
The Aryl Hydrocarbon Receptor Undergoes Chaperone-Mediated Autophagy in Triple-Negative Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
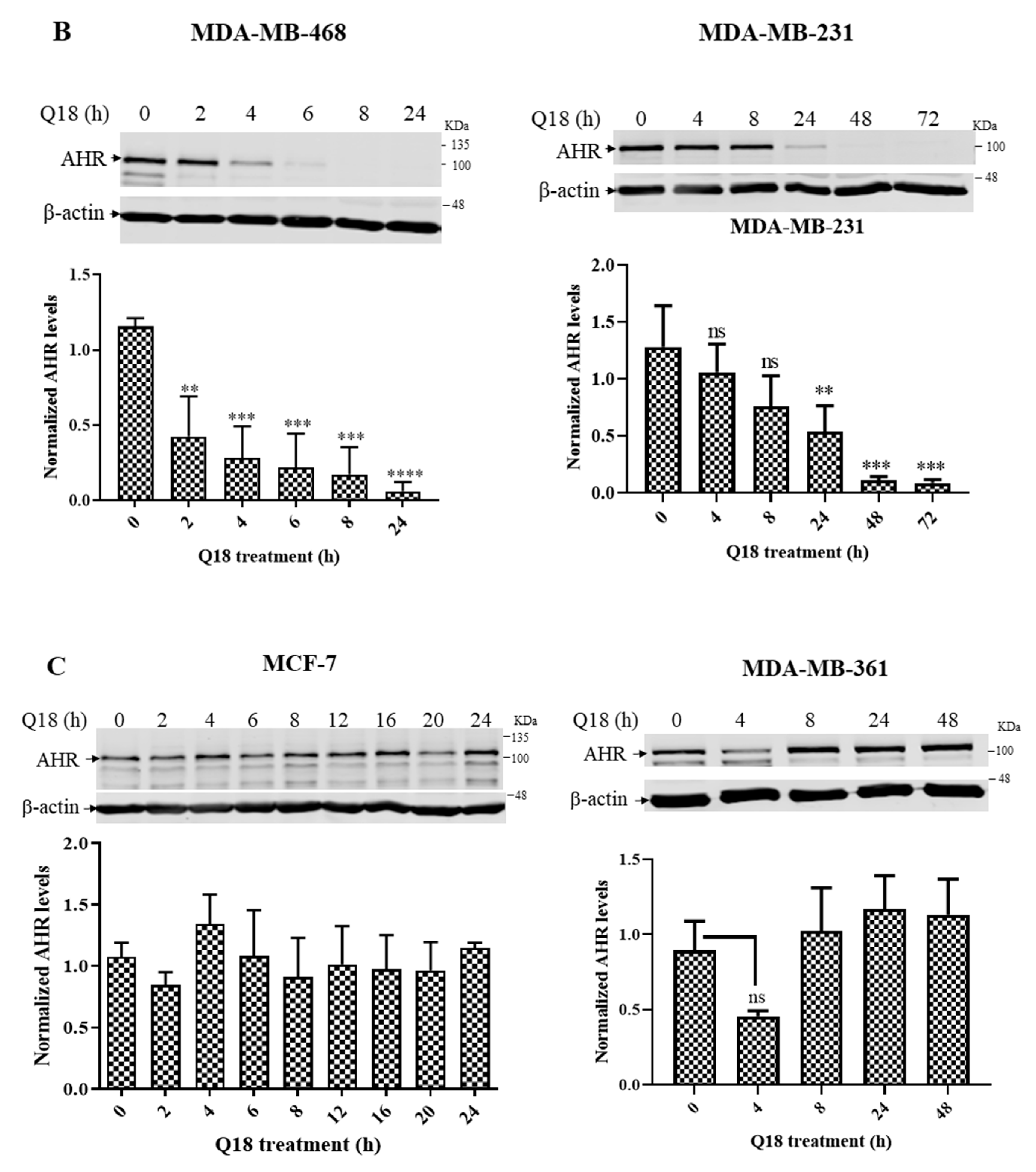
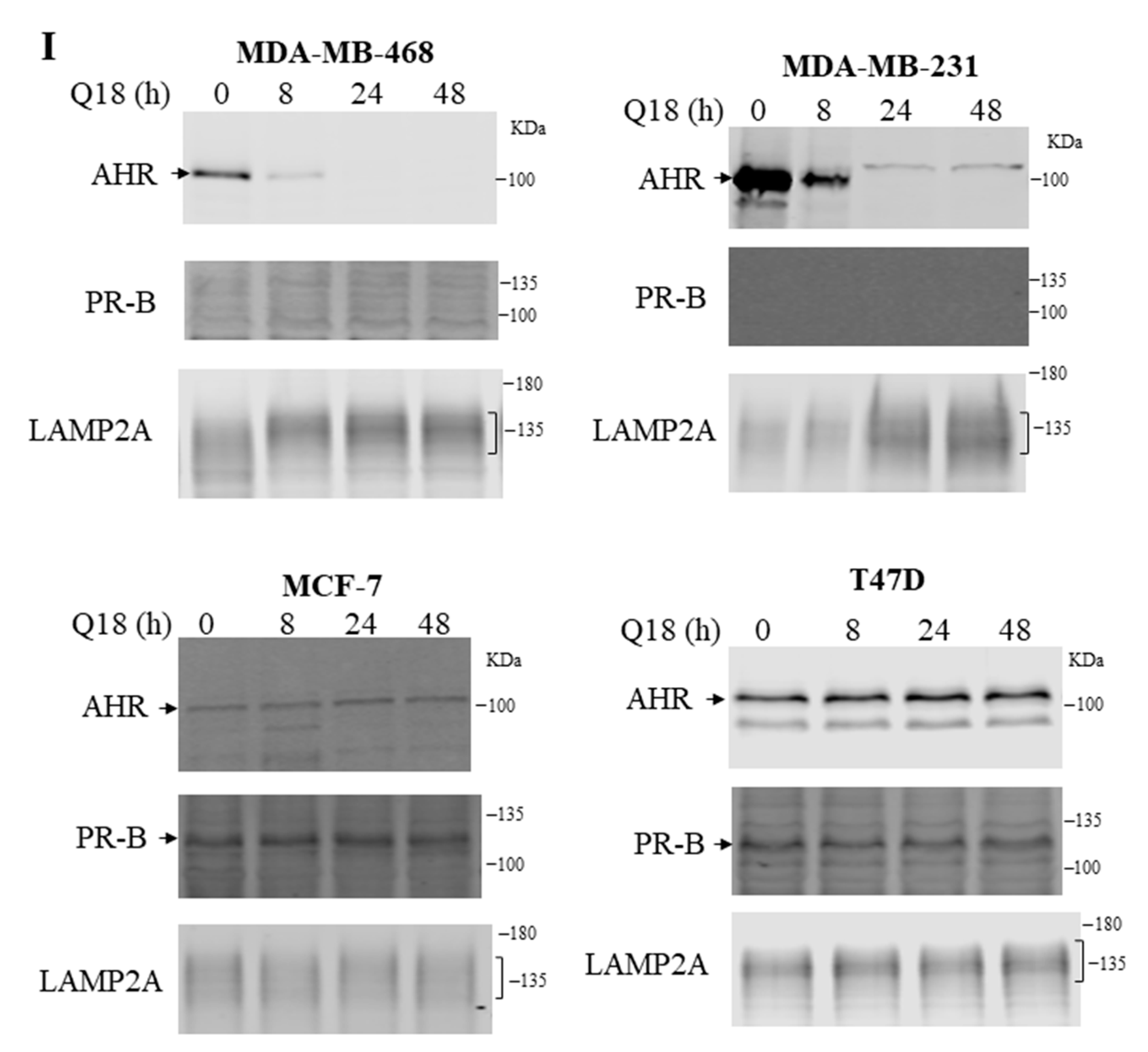
2.1. Suppression of the AHR Protein Levels in Triple-Negative, but Not in Non-Triple-Negative, Breast Cancer Cells by Q18
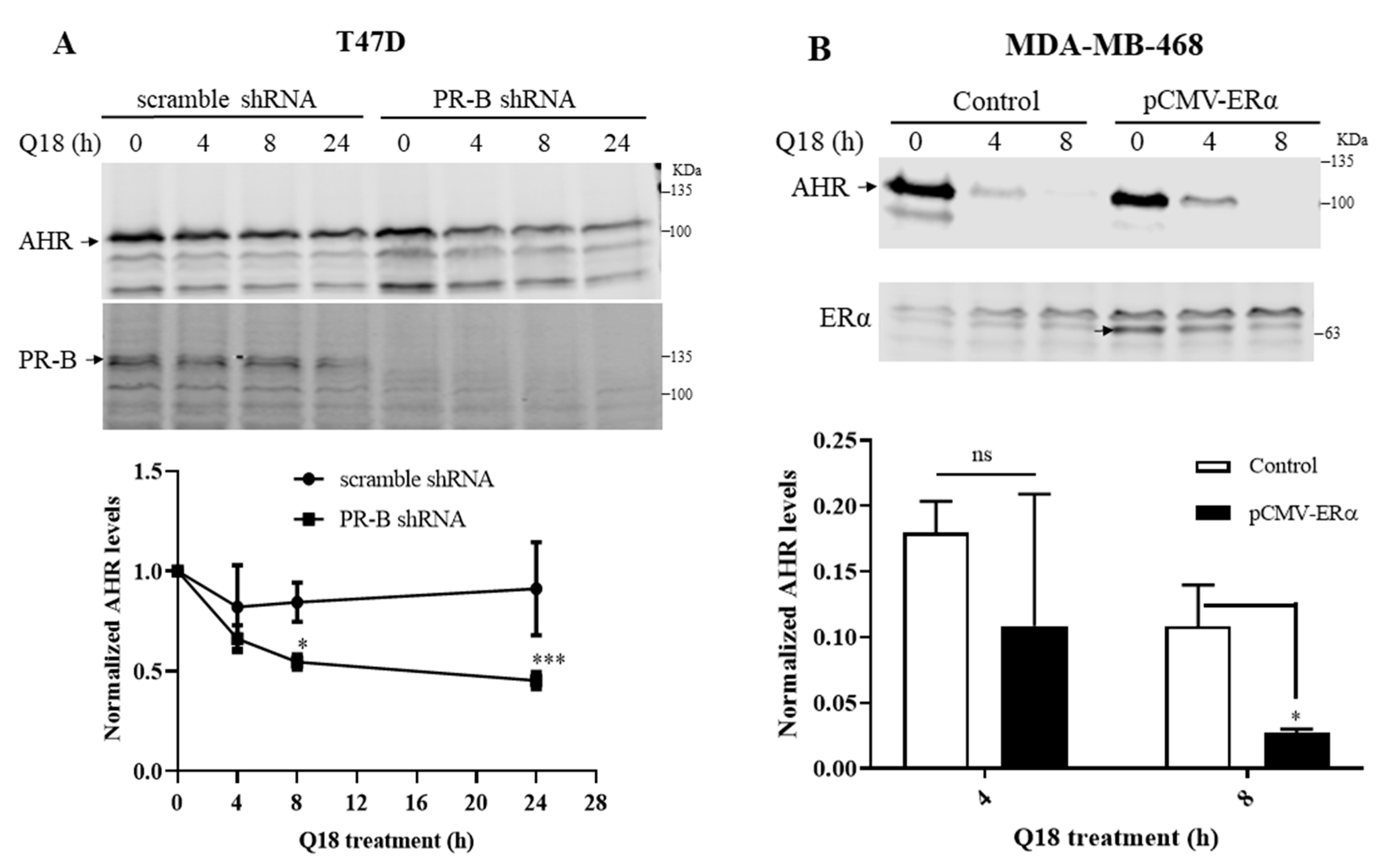
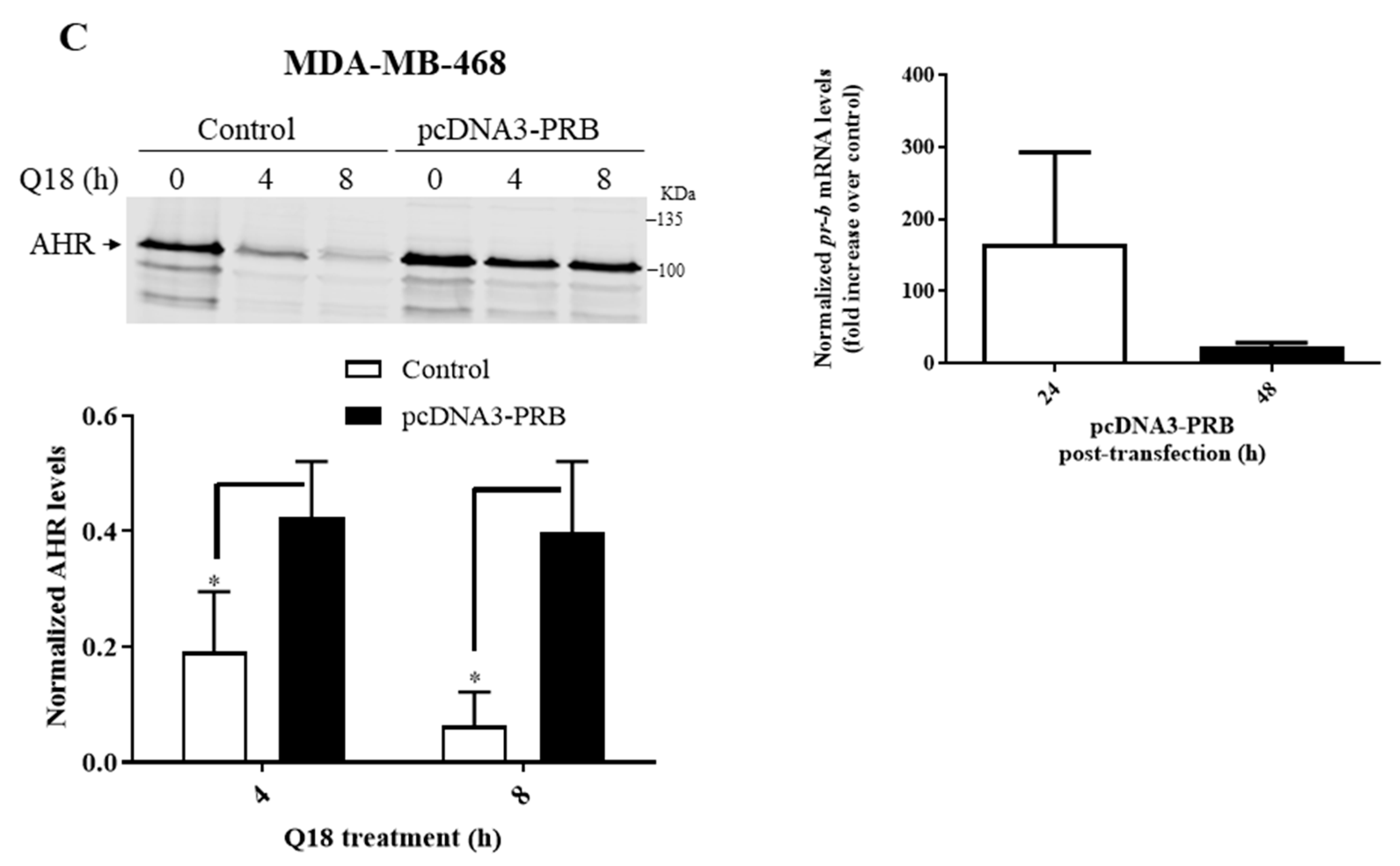
2.2. Degradation of AHR in Triple-Negative Breast Cancer Cells Is Dependent on Progesterone Receptor but Not Estrogen Receptor
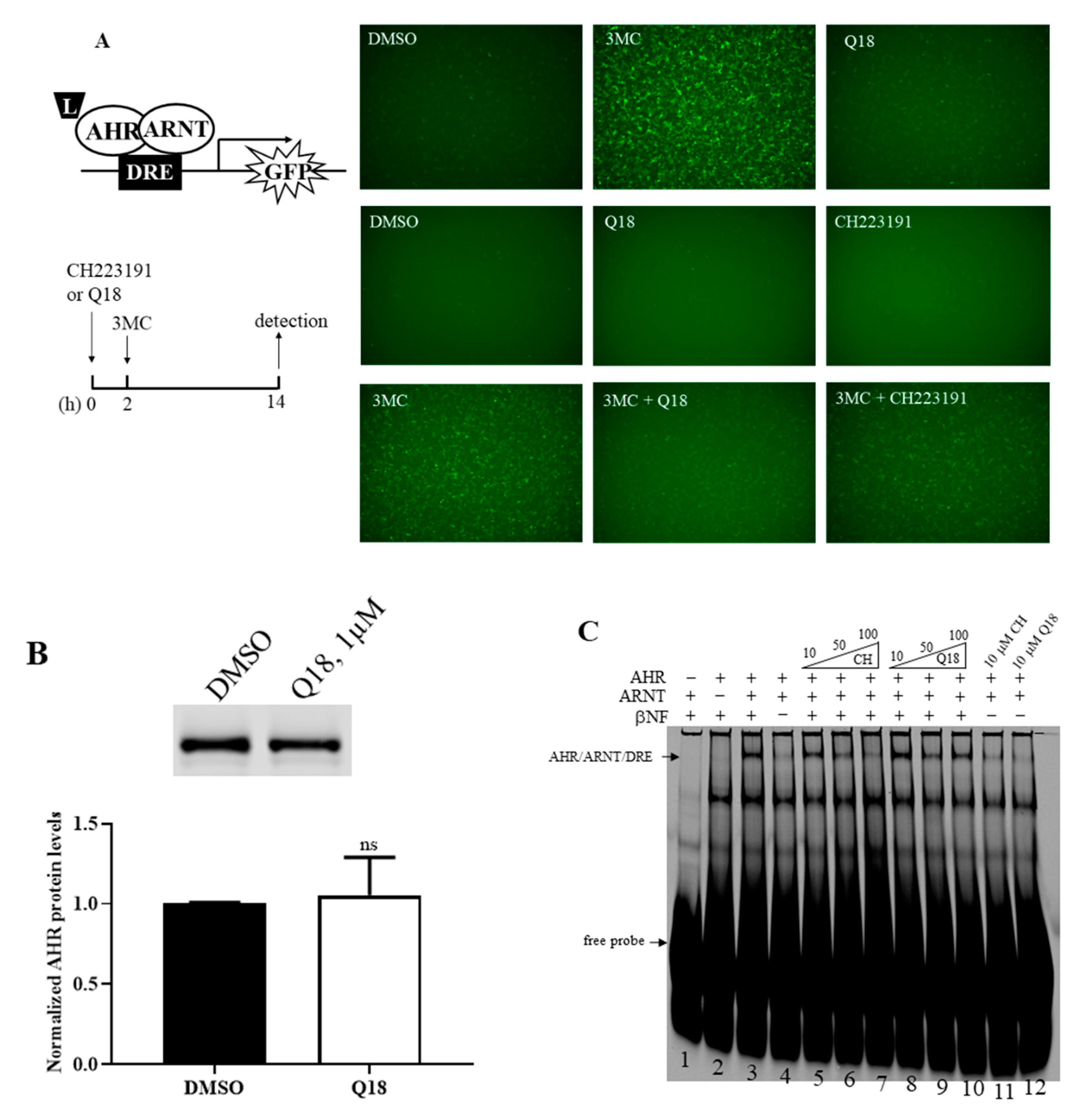
2.3. Q18 is an AHR Antagonist
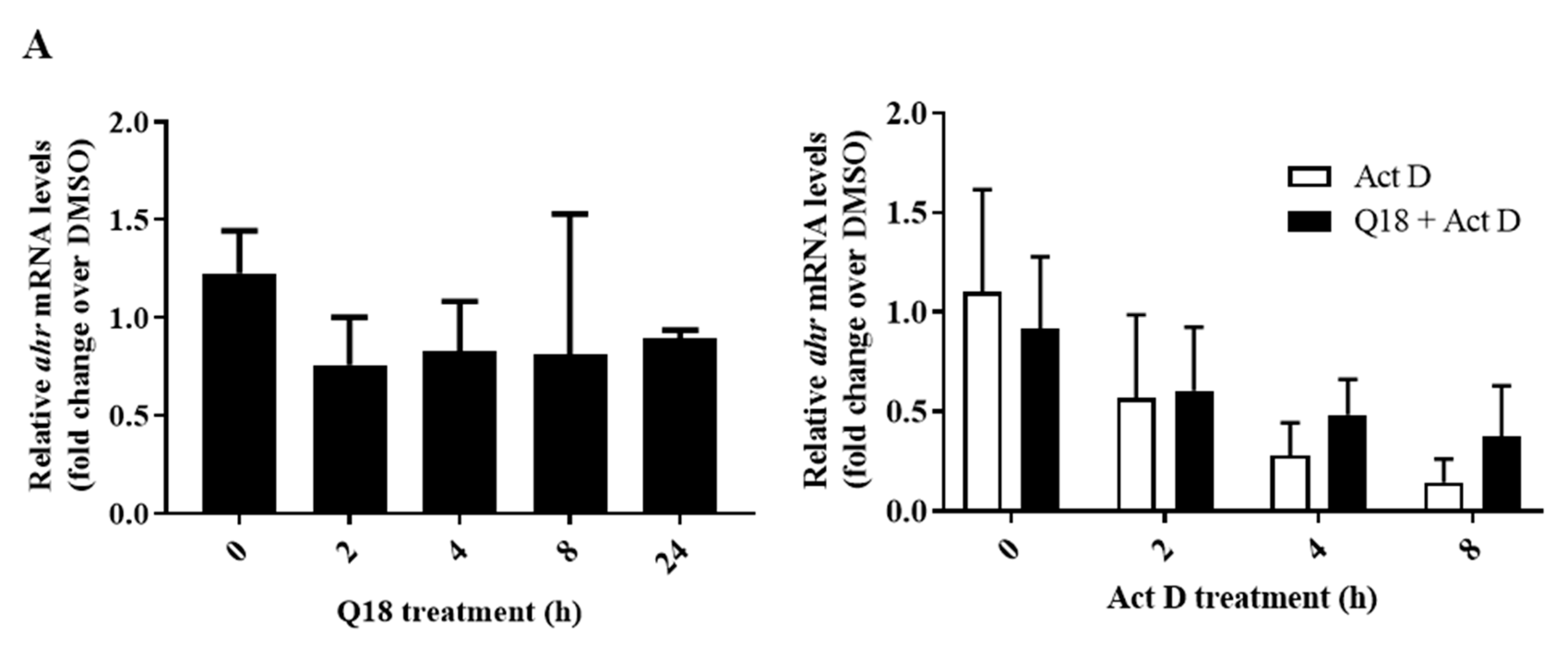
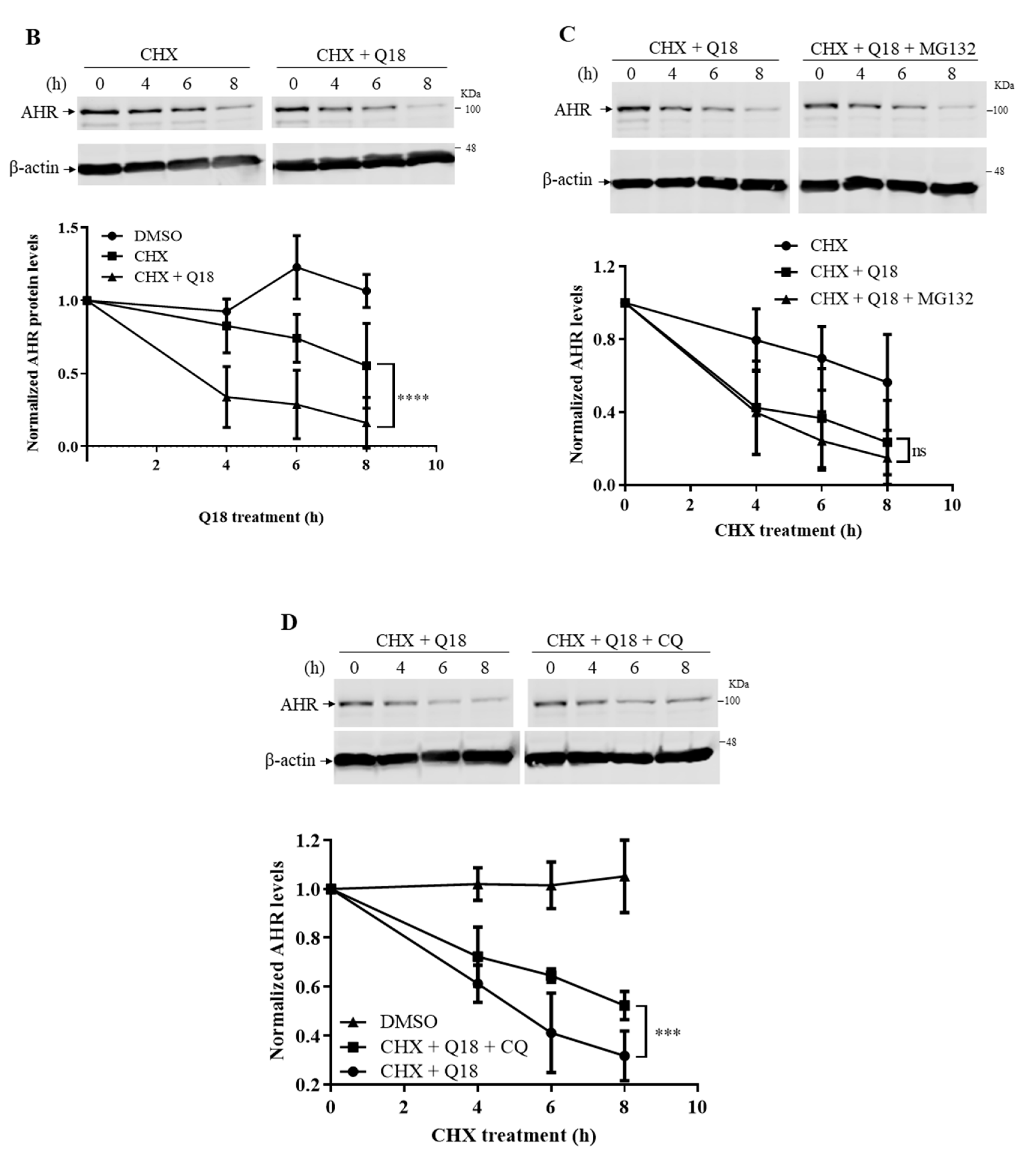
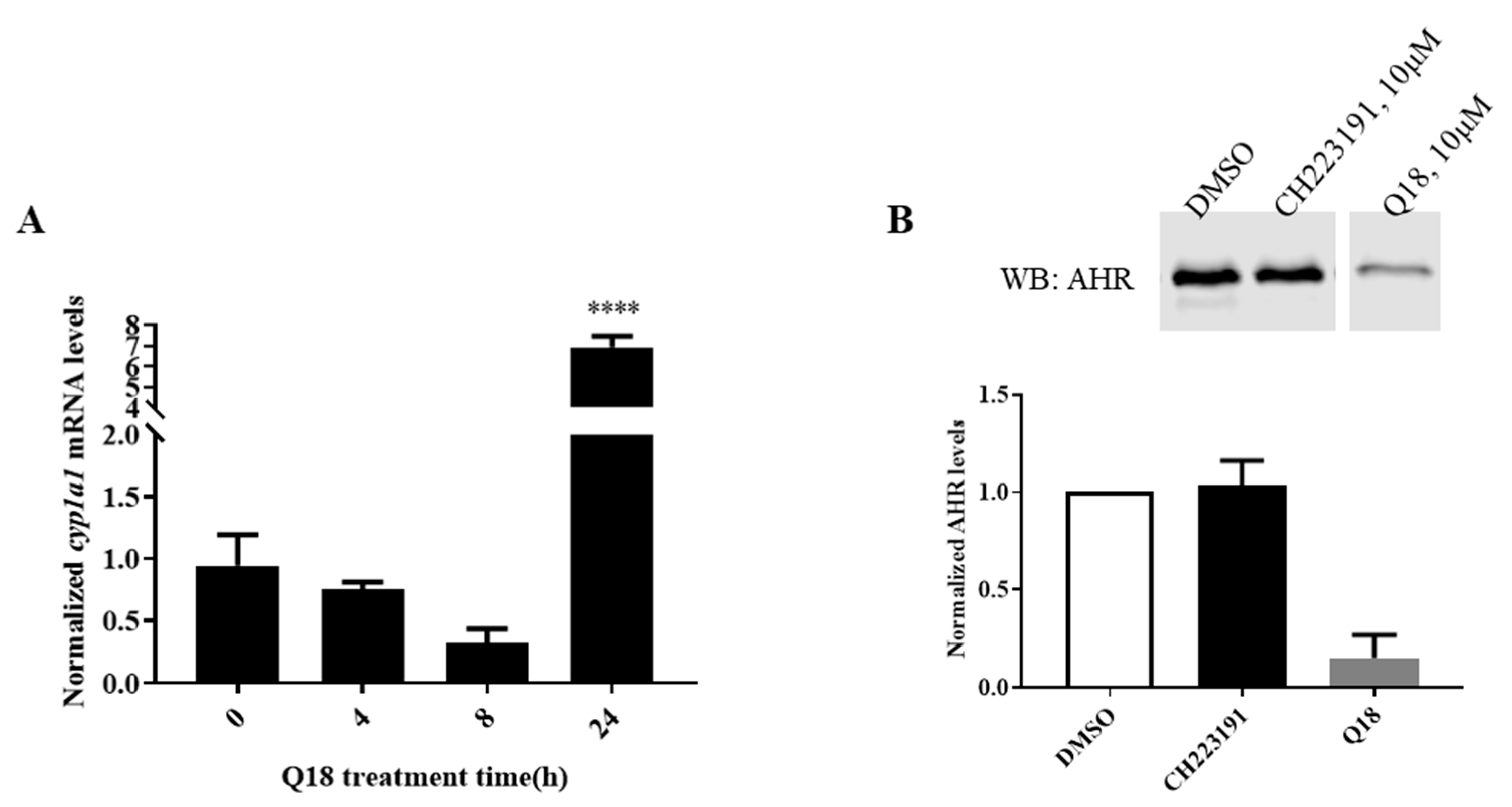
2.4. Q18 Promotes the Autophagy-Mediated AHR Protein Degradation and Does Not Affect the AHR Transcript Levels
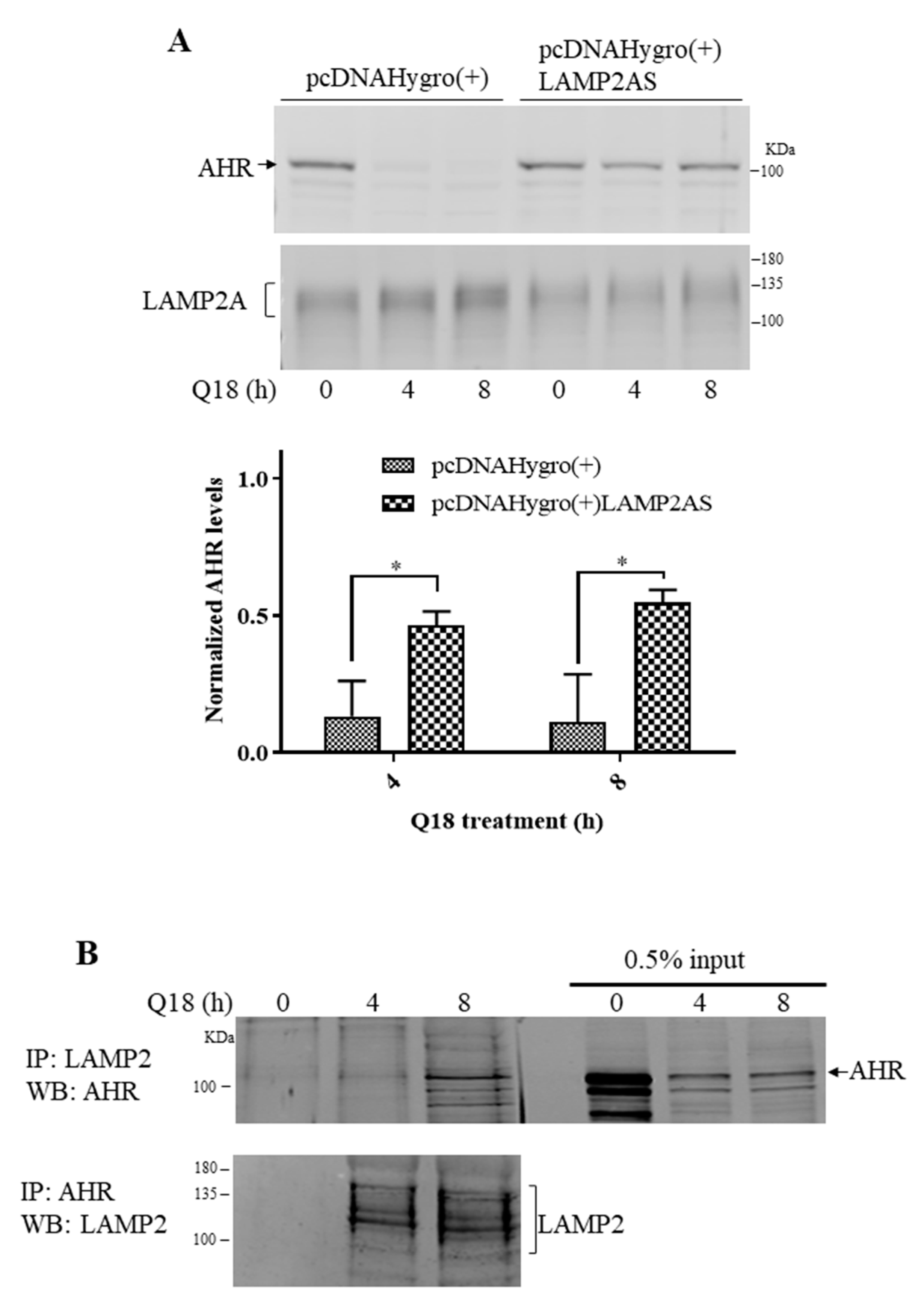
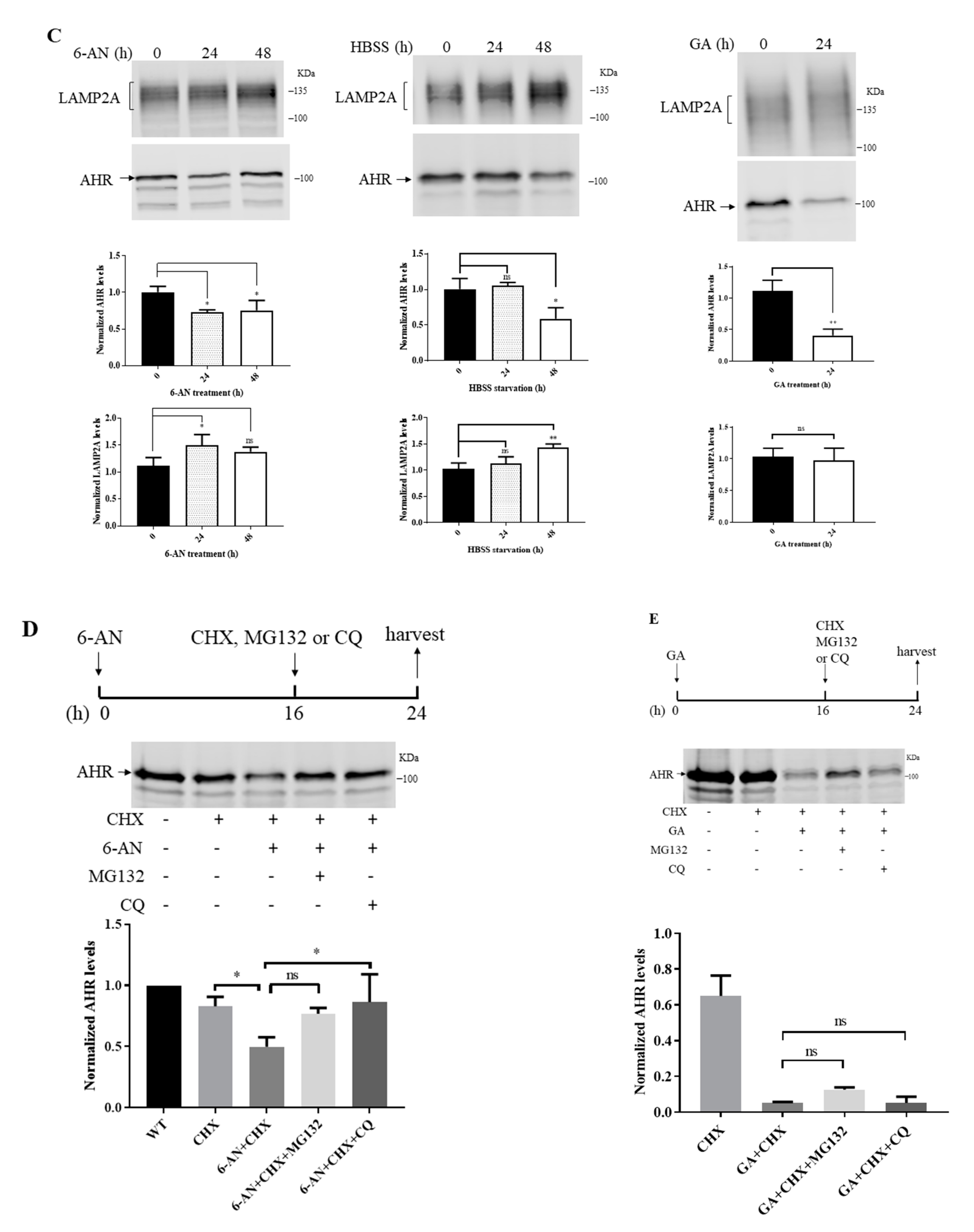
2.5. Degradation of AHR in Triple-Negative Breast Cancer Cells Is LAMP2A-Mediated
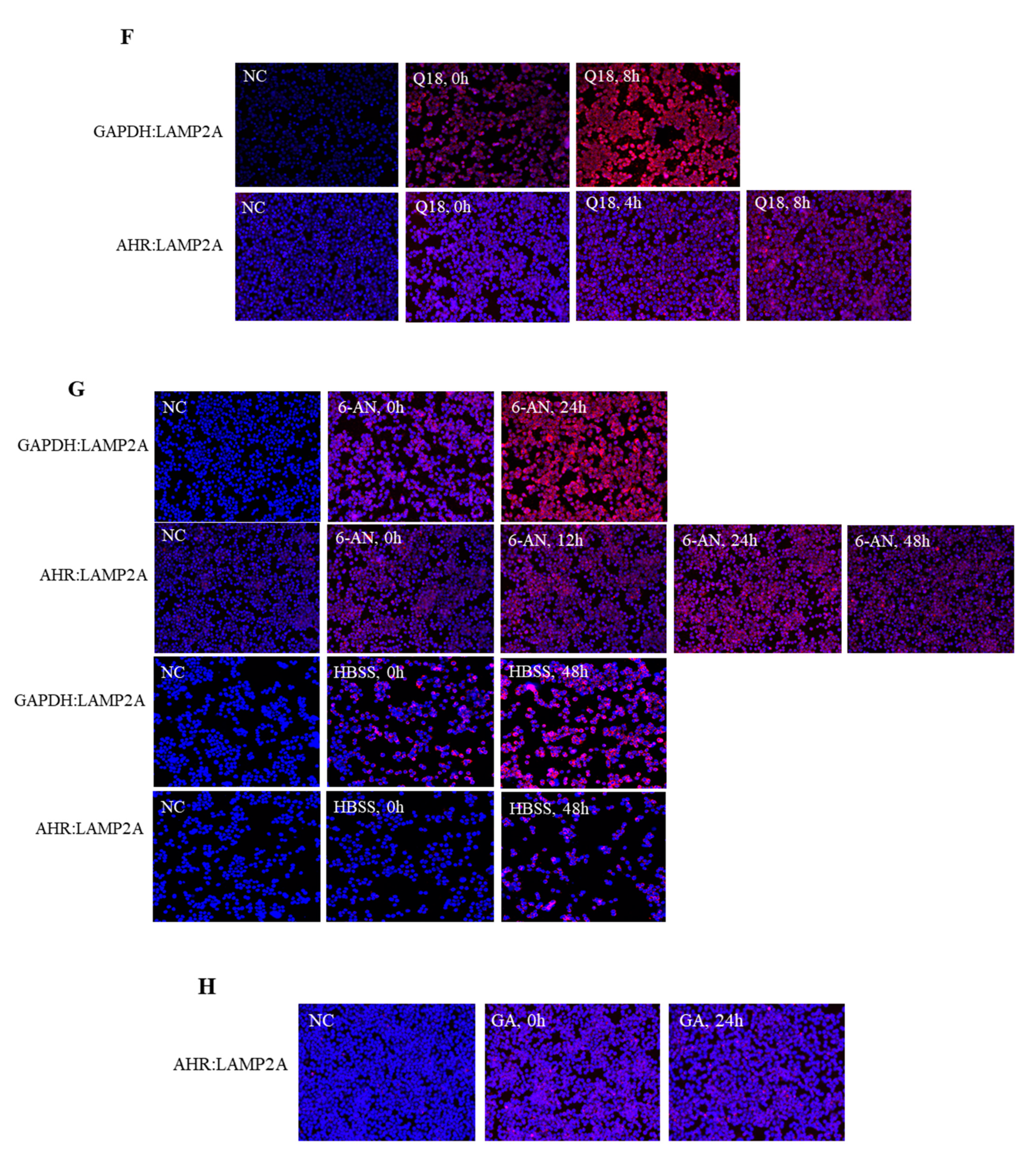
2.6. Treatment of Q18, 6-AN or Starvation in MDA-MB-468 Cells Causes Interaction between AHR and LAMP2A in Proximity Ligation Assay
2.7. Treatment of Q18 Increases the LAMP2A Expression in TNBC but Not in Non-TNBC Cells
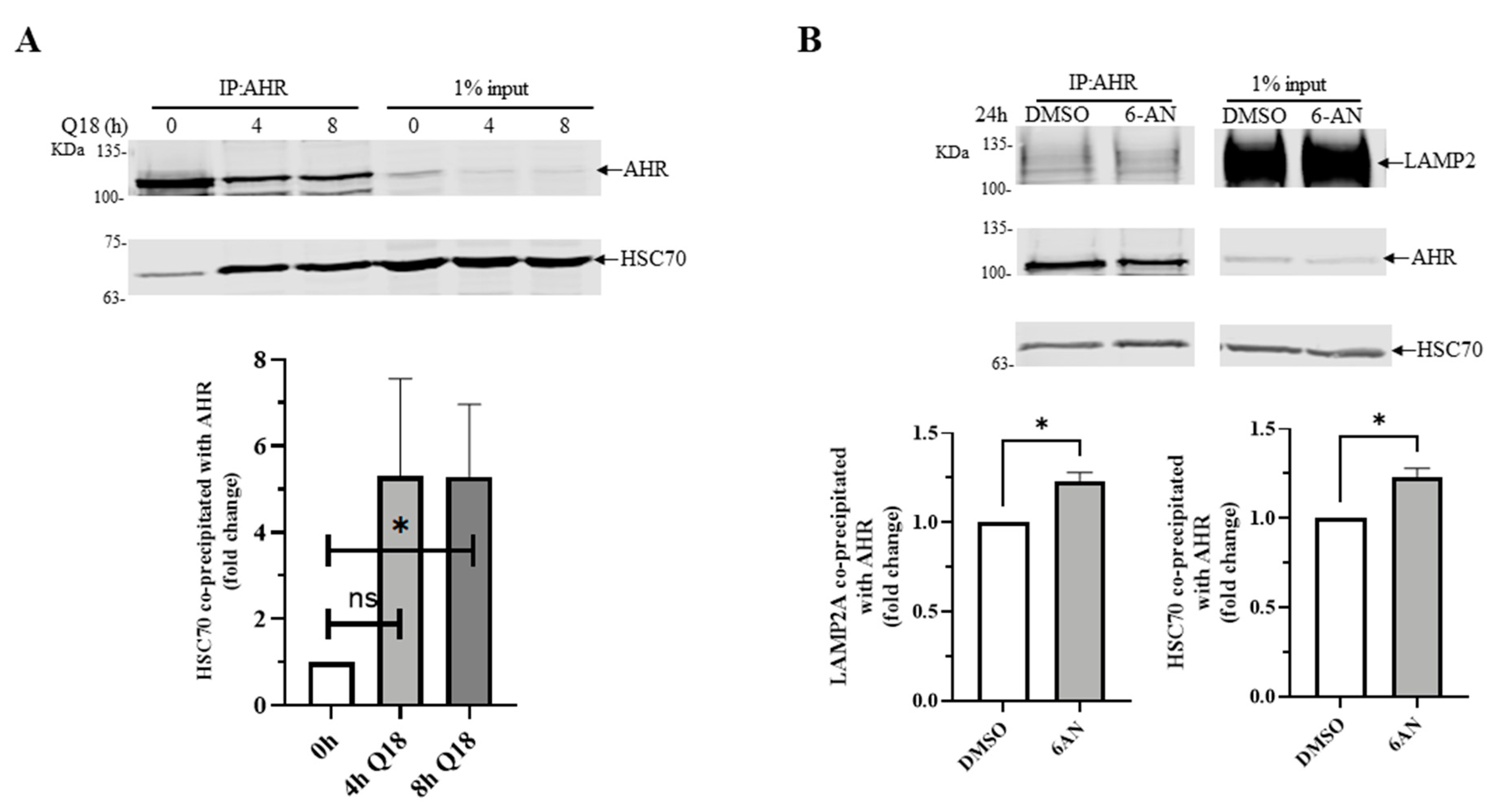
2.8. 6-AN Promotes AHR-HSC70 and AHR-LAMP2A Interaction in MDA-MB-468 Cells
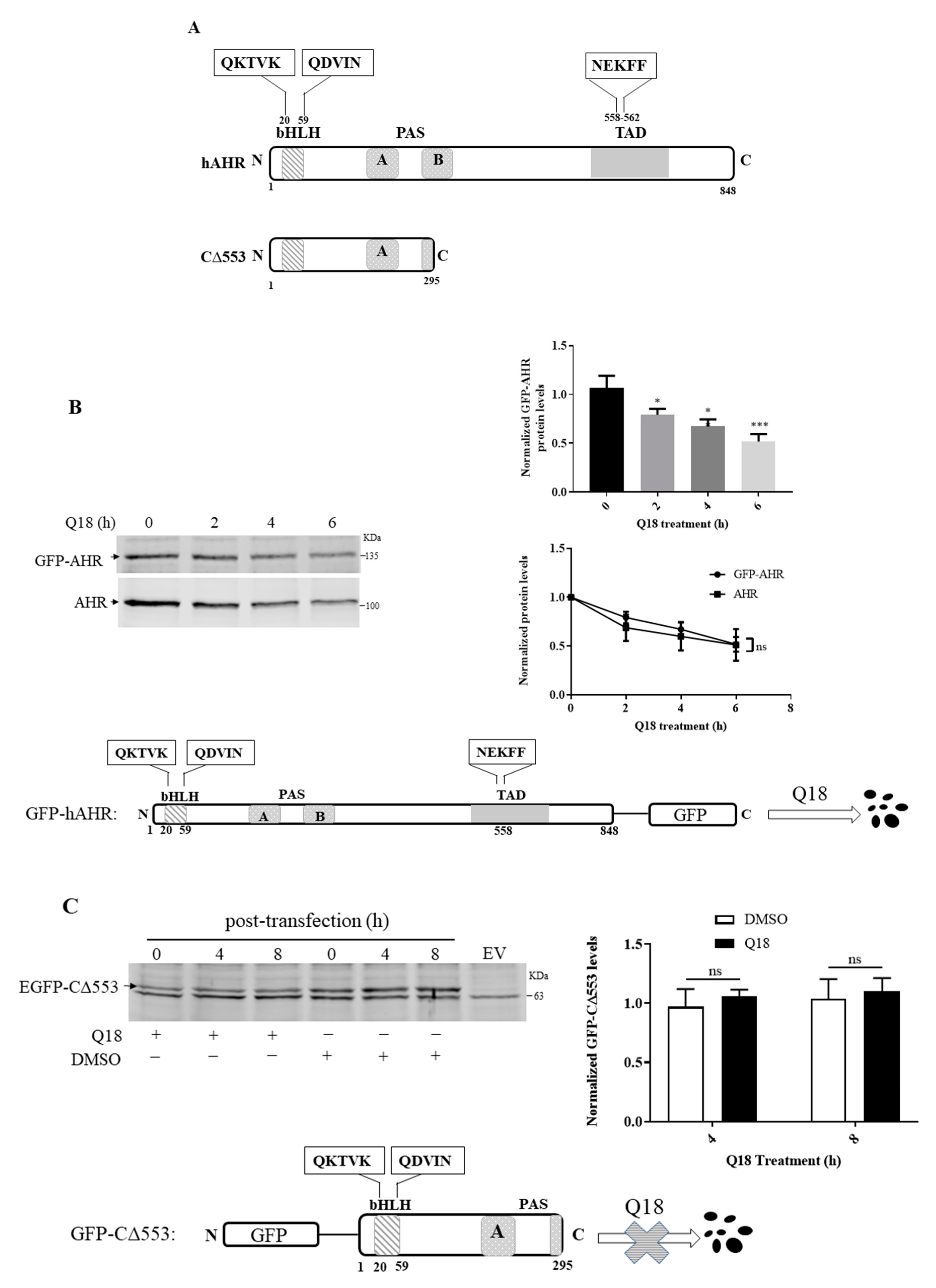
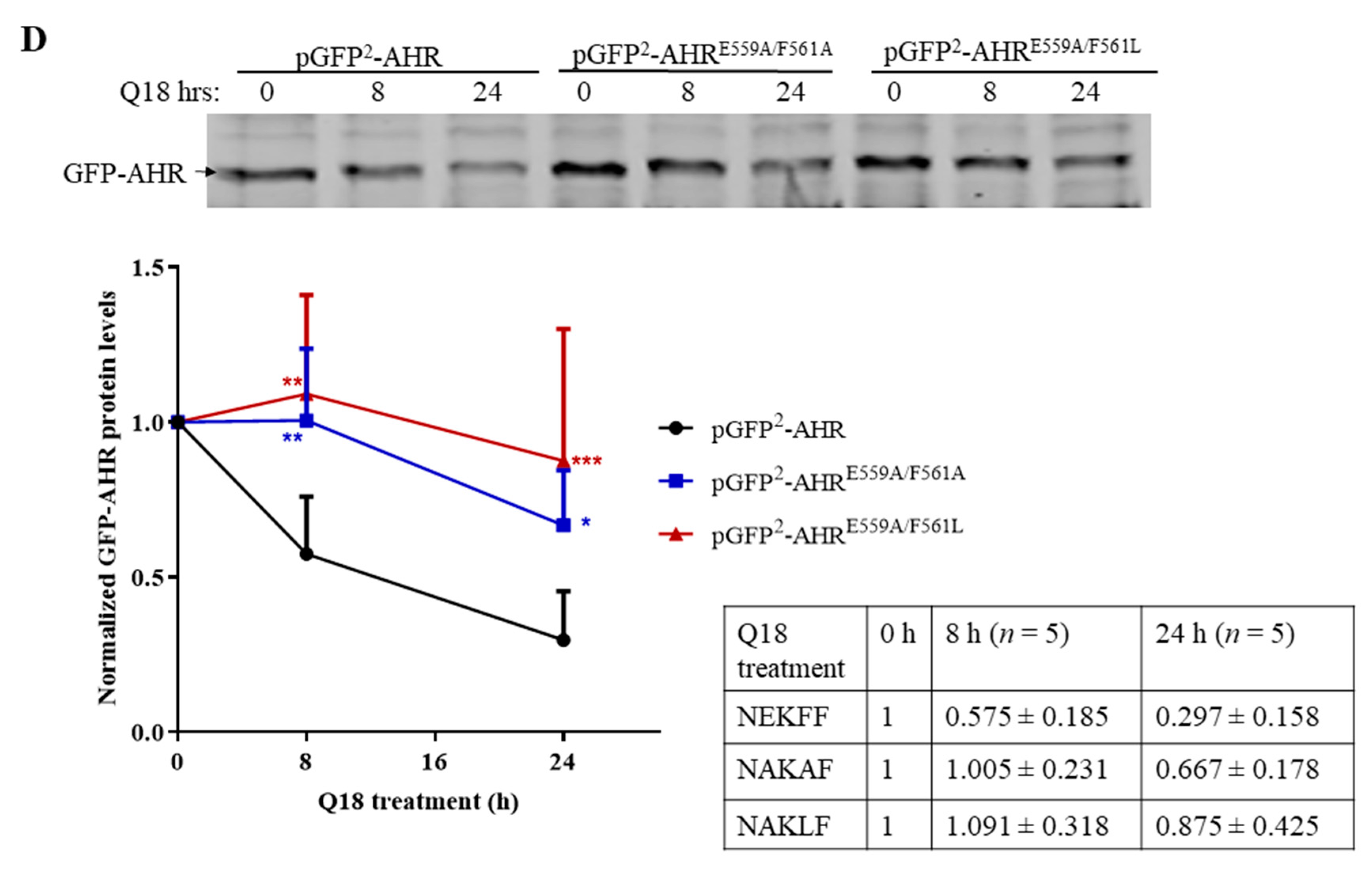
2.9. AHR Contains a CMA Signature Motif
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Western Blot Analysis
4.3. Reverse Transcription-Quantitative PCR
4.4. Generation of Lentivirus-Mediated Stable Knockdown Cells
4.5. Transient Transfection
4.6. Co-Immunoprecipitation
4.7. Proximity Ligation Assay
4.8. Site-Directed Mutagenesis
4.9. Gel Shift Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | aryl hydrocarbon receptor |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| HSP90 | heat shock protein 90 |
| HSC70 | heat shock cognate protein 70 |
| DRE | dioxin response element |
| CMA | chaperone-mediated autophagy |
| 3MC | 3-methylcholanthrene |
| βNF | β-naphthoflavone |
| HIF-1α | hypoxia inducible factor 1 alpha |
| CHIP | carboxy terminus of HSC70-interacting protein |
| LAMP2A | lysosomal associated membrane protein 2A |
| PR-B | progesterone receptor B |
| ERα | estrogen receptor alpha |
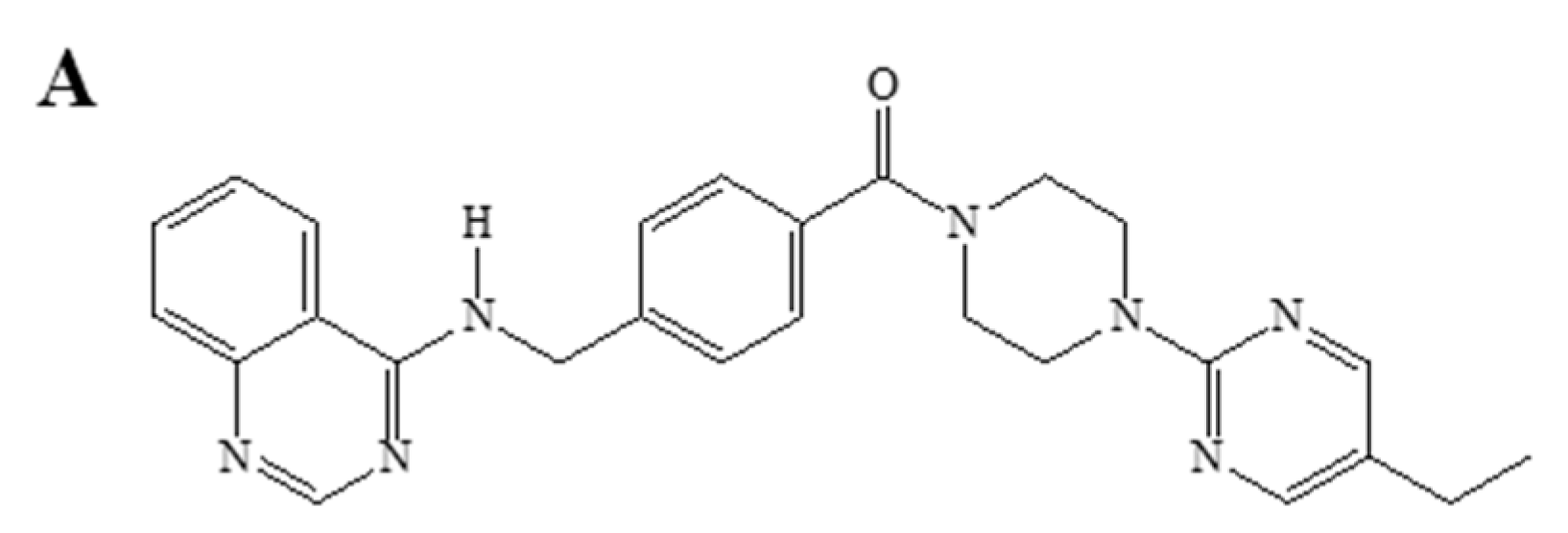
| Q18 | (4-(5-ethylpyrimidin-2-yl)piperazin-1-yl)(4-((quinazolin-4 ylamino)methyl)phenyl)methanone |
| CHX | cycloheximide |
| CQ | chloroquine |
| 6-AN | 6-amino-nicotinamide |
| GA | geldanamycin |
| CYP | cytochrome P450 |
| Act D | actinomycin D |
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 2000, 113, 4441–4450. [Google Scholar] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.-L.; Dice, J.F. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J. Biol. Chem. 1988, 263, 6797–6805. [Google Scholar] [CrossRef]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.J.; Fernandez-Salguero, P. The aryl hydrocarbon receptor: Studies using the AHR-null mice. Drug Metab. Dispos. 1998, 26, 1194–1198. [Google Scholar]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlezinger, J.J.; Liu, D.; Farago, M.; Seldin, D.C.; Belguise, K.; Sonenshein, G.E.; Sherr, D.H. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol. Chem. 2006, 387, 1175–1187. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Jin, U.H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef]
- Silginer, M.; Burghardt, I.; Gramatzki, D.; Bunse, L.; Leske, H.; Rushing, E.J.; Hao, N.; Platten, M.; Weller, M.; Roth, P. The aryl hydrocarbon receptor links integrin signaling to the TGF-beta pathway. Oncogene 2016, 35, 3260–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, E.M.; Bennink, M.R.; Bolling, S.F. Diet-relevant phytochemical intake affects the cardiac AhR and nrf2 transcriptome and reduces heart failure in hypertensive rats. J. Nutl. Biochem. 2013, 24, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Fan, Y.; Kurita, H.; Jiang, M.; Koch, S.; Rao, M.B.; Rubinstein, J.; Puga, A. Aryl hydrocarbon receptor ablation in cardiomyocytes protects male mice from heart dysfunction induced by NKX2.5 haploinsufficiency. Toxicol. Sci. 2017, 160, 74–82. [Google Scholar] [CrossRef]
- Wong, P.S.; Vogel, C.F.; Kokosinski, K.; Matsumura, F. Aryl hydrocarbon receptor activation in NCI-H441 cells and C57BL/6 mice: Possible mechanisms for lung dysfunction. Am. J. Respir. Cell Mol. Biol. 2010, 42, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.; Park, H.Y.; Suh, Y.S.; Yoon, E.H.; Kim, J.; Jang, W.H.; Lee, W.S.; Park, S.G.; Choi, I.W.; Choi, I.; et al. Inhibition of acute lethal pulmonary inflammation by the IDO-AhR pathway. Proc. Natl. Acad. Sci. USA 2017, 114, E5881–E5890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hatzakis, E.; Nichols, R.G.; Hao, R.; Correll, J.; Smith, P.B.; Chiaro, C.R.; Perdew, G.H.; Patterson, A.D. Metabolomics reveals that aryl hydrocarbon receptor activation by environmental chemicals induces systemic metabolic dysfunction in mice. Environ. Sci. Technol. 2015, 49, 8067–8077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belton, K.R.; Tian, Y.; Zhang, L.; Anitha, M.; Smith, P.B.; Perdew, G.H.; Patterson, A.D. Metabolomics reveals aryl hydrocarbon receptor activation induces liver and mammary gland metabolic dysfunction in lactating mice. J. Proteome Res. 2018, 17, 1375–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, M.; Kazmin, D.; Hu, P.; Thomas, R.S.; McDonnell, D.P.; Malek, G. Aryl hydrocarbon receptor knock-out exacerbates choroidal neovascularization via multiple pathogenic pathways. J. Pathol. 2015, 235, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.A.; Davis, S.S.; Rosko, A.; Nguyen, S.M.; Mitchell, K.P.; Mateen, S.; Neves, J.; Garcia, T.Y.; Mooney, S.; Perdew, G.H.; et al. A novel AhR ligand, 2AI, protects the retina from environmental stress. Sci. Rep. 2016, 6, 29025. [Google Scholar] [CrossRef]
- Carver, L.A.; Bradfield, C.A. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 1997, 272, 11452–11456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Whitlock, J.P., Jr. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 1997, 272, 8878–8884. [Google Scholar] [CrossRef] [Green Version]
- Petrulis, J.R.; Kusnadi, A.; Ramadoss, P.; Hollingshead, B.; Perdew, G.H. The hsp90 co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J. Biol. Chem. 2003, 278, 2677–2685. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskas, A.; Sundstrom, S.; Poellinger, L.; Pongratz, I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–2607. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Puga, A.; Vasiliou, V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann. N. Y. Acad. Sci. 1993, 685, 624–640. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef] [Green Version]
- Pollenz, R.S.; Buggy, C. Ligand-dependent and -independent degradation of the human aryl hydrocarbon receptor (hAHR) in cell culture models. Chem. Biol. Interact. 2006, 164, 49–59. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Wang, D.; Wang, Y.; Li, Y.; Uchizono, J.A.; Chan, W.K. p23 co-chaperone protects the aryl hydrocarbon receptor from degradation in mouse and human cell lines. Biochem. Pharmacol. 2012, 84, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Shallal, H.M.; Russu, W.A. Discovery, synthesis, and investigation of the antitumor activity of novel piperazinylpyrimidine derivatives. Eur. J. Med. Chem. 2011, 46, 2043–2057. [Google Scholar] [CrossRef]
- Zheng, Y.; Xie, J.; Huang, X.; Dong, J.; Park, M.S.; Chan, W.K. Binding studies using Pichia pastoris expressed human aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator proteins. Protein Expr. Purif. 2016, 122, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Shetty, P.V.; Bhagwat, B.Y.; Chan, W.K. p23 enhances the formation of the aryl hydrocarbon receptor-DNA complex. Biochem. Pharmacol. 2003, 65, 848–941. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.V.; Soares, A.R.; Ramalho, J.S.; Pereira, P.; Girao, H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by chaperone-mediated autophagy. Sci. Rep. 2015, 5, 10210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Li, C.H.; Cheng, Y.W.; Lee, C.C.; Liao, P.L.; Lin, C.H.; Huang, S.H.; Kang, J.J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, 41927. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Lee, J.E.; Myung, C.H.; Park, J.I.; Jo, C.S.; Hwang, J.S. Particulate matter-induced aryl hydrocarbon receptor regulates autophagy in keratinocytes. Biomol. Ther. 2019, 27, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bhakta, A.; Wang, S.; Li, Z.; Manley, S.; Huang, H.; Copple, B.; Ding, W.X. Role of hypoxia inducing factor-1beta in alcohol-induced autophagy, steatosis and liver injury in mice. PLoS ONE 2014, 9, e115849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chan, W.K. Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone. Int. J. Mol. Sci. 2020, 21, 3449. [Google Scholar] [CrossRef]
- Bento, C.F.; Fernandes, R.; Ramalho, J.; Marques, C.; Shang, F.; Taylor, A.; Pereira, P. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1alpha for degradation in the presence of methylglyoxal. PLoS ONE 2010, 5, e15062. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Hu, H.; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Li, G.; Shao, Q.; Yang, G.; Zheng, L.; Zhang, T.; Zhao, Y. CHIP: A new modulator of human malignant disorders. Oncotarget 2016, 7, 29864–29874. [Google Scholar] [CrossRef] [Green Version]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Phil. Trans. R. Soc. B 2018, 373, 20160530. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, M.J.; Peet, D.J.; Whitelaw, M.L. Defining the role for XAP2 in stabilization of the dioxin receptor. J. Biol. Chem. 2003, 278, 35878–35888. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.L.; Perdew, G.H. Carboxyl terminus of hsc70-interacting protein (CHIP) can remodel mature aryl hydrocarbon receptor (AhR) complexes and mediate ubiquitination of both the AhR and the 90 kDa heat-shock protein (hsp90) in vitro. Biochemistry 2007, 46, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Mota, A.L.; Evangelista, A.F.; Macedo, T.; Oliveira, R.; Scapulatempo-Neto, C.; Vieira, R.A.; Marques, M.M.C. Molecular characterization of breast cancer cell lines by clinical immunohistochemical markers. Oncol. Lett. 2017, 13, 4708–4712. [Google Scholar] [CrossRef] [Green Version]
- Rataj, F.; Moller, F.J.; Jahne, M.; Honscheid, P.; Zierau, O.; Vollmer, G.; Kretzschmar, G. Progesterone, as well as 17beta-estradiol, is important for regulating AHR battery homoeostasis in the rat uterus. Arch. Toxicol. 2015, 89, 393–404. [Google Scholar] [CrossRef]
- Hasan, A.; Fischer, B. Hormonal control of aryl hydrocarbon receptor (AhR) expression in the preimplantation rabbit uterus. Anat. Embryol. 2001, 204, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Kuil, C.W.; Brouwer, A.; Van der Saag, P.T.; Van der Burg, B. Interference between progesterone and dioxin signal transduction pathways. Different mechanisms are involved in repression by the progesterone receptor A and B isoforms. J. Biol. Chem. 1998, 273, 8829–8834. [Google Scholar] [CrossRef] [Green Version]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Slot, L.A.; Lauridsen, A.M.; Hendil, K.B. Intracellular protein degradation in serum-deprived human fibroblasts. Biochem. J. 1986, 237, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Dice, J.F. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.K.; Leonards, P.E.; Zhao, B.; Bergman, A.; Denison, M.S.; Van den Berg, M. Determination of in vitro relative potency (REP) values for mono-ortho polychlorinated biphenyls after purification with active charcoal. Toxicol. Lett. 2006, 165, 230–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yakkundi, P.; Chan, W.K. Down-regulation of p23 in normal lung epithelial cells reduces toxicities from exposure to benzo[a]pyrene and cigarette smoke condensate via an aryl hydrocarbon receptor-dependent mechanism. Toxicol. Sci. 2019, 167, 239–248. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Yang, Y.; Russu, W.A.; Chan, W.K. The Aryl Hydrocarbon Receptor Undergoes Chaperone-Mediated Autophagy in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1654. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041654
Chen J, Yang Y, Russu WA, Chan WK. The Aryl Hydrocarbon Receptor Undergoes Chaperone-Mediated Autophagy in Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(4):1654. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041654
Chicago/Turabian StyleChen, Jinyun, Yujie Yang, Wade A. Russu, and William K. Chan. 2021. "The Aryl Hydrocarbon Receptor Undergoes Chaperone-Mediated Autophagy in Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 4: 1654. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041654