
PRKAR1B-AS2 Long Noncoding RNA Promotes Tumorigenesis, Survival, and Chemoresistance via the PI3K/AKT/mTOR Pathway

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
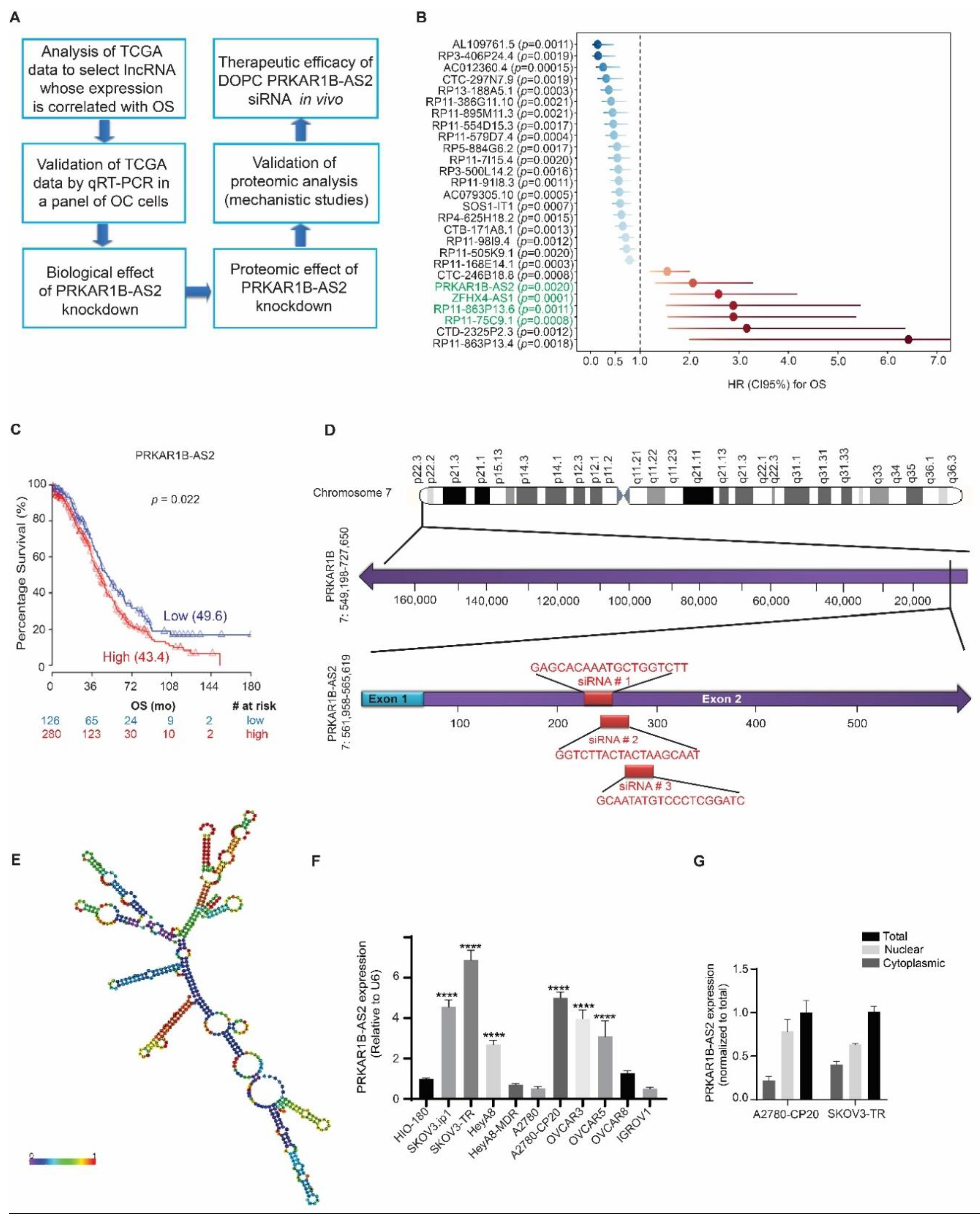
2.1. PRKAR1B-AS2 lncRNA Is Overexpressed in OC and Associated with Poor Survival
2.2. Silencing PRKAR1B-AS2 Inhibits Cell Viability and Improves the Response to Cisplatin In Vitro
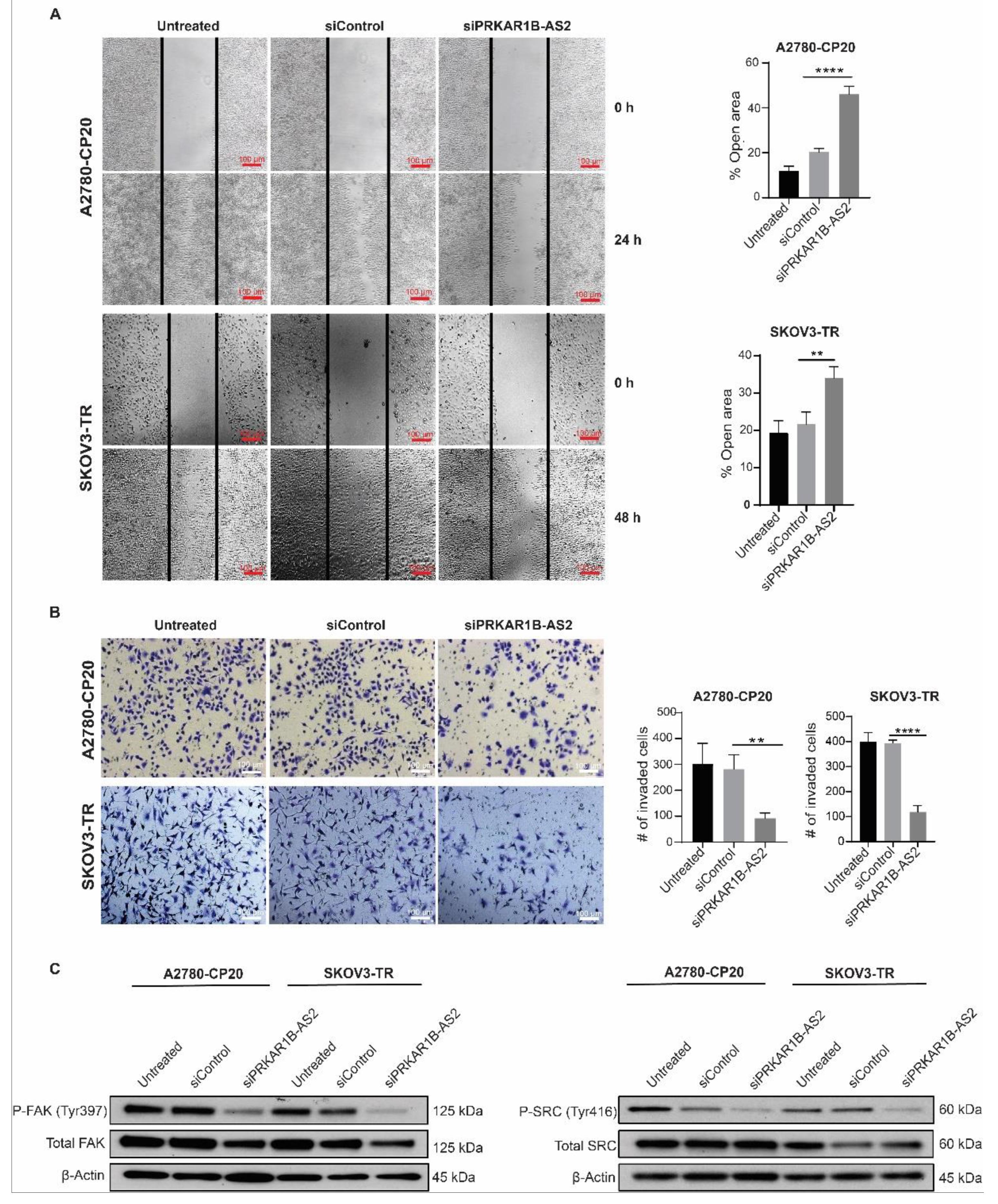
2.3. PRKAR1B-AS2 Knockdown Suppresses Migration and Invasion of OC Cells In Vitro
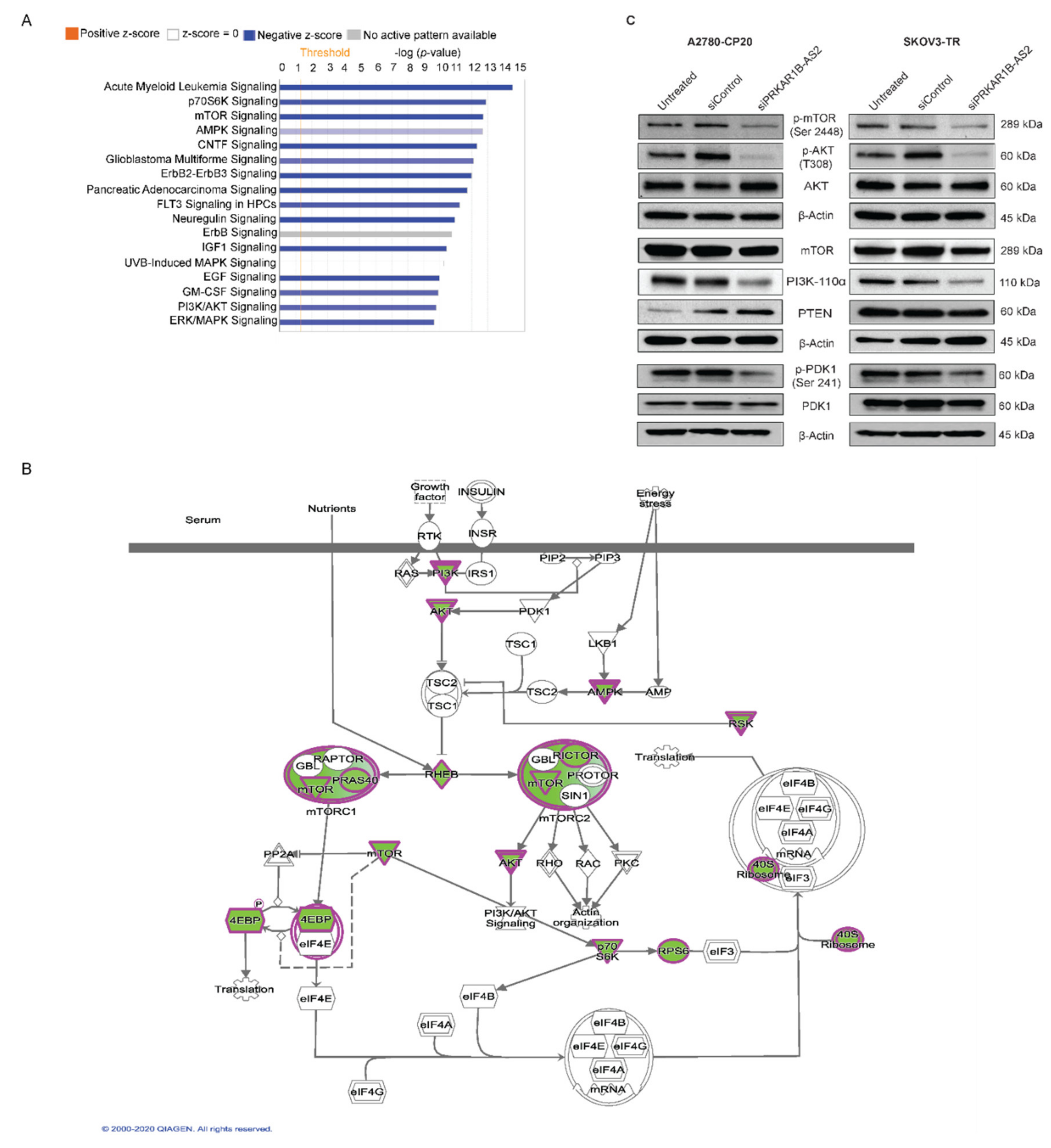
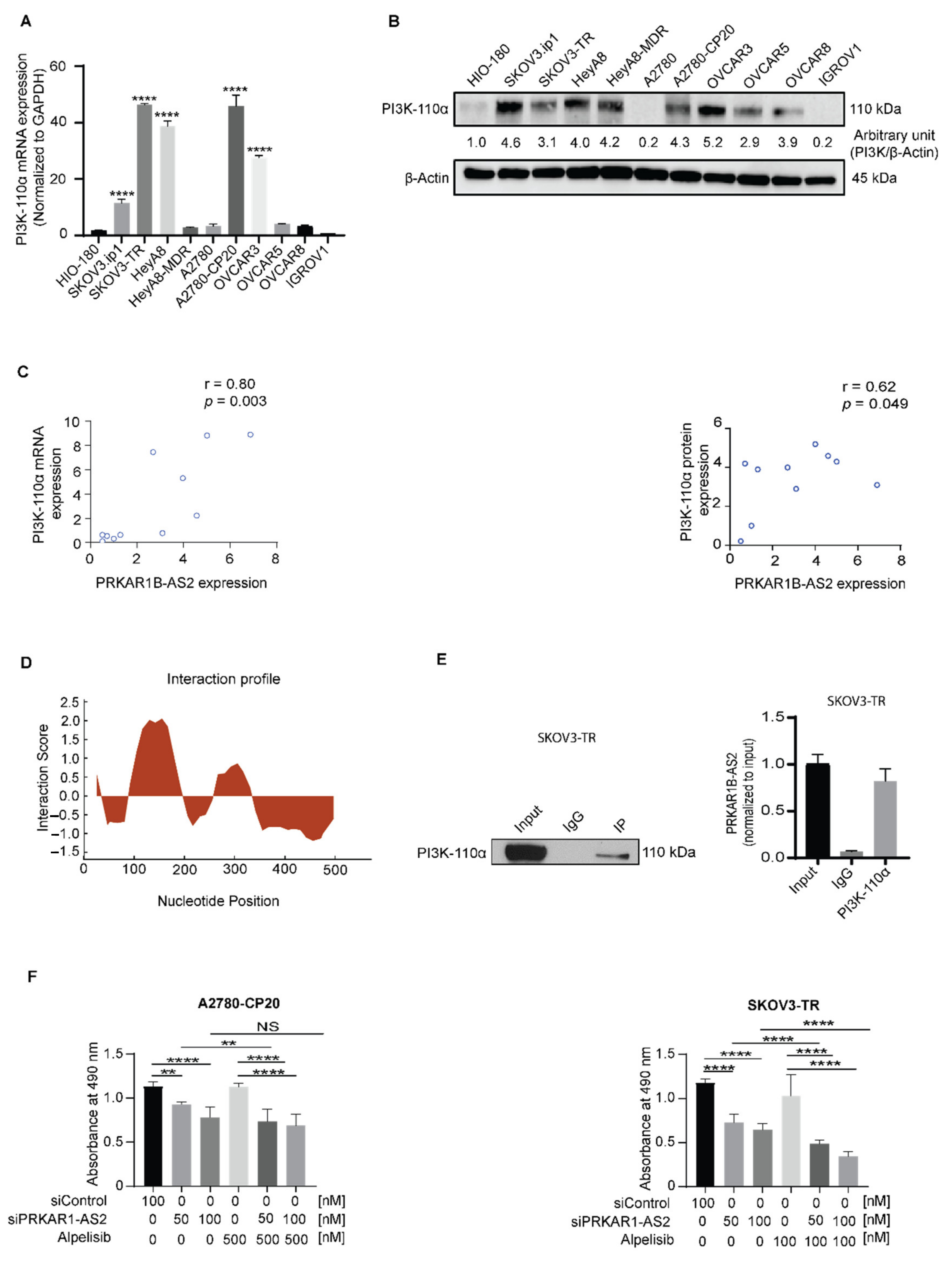
2.4. PRKAR1B-AS2 Regulates the PI3K/AKT/mTOR Signaling Pathway
2.5. PRKAR1B-AS2 Silencing Improves OC Sensitivity to Cisplatin In Vivo
3. Discussion
4. Materials and Methods
4.1. OS Analysis of lncRNA Expression in TCGA OC Cohort
4.2. LncRNA–Protein Interactions
4.3. Cell Culture, Reagents, and siRNA Transfection
4.4. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR Analysis
4.5. Cell Viability Assay
4.6. Colony Formation Assay
4.7. Wound Healing Assay
4.8. Invasion Assay
4.9. Annexin V/PI Assay
4.10. Cell Extracts and Western Blot Analysis
4.11. RIP Assay
4.12. RPPA Analysis
4.13. Liposomal Nanoparticle Preparation
4.14. OC Xenograft Mouse Model Studies
4.15. Immunohistochemistry
4.16. TUNEL Assay
4.17. In Situ Hybridization
4.18. Collection of Mouse Tumor Specimens for Quantitative Reverse Transcription PCR
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| lncRNA | Long non-coding RNA |
| mTOR | Mammalian target of rapamycin |
| OC | Ovarian cancer |
| OS | Overall survival |
| PDK1 | Phosphoinositide dependent kinase 1 |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol-3,4-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5- triphosphate |
| PRKAR1B | Protein Kinase CAMP-Dependent Type I Regulatory Subunit Beta |
| PTEN | Phosphatase and tensin homolog |
| RIP | RNA immunoprecipitation |
| RPPA | Reverse phase protein array |
| siRNA | Small interfering RNA |
| TCGA | The cancer genome atlas |
| TSC1/2 | Tuberous sclerosis complex 1 and 2 |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick-end labeling |
References
- DeSantis, C.E.; Miller, K.D.; Dale, W.; Mohile, S.G.; Cohen, H.J.; Leach, C.R.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Cancer statistics for adults aged 85 years and older, 2019. CA Cancer J. Clin. 2019, 69, 452–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesterov, A.; Lu, X.; Johnson, M.; Miller, G.J.; Ivashchenko, Y.; Kraft, A.S. Elevated AKT activity protects the prostate cancer cell line lncap from trail-induced apoptosis. J. Biol. Chem. 2001, 276, 10767–10774. [Google Scholar] [CrossRef] [Green Version]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage iii and stage iv ovarian cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A.; Brady, M.F.; McGuire, W.P.; Harper, P.G.; Alberts, D.S.; Friedlander, M.; Colombo, N.; Fowler, J.M.; Argenta, P.A.; De Geest, K. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: A phase iii trial of the gynecologic cancer intergroup. J. Clin. Oncol. 2009, 27, 1419. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, A.; Lück, H.-J.; Meier, W.; Adams, H.-P.; Mobus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schröder, W. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573. [Google Scholar] [CrossRef]
- Johnson, S.; Shen, D.-W.; Pastan, I.; Gottesman, M.; Hamilton, T. Cross-resistance, cisplatin accumulation, and platinum–DNA adduct formation and removal in cisplatin-sensitive and-resistant human hepatoma cell lines. Exp. Cell Res. 1996, 226, 133–139. [Google Scholar] [CrossRef]
- Peng, D.-J.; Wang, J.; Zhou, J.-Y.; Wu, G.S. Role of the akt/mtor survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.L.; Cantley, L.C. Pi3k pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Kriplani, N.; Hermida, M.A.; Brown, E.R.; Leslie, N.R. Class i pi 3-kinases: Function and evolution. Adv. Biol. Regul. 2015, 59, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The akt-mtor tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Plastaras, J.P.; Dorsey, J.F.; Carroll, K.; Kim, S.-H.; Birnbaum, M.J.; El-Deiry, W.S. Role of pi3k/akt signaling in trail-and radiation-induced gastrointestinal apoptosis. Cancer Biol. Ther. 2008, 7, 2047–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, K.; Srivastava, R.K. Role of the phosphatidylinositol 3′-kinase/pten/akt kinase pathway in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in non-small cell lung cancer cells. Cancer Res. 2002, 62, 4929–4937. [Google Scholar] [PubMed]
- Musa, F.; Schneider, R. Targeting the pi3k/akt/mtor pathway in ovarian cancer. TCR 2015, 4, 97–106. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F. Landscape of transcription in human cells. Nature 2012, 489, 101. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M. The emerging role of lncrnas in cancer. Nat. Med. 2015, 21, 1253. [Google Scholar] [CrossRef]
- Bernstein, E.; Allis, C.D. Rna meets chromatin. Genes Dev. 2005, 19, 1635–1655. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, J.; Pandey, G.K.; Kanduri, C. Regulation of the mammalian epigenome by long noncoding rnas. Biochim. Biophys. Acta 2009, 1790, 936–947. [Google Scholar] [CrossRef]
- Li, S.; Pan, X.; Yang, S.; Ma, A.; Yin, S.; Dong, Y.; Pei, H.; Bi, X.; Li, W. Lncrna malat1 promotes oxidized low-density lipoprotein-induced autophagy in huvecs by inhibiting the pi3k/akt pathway. J. Cell Biochem. 2019, 120, 4092–4101. [Google Scholar] [CrossRef]
- Pauli, A.; Rinn, J.L.; Schier, A.F. Non-coding rnas as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncrnas in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, R.; Almeida, M.I.E.; Colombatti, A.; Calin, G.A. Long non-coding rnas and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harries, L.W. Long non-coding RNAs and human disease. Biochem. Soc. Trans. 2012, 40, 902–906. [Google Scholar] [CrossRef]
- Yan, B.; Wang, Z. Long noncoding rna: Its physiological and pathological roles. DNA Cell Biol. 2012, 31, S-34–S-41. [Google Scholar] [CrossRef] [PubMed]
- Oncul, S.; Amero, P.; Rodriguez-Aguayo, C.; Calin, G.A.; Sood, A.K.; Lopez-Berestein, G. Long non-coding rnas in ovarian cancer: Expression profile and functional spectrum. RNA Biol. 2019, 17, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zheng, L.L.; Wu, J.; Sun, W.J.; Wang, Z.L.; Zhou, H.; Qu, L.H.; Yang, J.H. Discovery of protein-lncrna interactions by integrating large-scale clip-seq and rna-seq datasets. Front. Bioeng. Biotechnol. 2014, 2, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.L.; Chen, L.C.; Shen, T.L. Emerging roles of focal adhesion kinase in cancer. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Creighton, C.J.; Huang, S. Reverse phase protein arrays in signaling pathways: A data integration perspective. Drug Des. Dev. Ther. 2015, 9, 3519–3527. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/AKT pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chen, D.; Liu, H.; Yang, K. Increased expression of lncrna casc9 promotes tumor progression by suppressing autophagy-mediated cell apoptosis via the AKT/mTOR pathway in oral squamous cell carcinoma. Cell Death Dis. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The pi3k/akt/mtor pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the pi3k/akt pathway in primary ovarian cancer predict patient outcome. Genes Chromosom. Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (occp): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Qi, L.; Cai, Y.; Yang, P.; Xuan, G.; Jiang, Y. Hulc long noncoding rna silencing suppresses angiogenesis by regulating esm-1 via the PI3K/AKT/mTOR signaling pathway in human gliomas. Oncotarget 2016, 7, 14429–14440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.B.; Kong, R.; Yin, D.D.; You, L.H.; Sun, M.; Han, L.; Xu, T.P.; Xia, R.; Yang, J.S.; De, W.; et al. Long noncoding RNA anril indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of mir-99a/mir-449a. Oncotarget 2014, 5, 2276–2292. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Shukla, S.; Maclennan, G.T.; Hartman, D.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of pi3k-akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef]
- Roelants, C.; Giacosa, S.; Pillet, C.; Bussat, R.; Champelovier, P.; Bastien, O.; Guyon, L.; Arnoux, V.; Cochet, C.; Filhol, O. Combined inhibition of pi3k and src kinases demonstrates synergistic therapeutic efficacy in clear-cell renal carcinoma. Oncotarget 2018, 9, 30066–30078. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N., Jr.; Chavez-Reyes, A.; Bucana, C.; Schmandt, R.; Deavers, M.T.; Lopez-Berestein, G.; Sood, A.K. Therapeutic epha2 gene targeting in vivo using neutral liposomal small interfering rna delivery. Cancer Res. 2005, 65, 6910–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlon, K.J.; Wang, W.Z.; Han, H. Cyclosporin a suppresses cisplatin-induced oncogene expression in human cancer cells. Cancer Treat. Rev. 1990, 17 (Suppl. A), 27–35. [Google Scholar] [CrossRef]
- Wang, H.; Fang, L.; Jiang, J.; Kuang, Y.; Wang, B.; Shang, X.; Han, P.; Li, Y.; Liu, M.; Zhang, Z.; et al. The cisplatin-induced lncrna pandar dictates the chemoresistance of ovarian cancer via regulating sfrs2-mediated p53 phosphorylation. Cell Death Dis. 2018, 9, 1103. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, A.; Mevel, R.; Thibault, B.; Courtais, E.; Chantalat, E.; Delord, J.P.; Couderc, B.; Guillermet-Guibert, J.; Martinez, A. Effect of combined inhibition of p110 alpha pi3k isoform and stat3 pathway in ovarian cancer platinum-based resistance. Oncotarget 2018, 9, 27220–27232. [Google Scholar] [CrossRef] [Green Version]
- Carden, C.P.; Stewart, A.; Thavasu, P.; Kipps, E.; Pope, L.; Crespo, M.; Miranda, S.; Attard, G.; Garrett, M.D.; Clarke, P.A.; et al. The association of pi3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol. Cancer Ther. 2012, 11, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Mabuchi, S.; Kawase, C.; Altomare, D.A.; Morishige, K.; Sawada, K.; Hayashi, M.; Tsujimoto, M.; Yamoto, M.; Klein-Szanto, A.J.; Schilder, R.J.; et al. Mtor is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin. Cancer Res. 2009, 15, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Cheaib, B.; Auguste, A.; Leary, A. The pi3k/akt/mtor pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific pi3kα inhibitor nvp-byl719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-kinase α-selective inhibition with alpelisib (byl719) in pik3ca-altered solid tumors: Results from the first-in-human study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific pi3k inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting pi3k in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Dar, M.S.; Dar, M.J. P110α and p110β isoforms of pi3k signaling: Are they two sides of the same coin? FEBS Lett. 2016, 590, 3071–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L. Linking long noncoding rna localization and function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long noncoding rnas: At the intersection of cancer and chromatin biology. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Vance, K.W.; Ponting, C. P, Transcriptional regulatory functions of nuclear long noncoding rnas. Trends Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear long noncoding rnas: Key regulators of gene expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; McClusky, W.G.; Abdelmohsen, K.; Gorospe, M. Cytoplasmic functions of long noncoding rnas. Wiley Interdiscip. Rev. RNA 2018, 9, e1471. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.-H.; Wu, Q.-N.; Jin, Y.; Wang, D.-S.; Chen, Y.-X.; Liu, J.; Luo, X.-J.; Meng, Q.; Pu, H.-Y.; et al. Lncrna linris stabilizes igf2bp2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer 2019, 18, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Yan, T.; Bao, Y.; Shen, C.; Yu, C.; Zhu, X.; Tian, X.; Guo, F.; Liang, Q.; Liu, Q.; et al. Lncrna glcc1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-myc. Nat. Commun. 2019, 10, 3499. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Aguayo, C.; Bayraktar, E.; Ivan, C.; Aslan, B.; Mai, J.; He, G.; Mangala, L.S.; Jiang, D.; Nagaraja, A.S.; Ozpolat, B.; et al. PTGER3 induces ovary tumorigenesis and confers resistance to cisplatin therapy through up-regulation ras-mapk/erk-ets1-elk1/cftr1 axis. EBioMedicine 2019, 40, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Aguayo, C.; Monroig, P.D.C.; Redis, R.S.; Bayraktar, E.; Almeida, M.I.; Ivan, C.; Fuentes-Mattei, E.; Rashed, M.H.; Chavez-Reyes, A.; Ozpolat, B.; et al. Regulation of hnrnpa1 by micrornas controls the mir-18a-k-ras axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017, 3, 17029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Rodriguez-Aguayo, C.; Xu, R.; Gonzalez-Villasana, V.; Mai, J.; Huang, Y.; Zhang, G.; Guo, X.; Bai, L.; Qin, G.; et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin. Cancer Res. 2013, 19, 1806–1815. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Name | Sequence (Sense) 5′ to 3′ | Sequence (Antisense) 5′ to 3′ |
|---|---|---|
| PRKAR1B-AS2 siRNA 1 | GAGCACAAAUGCUGGUCUUdTdT | AAGACCAGCAUUUGUGCUCdTdT |
| PRKAR1B-AS2 siRNA 2 | GGUCUUACUACUAAGCAAUdTdT | AUUGCUUAGUAGUAAGACCdTdT |
| PRKAR1B-AS2 siRNA 3 | GCAAUAUGUCCCUCGGAUCdTdT | GAUCCGAGGGACAUAUUGCdTdT |
| Gene Symbol | Forward Primer | Reverse Primer |
|---|---|---|
| PRKAR1B-AS2 | 5′-TTGGACACTGCCCATCTT-CC3′ | 5′-TGCAGCCACGGGATGTTT-AT3′ |
| PRKAR1B-AS2 | 5′-CCTCGGATCCAAAGGAGA-GC-3′ | 5′-AAGATGGGCAGTGTCCAA-GG-3′ |
| PIK3CA | 5′-GAGTAACAGACTAGCTAGAGAC-3′ | 5′-AGAAAATCTTTCTCCTGCTC-3′ |
| GAPDH | 5′-GACAGTCAGCCGCATCTT-CT-3′ | 5′-GCGCCCAATACGACCAAA-TC-3′ |
| U6 snRNA | 5′-CTCGCTTCGGCAGCACA-3′ | 5′-GGAACGCTTCACGAATTT-GC-3′ |
| Primary Antibody | Purchasing Company (Catalog #) | Dilution Factor | Secondary Antibody | Purchasing Company | Dilution Factor |
|---|---|---|---|---|---|
| PI3 Kinase p110α (C73F8) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (4249) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| PI3 Kinase p110α Antibody | Cell Signaling Technology, Danvers, MA, USA (4255) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Phospho-mTOR (Ser2448) Antibody | Cell Signaling Technology, Danvers, MA, USA (2971) | 1:500 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| mTOR (7C10) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (2983) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Phospho-Akt (Ser473) (D9E) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (4060) | 1:400 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Akt (pan) (40D4) Mouse mAb | Cell Signaling Technology, Danvers, MA, USA (2920) | 1:1000 | Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7076) | 1:2000 |
| PDK1 Antibody | Cell Signaling Technology, Danvers, MA, USA (3062) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Phospho-PDK1 (Ser241) (C49H2) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (3438) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| PTEN (D4.3) XP® Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (9188) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| p70 S6 Kinase (49D7) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (2708) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Phospho-p70 S6 Kinase (Thr389) (1A5) Mouse mAb | Cell Signaling Technology, Danvers, MA, USA (9206) | 1:500 | Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7076) | 1:2000 |
| Phospho-Src Family (Tyr416) (D49G4) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (6943) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Src (36D10) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (2109) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Cleaved Caspase-9 (Asp315) Antibody (Human Specific) | Cell Signaling Technology, Danvers, MA, USA (9505) | 1:5000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Caspase-9 (C9) Mouse mAb | Cell Signaling Technology, Danvers, MA, USA (9508) | 1:1000 | Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7076) | 1:2000 |
| Cleaved Caspase-3 (Asp175) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (9661) | 1:300 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Caspase-3 (D3R6Y) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (14220) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Phospho-FAK (Tyr397) (D20B1) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (8556) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Cleaved Caspase-7 (Asp198) (D6H1) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (8438) | 1:400 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Caspase-7 (D2Q3L) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (12827) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Cleaved PARP (Asp214) (D64E10) XP® Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (5625) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| PARP (46D11) Rabbit mAb | Cell Signaling Technology, Danvers, MA, USA (9532) | 1:1000 | Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology, Danvers, MA, USA (7074) | 1:2000 |
| Anti-beta Actin Mouse Antibody | Abcam (ab6276) | 1:10000 | Anti-mouse IgG, HRP-linked | CST, Danvers, USA (7076) | 1:2000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed, A.M.; Bayraktar, E.; Amero, P.; Salama, S.A.; Abdelaziz, A.H.; Ismail, R.S.; Zhang, X.; Ivan, C.; Sood, A.K.; Lopez-Berestein, G.; et al. PRKAR1B-AS2 Long Noncoding RNA Promotes Tumorigenesis, Survival, and Chemoresistance via the PI3K/AKT/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041882
Elsayed AM, Bayraktar E, Amero P, Salama SA, Abdelaziz AH, Ismail RS, Zhang X, Ivan C, Sood AK, Lopez-Berestein G, et al. PRKAR1B-AS2 Long Noncoding RNA Promotes Tumorigenesis, Survival, and Chemoresistance via the PI3K/AKT/mTOR Pathway. International Journal of Molecular Sciences. 2021; 22(4):1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041882
Chicago/Turabian StyleElsayed, Abdelrahman M., Emine Bayraktar, Paola Amero, Salama A. Salama, Abdelaziz H. Abdelaziz, Raed S. Ismail, Xinna Zhang, Cristina Ivan, Anil K. Sood, Gabriel Lopez-Berestein, and et al. 2021. "PRKAR1B-AS2 Long Noncoding RNA Promotes Tumorigenesis, Survival, and Chemoresistance via the PI3K/AKT/mTOR Pathway" International Journal of Molecular Sciences 22, no. 4: 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041882