
Histone Acetylation Domains Are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
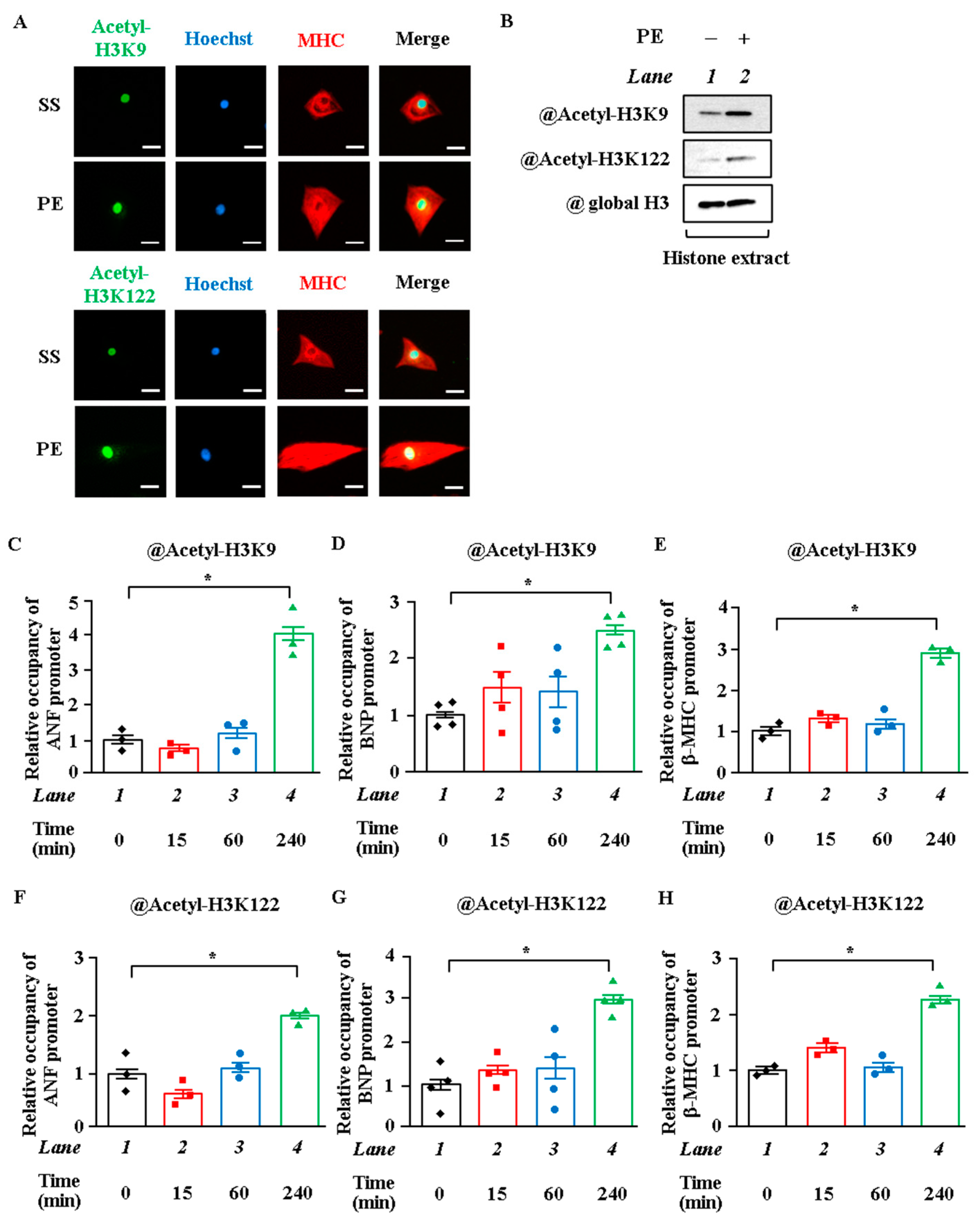
2.1. Acetylation Levels of H3K9 and H3K122 Were Elevated by Hypertrophic Stimulation in Cardiomyocytes
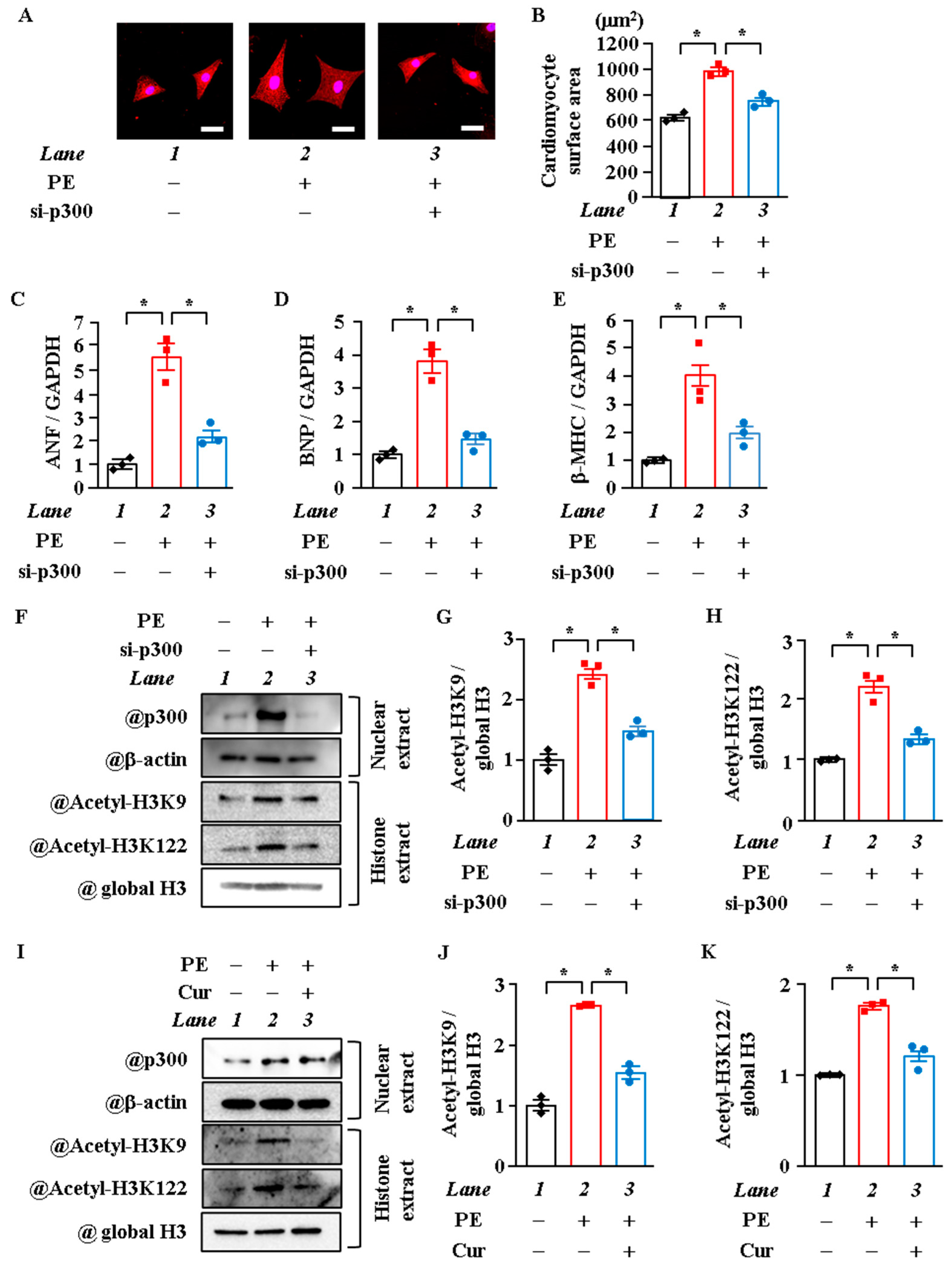
2.2. P300-HAT Inhibition Suppressed H3K9 Acetylation, H3K122 Acetylation, and Cardiomyocyte Hypertrophy
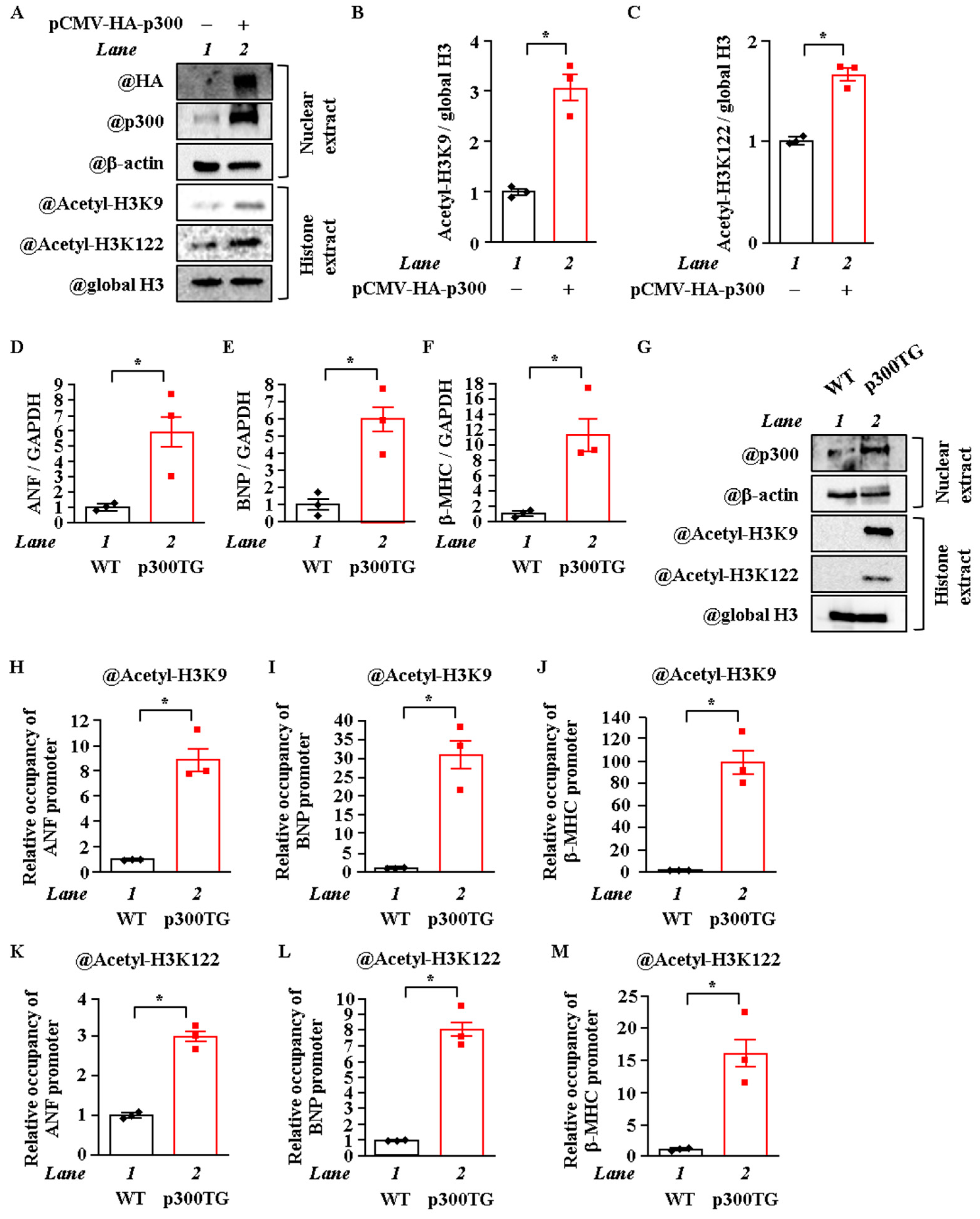
2.3. Overexpression of P300 Enhanced H3K9 and H3K122 Acetylation
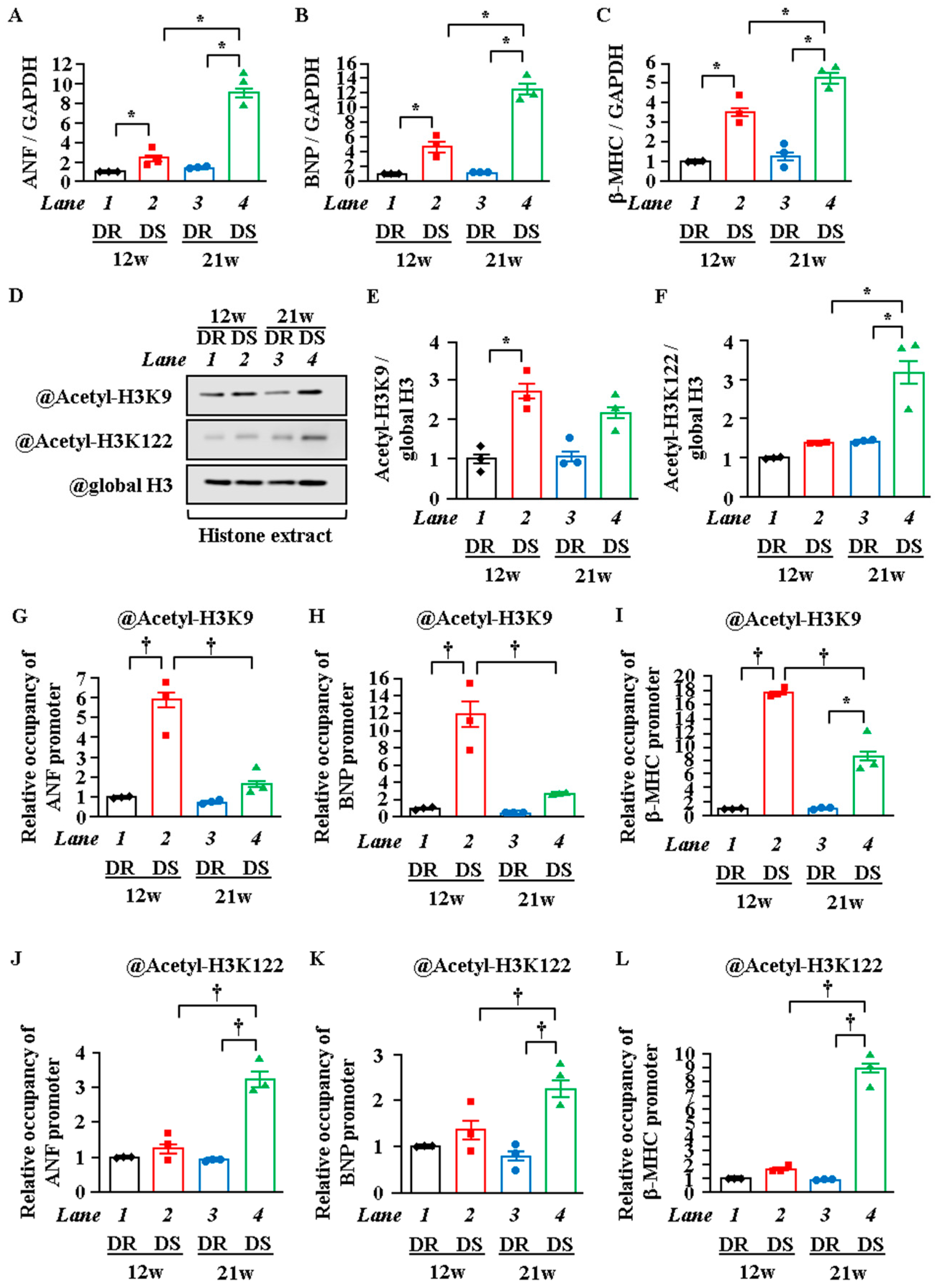
2.4. H3K122 Acetylation Was Enhanced in Heart Failure
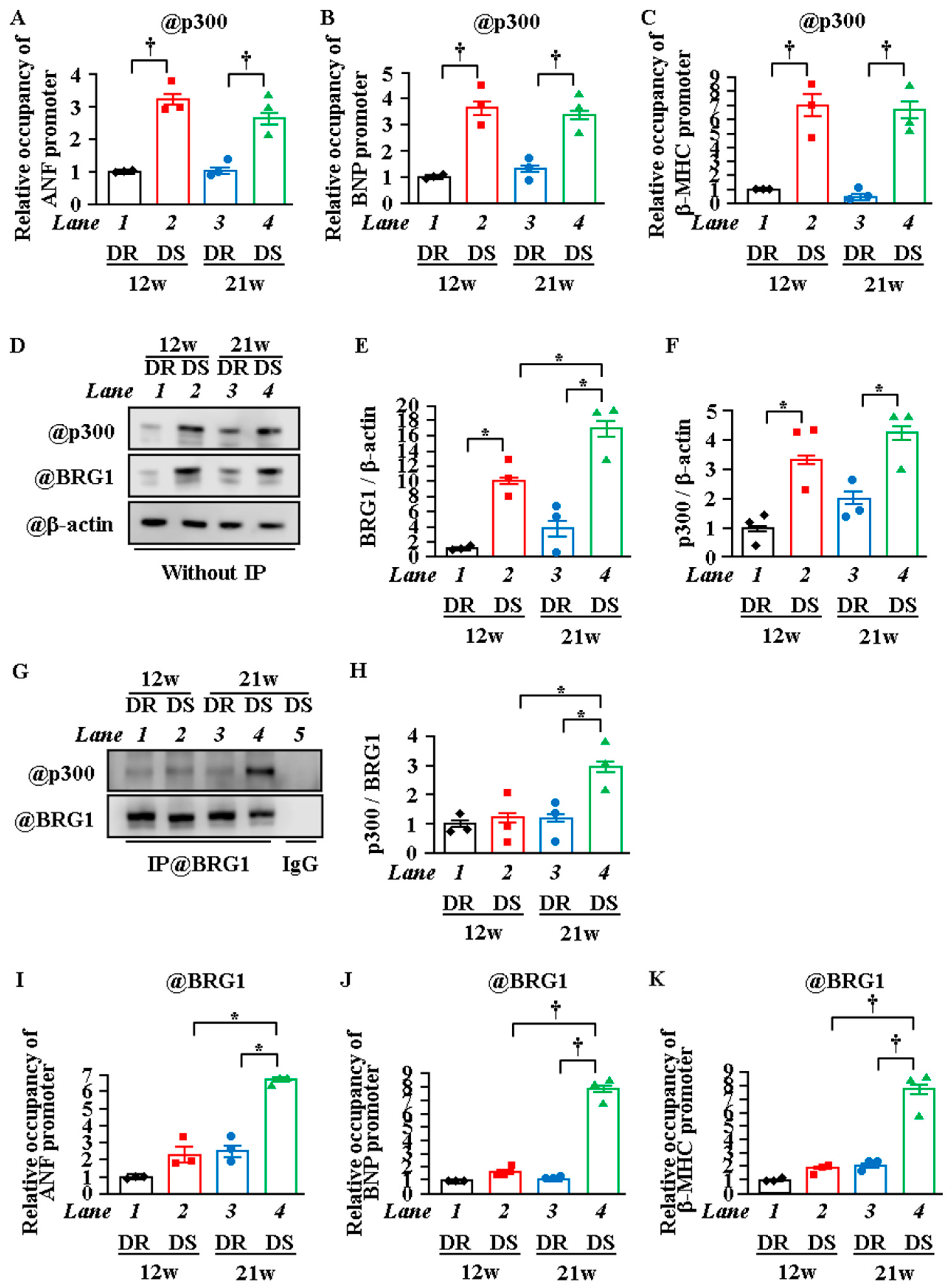
2.5. Interaction between P300 and BRG1 Was Enhanced, and the Recruitment of BRG1 Was Increased in Heart Failure
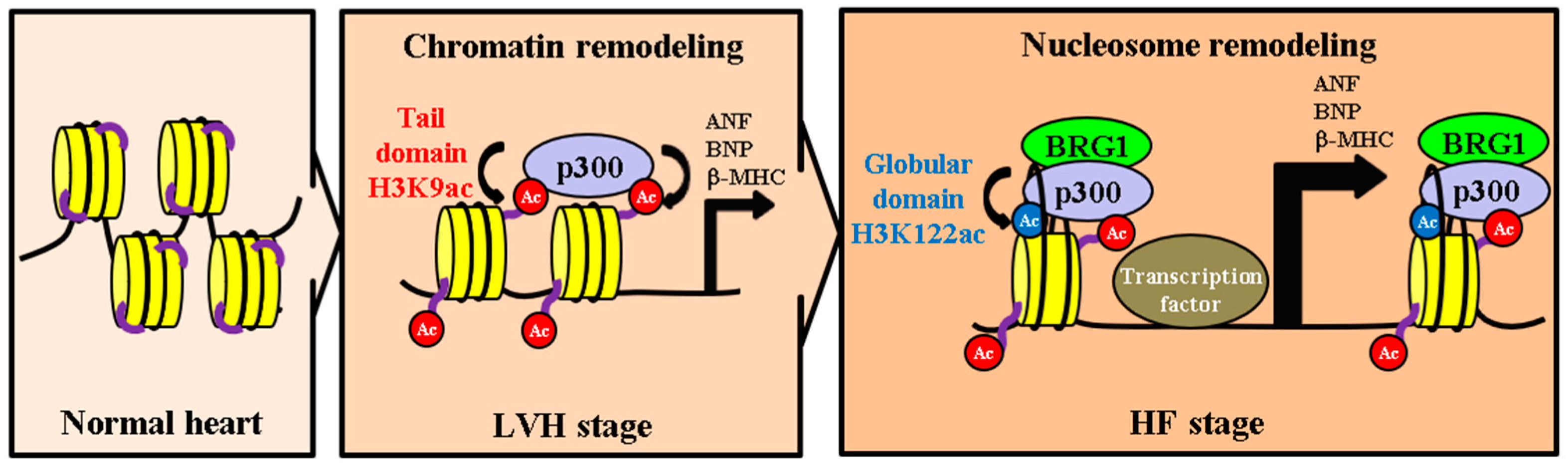
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Neonatal Rat Ventricular Cardiomyocyte Culture
4.3. Echocardiography
4.4. Transfection
4.5. Immunoblot Analysis of Histones
4.6. Immunoprecipitation and Western Blotting
4.7. Quantitative RT-PCR
4.8. Immunofluorescence Staining and Measurement of Surface Area of Cardiomyocytes
4.9. Chromatin Immunoprecipitation Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef]
- Borow, K.M.; Yaroshinsky, A.; Greenberg, B.; Perin, E.C. Phase 3 DREAM-HF Trial of Mesenchymal Precursor Cells in Chronic Heart Failure. Circ. Res. 2019, 125, 265–281. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell. 2008, 31, 449–461. [Google Scholar] [CrossRef]
- Greco, C.M.; Condorelli, G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat. Rev. Cardiol. 2015, 12, 488–497. [Google Scholar] [CrossRef]
- Papait, R.; Greco, C.; Kunderfranco, P.; Latronico, M.V.; Condorelli, G. Epigenetics: A new mechanism of regulation of heart failure? Basic Res. Cardiol. 2013, 108, 361. [Google Scholar] [CrossRef] [PubMed]
- Olio, F.D.; Trinchera, M. Epigenetic Bases of Aberrant Glycosylation in Cancer. Int. J. Mol. Sci. 2017, 18, 998. [Google Scholar]
- Nakagawa, Y.; Kuwahara, K.; Takemura, G.; Akao, M.; Kato, M.; Arai, Y.; Takano, M.; Harada, M.; Murakami, M.; Nakanishi, M.; et al. p300 plays a critical role in maintaining cardiac mitochondrial function and cell survival in postnatal hearts. Circ. Res. 2009, 105, 746–754. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kawamura, T.; Morimoto, T.; Ono, K.; Wada, H.; Kawase, Y.; Matsumori, A.; Nishio, R.; Kita, T.; Hasegawa, K. Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation 2006, 113, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Yanazume, T.; Hasegawa, K.; Morimoto, T.; Kawamura, T.; Wada, H.; Matsumori, A.; Kawase, Y.; Hirai, M.; Kita, T. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell. Biol. 2003, 23, 3593–3606. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.Q.; Shehadeh, L.A.; Mitrani, J.M.; Pessanha, M.; Slepak, T.I.; Webster, K.A.; Bishopric, H.N. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 2008, 118, 934–946. [Google Scholar] [CrossRef]
- Shikama, N.; Lutz, W.; Kretzschmar, R.; Sauter, N.; Roth, J.F.; Marino, S.; Wittwer, J.; Scheidweiler, A.; Eckner, R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003, 22, 5175–5185. [Google Scholar] [CrossRef]
- Morimoto, T.; Sunagawa, Y.; Kawamura, T.; Takaya, T.; Wada, H.; Nagasawa, A.; Komeda, M.; Fujita, M.; Shimatsu, A.; Kita, T.; et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J. Clin. Investig. 2008, 118, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Peng, B.; Luo, X.; Sun, H.; Peng, C. Anacardic acid attenuates pressure-overload cardiac hypertrophy through inhibiting histone acetylases. J. Cell. Mol. Med. 2019, 23, 2744–2752. [Google Scholar] [CrossRef]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, K.A. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115, 311–324. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Fry, C.J.; Peterson, C.L. Transcription. Unlocking the gates to gene expression. Science 2002, 295, 1847–1848. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Shah, R.C.; Chang, M.J.; Benveniste, E.N. Coordination of cell signaling, chromatin remodeling, histone modifications, and regulator recruitment in human matrix metalloproteinase 9 gene transcription. Mol. Cell Biol. 2004, 24, 5496–5509. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef]
- Rhee, H.S.; Bataille, A.R.; Zhang, L.; Pugh, B.F. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 2014, 159, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, A.R.; Ongaigui, C.L.K.; Boxer, D.L.; Chang, Y.H.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef]
- Li, N.; Li, Y.; Lv, J.; Zheng, X.; Wen, H.; Shen, H.; Zhu, G.; Chen, T.Y.; Dhar, S.S.; Kan, P.Y.; et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol. Cell 2016, 63, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Molina-Serrano, D.; Kirmizis, A. Beyond the histone tail: Acetylation at the nucleosome dyad commands transcription. Nucleus 2013, 4, 343–348. [Google Scholar] [CrossRef]
- Tropberger, P.; Pott, S.; Keller, C.; Kamieniarz-Gdula, K.; Caron, M.; Richter, F.; Li, G.; Mittler, G.; Liu, T.E.; Bühler, M.; et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013, 152, 859–872. [Google Scholar] [CrossRef]
- Kelly, A.D.; Issa, J.J. The promise of epigenetic therapy: Reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 2017, 42, 68–77. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Bruggeman, E.C.; Wen, Z.; Yao, B. Epigenetic Regulations in Neuropsychiatric Disorders. Front. Genet. 2019, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.D.; Spitler, K.M.; Grueter, C.E. Disruption of cardiac Med1 inhibits RNA polymerase II promoter occupancy and promotes chromatin remodeling. Am. J. Physiol. Circ. Physiol. 2019, 316, H314–H325. [Google Scholar] [CrossRef]
- Toth, M.; Boros, I.M.; Balint, E. Elevated level of lysine 9-acetylated histone H3 at the MDR1 promoter in multidrug-resistant cells. Cancer Sci. 2012, 103, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.-J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [CrossRef]
- Luebben, W.R.; Sharma, N.; Nyborg, J.K. Nucleosome eviction and activated transcription require p300 acetylation of histone H3 lysine 14. Proc. Natl. Acad. Sci. USA 2010, 107, 19254–19259. [Google Scholar] [CrossRef]
- Kaburagi, S.; Hasegawa, K.; Morimoto, T.; Araki, M.; Sawamura, T.; Masaki, T.; Sasayama, S. The role of endothelin-converting enzyme-1 in the development of alpha1-adrenergic-stimulated hypertrophy in cultured neonatal rat cardiac myocytes. Circulation 1999, 99, 292–298. [Google Scholar] [CrossRef]
- Morimoto, T.; Hasegawa, K.; Kaburagi, S.; Kakita, T.; Wada, H.; Yanazume, T.; Sasayama, S. Phosphorylation of GATA-4 is involved in alpha 1-adrenergic agonist-responsive transcription of the endothelin-1 gene in cardiac myocytes. J. Biol. Chem. 2000, 275, 13721–13726. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Kamikawa, Y.F.; Donohoe, M.E. Brd4’s Bromodomains Mediate Histone H3 Acetylation and Chromatin Remodeling in Pluripotent Cells through P300 and Brg1. Cell Rep. 2018, 25, 1756–1771. [Google Scholar] [CrossRef]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.P.; Tan, W.L.W.; Anene-Nzelu, C.G.; Lee, C.J.M.; Li, P.Y.; Luu, T.D.A.; Chan, X.C.; Tiang, Z.; Ng, L.S.; Huang, X.; et al. Robust CTCF-Based Chromatin Architecture Underpins Epigenetic Changes in the Heart Failure Stress-Gene Response. Circulation 2019, 139, 1937–1956. [Google Scholar] [CrossRef]
- Bär, C.; Chatterjee, S.; Thum, T. Long Noncoding RNAs in Cardiovascular Pathology, Diagnosis, and Therapy. Circulation 2016, 134, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. Epigenetic regulation of the cardiovascular system: Introduction to a review series. Circ. Res. 2010, 107, 324–326. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Hang, C.T.; Yang, J.; Chang, C.P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 2011, 108, 378–396. [Google Scholar] [CrossRef]
- Yan, Y.; Seto, E. Brd4-p300 inhibition downregulates Nox4 and accelerates lung fibrosis resolution in aged mice. JCI Insight 2020, 5, 137127. [Google Scholar]
- Kathiriya, I.S.; Nora, E.P.; Bruneau, B.G. Investigating the transcriptional control of cardiovascular development. Circ. Res. 2015, 116, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced histone acetylation and transcription: A dynamic perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef]
- Tropberger, P.; Schneider, R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 657–661. [Google Scholar] [CrossRef]
- Fenley, A.T.; Adams, D.A.; Onufriev, A.V. Charge state of the globular histone core controls stability of the nucleosome. Biophys. J. 2010, 99, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Gatchalian, J.; Liao, J.; Maxwell, M.B.; Hargreaves, D.C. Control of Stimulus-Dependent Responses in Macrophages by SWI/SNF Chromatin Remodeling Complexes. Trends Immunol. 2020, 41, 126–140. [Google Scholar] [CrossRef]
- Bevilacqua, A.; Willis, M.S.; Bultman, S.J. SWI/SNF chromatin-remodeling complexes in cardiovascular development and disease. Cardiovasc. Pathol. 2014, 23, 85–91. [Google Scholar] [CrossRef]
- Vieira, J.M.; Howard, S.; Campo, C.V.D.; Bollini, S.; Dubé, K.N.; Masters, M.; Barnette, N.D.; Rohling, M.; Sun, X.; Hankinset, L.E.; et al. BRG1-SWI/SNF-dependent regulation of the Wt1 transcriptional landscape mediates epicardial activity during heart development and disease. Nat. Commun. 2017, 8, 16034. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef]
- Hang, C.T.; Yang, J.; Han, P.; Cheng, H.L.; Shang, C.; Ashley, E.; Zhou, B.; Chang, C.P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, A.; Joe, B.; de la Serna, I.L. SWI/SNF chromatin remodeling enzymes are associated with cardiac hypertrophy in a genetic rat model of hypertension. J. Cell. Physiol. 2013, 228, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.; Gebuhr, T.; Yee, D.; Mantia, C.L.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Chang, L.; Kiriazis, H.; Gao, X.M.; Du, X.J.; Osta, A.E. Cardiac genes show contextual SWI/SNF interactions with distinguishable gene activities. Epigenetics 2011, 6, 760–768. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Okabe, J.; Chang, L.; Su, Y.; Du, X.J.; Osta, A.E. The primary microRNA-208b interacts with Polycomb-group protein, Ezh2, to regulate gene expression in the heart. Nucleic Acids Res. 2014, 42, 790–803. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Keating, S.T.; Du, X.J.; Osta, A.E. Chromatin modifications remodel cardiac gene expression. Cardiovasc. Res. 2014, 103, 7–16. [Google Scholar] [CrossRef]
- Jain, S.; Wei, J.; Mitrani, L.R.; Bishopric, N.H. Auto-acetylation stabilizes p300 in cardiac myocytes during acute oxidative stress, promoting STAT3 accumulation and cell survival. Breast Cancer Res. Treat. 2012, 135, 103–114. [Google Scholar] [CrossRef]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res. 2007, 17, 324–332. [Google Scholar] [CrossRef]
- Trotter, K.W.; Archer, T.K. The BRG1 transcriptional coregulator. Nucl. Recept. Signal. 2008, 6, e004. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T.; Matsumori, A.; Kihara, Y.; Inoko, M.; Ono, K.; Iwanaga, Y.; Yamada, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ. Res. 1997, 81, 664–671. [Google Scholar] [CrossRef]
- Iwanaga, Y.; Kihara, Y.; Hasegawa, K.; Inagaki, K.; Yoneda, T.; Kaburagi, S.; Araki, M.; Sasayama, S. Cardiac endothelin-1 plays a critical role in the functional deterioration of left ventricles during the transition from compensatory hypertrophy to congestive heart failure in salt-sensitive hypertensive rats. Circulation 1998, 98, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Katanasaka, Y.; Sunagawa, Y.; Miyazaki, Y.; Funamoto, M.; Wada, H.; Hasegawa, K.; Morimoto, T. Tyrosine phosphorylation of RACK1 triggers cardiomyocyte hypertrophy by regulating the interaction between p300 and GATA4. Biochim. Biophys. Acta 2016, 1862, 1544–1557. [Google Scholar] [CrossRef]
- Sunagawa, Y.; Morimoto, T.; Takaya, T.; Kaichi, S.; Wada, H.; Kawamura, T.; Fujita, M.; Shimatsu, A.; Kita, T.; Hasegawa, K. Cyclin-dependent kinase-9 is a component of the p300/GATA4 complex required for phenylephrine-induced hypertrophy in cardiomyocytes. J. Biol. Chem. 2010, 285, 9556–9568. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funamoto, M.; Sunagawa, Y.; Katanasaka, Y.; Shimizu, K.; Miyazaki, Y.; Sari, N.; Shimizu, S.; Mori, K.; Wada, H.; Hasegawa, K.; et al. Histone Acetylation Domains Are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats. Int. J. Mol. Sci. 2021, 22, 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041771
Funamoto M, Sunagawa Y, Katanasaka Y, Shimizu K, Miyazaki Y, Sari N, Shimizu S, Mori K, Wada H, Hasegawa K, et al. Histone Acetylation Domains Are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats. International Journal of Molecular Sciences. 2021; 22(4):1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041771
Chicago/Turabian StyleFunamoto, Masafumi, Yoichi Sunagawa, Yasufumi Katanasaka, Kana Shimizu, Yusuke Miyazaki, Nurmila Sari, Satoshi Shimizu, Kiyoshi Mori, Hiromichi Wada, Koji Hasegawa, and et al. 2021. "Histone Acetylation Domains Are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats" International Journal of Molecular Sciences 22, no. 4: 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041771