986, 13, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Dongré, A.R.; Jones, J.L.; Somogyi, Á.; Wysocki, V.H. Influence of Peptide Composition, Gas-Phase Basicity, and Chemical Modification on Fragmentation Efficiency: Evidence for the Mobile Proton Model. J. Am. Chem. Soc. 1996, 118, 8365–8374. [Google Scholar] [CrossRef]
- Good, D.M.; Wirtala, M.; McAlister, G.C.; Coon, J.J. Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteom. 2007, 6, 1942–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikesh, L.M.; Ueberheide, B.; Chi, A.; Coon, J.J.; Syka, J.E.P.; Shabanowitz, J.; Hunt, D.F. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 2006, 1764, 1811–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, C.H.; Chung, C.K.; Yin, S.; Ramachandran, P.; Loo, J.A.; Beauchamp, J.L. Probing the mechanism of electron capture and electron transfer dissociation using tags with variable electron affinity. J. Am. Chem. Soc. 2009, 131, 5444–5459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.W.; Mikhailov, V.A.; Iniesta, J.; Cooper, H.J. Electron Capture Dissociation Mass Spectrometry of Tyrosine Nitrated Peptides. J. Am. Soc. Mass Spectrom. 2010, 21, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Prokai, L. Conversion of 3-nitrotyrosine to 3-aminotyrosine residues facilitates mapping of tyrosine nitration in proteins by electrospray ionization-tandem mass spectrometry using electron capture dissociation. J. Mass Spectrom. 2012, 47, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Heinis, T.; Chowdhury, S.; Scott, S.L.; Kebarle, P. Electron affinities of benzo-, naphtho-, and anthraquinones determined from gas-phase equilibria measurements. J. Am. Chem. Soc. 1988, 110, 400–407. [Google Scholar] [CrossRef]
- Smith, R.G. Characterization of Anthracycline Antibiotics by Desorption Chemical Ionization Mass Spectrometry. Anal. Chem. 1982, 54, 2006–2008. [Google Scholar] [CrossRef]
- Bachur, N.R. Anthracycline antibiotic pharmacology and metabolism. Cancer Treat. Rep. 1979, 63, 817–820. [Google Scholar] [PubMed]
- Steckel, A.; Uray, K.; Kalló, G.; Csosz, É.; Schlosser, G. Investigation of Neutral Losses and the Citrulline Effect for Modified H4 N-Terminal Pentapeptides. J. Am. Soc. Mass Spectrom. 2020, 31, 565–573. [Google Scholar] [CrossRef]
- Yu, Q.; Wang, B.; Chen, Z.; Urabe, G.; Glover, M.S.; Shi, X.; Guo, L.; Kent, K.C.; Li, L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Frese, C.K.; Zhou, H.; Taus, T.; Altelaar, A.F.M.; Mechtler, K.; Heck, A.J.R.; Mohammed, S. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Yergey, A.L.; Coorssen, J.R.; Backlund, P.S.; Blank, P.S.; Humphrey, G.A.; Zimmerberg, J.; Campbell, J.M.; Vestal, M.L. De Novo Sequencing of Peptides using MALDI/TOF-TOF. J. Am. Soc. Mass Spectrom. 2002, 13, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Suckau, D.; Resemann, A.; Schuerenberg, M.; Hufnagel, P.; Franzen, J.; Holle, A. A novel MALDI LIFT-TOF/TOF mass spectrometer for proteomics. Anal. Bioanal. Chem. 2003, 376, 952–965. [Google Scholar] [CrossRef]
- Demeure, K.; Gabelica, V.; De Pauw, E.A. New advances in the understanding of the in-source decay fragmentation of peptides in MALDI-TOF-MS. J. Am. Soc. Mass Spectrom. 2010, 21, 1906–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köcher, T.; Engström, Å.; Zubarev, R.A. Fragmentation of Peptides in MALDI In-Source Decay Mediated by Hydrogen Radicals. Anal. Chem. 2005, 77, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Macht, M.; Asperger, A.; Deininger, S.O. Comparison of laser-induced dissociation and high-energy collision-induced dissociation using matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI-TOF/TOF) for peptide and protein identification. Rapid Commun. Mass Spectrom. 2004, 18, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Khatun, J.; Ramkissoon, K.; Giddings, M.C. Fragmentation characteristics of collision-induced dissociation in MALDI TOF/TOF mass spectrometry. Anal. Chem. 2007, 79, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Identification and Design of Peptides as a New Drug Delivery System for the Brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohalm, M.; Hassman, M.; Kosata, B.; Kodicek, M. mMass data miner: An open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | m/z Calc. for [M+2H]2+ | m/z Meas. 1 for [M+2H]2+ | Deviation (ppm) | Rt (min) 2 | |
|---|---|---|---|---|---|
| 1 | H-NleEHFRWGK-NH2 | 536.2960 | 536.2954 | 1.1 | 6.0 |
| 2 | Dau = Aoa-NleEHFRWGK-NH2 | 827.3884 | 827.3876 | 1.0 | 8.4 |
| 3 | H-NleEHFRWGK(Dau = Aoa)-NH2 | 827.3884 | 827.3875 | 1.1 | 8.1 |
| 4 | H-SYSNleEHFRWGKPV-NH2 | 802.9202 | 802.9192 | 1.2 | 7.2 |
| 5 | Dau = Aoa-SYSNleEHFRWGKPV-NH2 | 1094.0127 | 1094.0114 | 1.2 | 8.6 |
| 6 | H-SYSNleEHFRWGK(Dau = Aoa)PV-NH2 | 1094.0127 | 1094.0115 | 1.1 | 8.6 |
| Compound | z | Low E HCD (NCE ≤ 20%) | High E HCD (NCE ≥ 25%) | ETD | EThcD |
|---|---|---|---|---|---|
| 1; (4) | 2 | +; (−) | + +; (+ + +) | + + | + + a |
| 3 | + + +; (+ +) | + + +; (+ +) | + + +; (+ +) | + + + (+ +) a | |
| 2; (5) Dau at N-terminus | 2 | − | + + | − | +; (+ +) b |
| 3 | +; (−) | + + | − | −; (+) b | |
| 3; (6) Dau at Lys | 2 | − | +; (+ +) | − | − c |
| 3 | − | +; (+ +) | − | − d |
| Compound | ISD | LID | CID |
|---|---|---|---|
| 1; (4) | − | + +; (+ + +) | + + +; (+ +) |
| 2; (5) Dau at N-terminus | − | + | + |
| 3; (6) Dau at Lys | − | + +; (+) | + +; (+) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borbély, A.; Pethő, L.; Szabó, I.; Al-Majidi, M.; Steckel, A.; Nagy, T.; Kéki, S.; Kalló, G.; Csősz, É.; Mező, G.; et al. Structural Characterization of Daunomycin-Peptide Conjugates by Various Tandem Mass Spectrometric Techniques. Int. J. Mol. Sci. 2021, 22, 1648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041648
Borbély A, Pethő L, Szabó I, Al-Majidi M, Steckel A, Nagy T, Kéki S, Kalló G, Csősz É, Mező G, et al. Structural Characterization of Daunomycin-Peptide Conjugates by Various Tandem Mass Spectrometric Techniques. International Journal of Molecular Sciences. 2021; 22(4):1648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041648
Chicago/Turabian StyleBorbély, Adina, Lilla Pethő, Ildikó Szabó, Mohammed Al-Majidi, Arnold Steckel, Tibor Nagy, Sándor Kéki, Gergő Kalló, Éva Csősz, Gábor Mező, and et al. 2021. "Structural Characterization of Daunomycin-Peptide Conjugates by Various Tandem Mass Spectrometric Techniques" International Journal of Molecular Sciences 22, no. 4: 1648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041648