
The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance


 , and
, and
Abstract
:1. Introduction
2. The Etiology of HCC
2.1. Virus Infections Initiate HCC
2.2. Obesity and NAFLD Cause HCC
2.3. Other Factors That Cause HCC
3. IGF/IGF-1 Signaling in HCC
3.1. Abnormal IGF/IGF-1 Signaling in HCC
3.1.1. Increases in IGF-1/IGF-2 Secretion
3.1.2. Increases in IGF-1R Expression
3.1.3. Reduction in IGFBP Secretion
3.1.4. Increases in IGFBP Protease Secretion
3.1.5. Virus Infection Promotes Dysregulation of IGF/IGF-1R Signaling in HCC
3.1.6. Microenvironment Regulates IGF/IGF-1R Signaling in HCC
3.2. IGF/IGF-1R Signaling Induces Expressions of Stemness-Related Proteins in HCC
3.2.1. IGF/IGF-1R Signaling Induces Expression of Cancer Stemness-Related Transcription Factors
3.2.2. IGF/IGF-1R Signaling Induces Expressions of Stemness-Related Cell Surface Markers
3.2.3. IGF/IGF-1R Signaling Supports the Stem Cell Niche in HCC
3.2.4. HBV Replication Promotes IGF/IGF-1R Signaling-Mediated Cancer Stemness Properties in HCC
3.2.5. IGF/IGF-1R Signaling Induces Cancer Stemness Properties through Crosstalk with Cytokine Signaling
3.3. IGF/IGF-1R Signaling Induces the EMT in HCC
4. The Stemness of HCC Contributes to the Limitation of Targeted Therapies
4.1. Current Targeted Treatments for HCC
4.2. Treatment Limitations and Acquired Drug Resistance in HCC
5. IGF/IGF-1R Signaling Responses for Targeted-Drugs Resistance in HCC
5.1. IGF/IGF-1 Signaling in Sorafenib Resistance
5.2. IGF/IGF-1 Signaling in Other Targeted-Drug Resistances
6. IGF/IGF-1 Signaling as a Potential Target for the Treatment of Advanced HCC
6.1. Former Trials Targeting IGF/IGF-1R Signaling in HCC
6.2. Reasons for Failing Trials and Potential Improvement of IGF/IGF-1R Targeted Therapies in HBV-HCC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFR | Albumin-to-Fibrinogen Ratio |
| AIF-1 | Allograft Inflammatory Factor-1 |
| Akt | Ak Strain Transforming |
| ALDH | Aldehyde Dehydrogenase |
| Bax | BCL2 Associated X |
| BC-HCC | Hepatitis B and C-Hepatocellular Carcinoma |
| Bcl2 | BCL2 Apoptosis Regulator |
| CAF | Cancer-Associated Fibroblasts |
| c-KIT | KIT Proto-Oncogene |
| CM | Conditioned Media |
| CRISPR/Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats Cas9 |
| CSC | Cancer Stem Cell |
| DAAs | Direct-Acting Antivirus |
| DENA | Diethylnitrosamine |
| DNA | Deoxyribonucleic Acid |
| DNMT | DNA Methyltransferase |
| EMT | Epithelial-Mesenchymal Transition |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| FDA | U.S. Food and Drug Administration |
| FGF | Fibroblast Growth Factor |
| FLT3 | FMS-Like Tyrosine Kinase 3 |
| GH | Growth Hormone |
| HBeAg | Hepatitis B E-Antigen |
| HBV | Hepatitis B Virus |
| HBx | Hepatitis B Virus X Protein |
| HCC | Hepatocellular Carcinoma |
| HCV | Hepatitis C Virus |
| hfMSC | Human Fetal Mesenchymal Stem Cell |
| HIF | Hypoxia-Inducible Factor |
| HIV | Human Immunodeficiency Virus |
| IGF-1/2 | Insulin-Like Growth Factor 1/2 |
| IGF-1R | Insulin-Like Growth Factor 1 Receptor |
| IGFBP | Insulin-Like Growth Factor Binding Protein |
| IL-6 | Interleukin 6 |
| INSR-A | Insulin Receptor-A |
| Jak | Janus Kinase |
| KPS | Karnofsky Performance Status Scale |
| lncRNA | Long Non-Coding RNA |
| MAPK (Erk) | Mitogen-Activated Protein Kinase |
| MAPKK (Mek) | Mitogen-Activated Protein Kinase Kinase |
| MER | Mer Receptor Tyrosine Kinase |
| MET | Receptor Tyrosine Kinase |
| miRNA | MicroRNA |
| MMP | Matrix Metalloproteinases |
| mRNA | Messenger RNA |
| MSC | Mesenchymal Stem Cell |
| mTOR | Mechanistic Target of Rapamycin |
| NANOG | Nanog Homeobox |
| NBNC | Non-B Non-C Hepatocellular Carcinoma |
| NFκB | Nuclear Factor Kappa Beta |
| NGF | Nerve Growth Factor |
| OCT4 | Octamer-Binding Transcription Factor 4 |
| ORF | Open Reading Frame |
| OS | Overall Survival |
| OV6 | Oval Cell Marker Antibody 6 |
| PD1 | Programmed Cell Death Protein 1 |
| PDGFR | Platelet Derived Growth Factor Receptor |
| PFS | Progression Free Survival |
| PI-3K | Phosphatidylinositol-3-Kinase |
| PROM1 (CD133) | Prominin-1 |
| PSA | Prostate-Specific Antigen |
| Raf | Rapidly Accelerated Fibrosarcoma |
| Rb pathway | Retinoblastoma Pathway |
| REACH | Research to Assess Svs [Spiration® Valve System] |
| RET | Rearranged During Transfection |
| rhGH | Recombinant Human GH |
| ROS1 | Ros Proto-Oncogene 1 |
| RUNX | Runt-Related Transcription Factor |
| SCF | Stem Cell Factor |
| SHARP | Study of Heart and Renal Protection |
| SiRNA | Silent RNA |
| SOX2 | Sex Determining Region Y-Box2 |
| STATs | Signal Transducer and Activator of Transcription |
| TAZ | Tafazzin |
| TBX5 | T-Box Transcription Factor 5 |
| TCGA | The Cancer Genome Atlas |
| TEAD | Tea Domain Family Member |
| TGF-β | Transforming Growth Factor Beta-1 |
| TIA-1 | T-Cell-Restricted Intracellular Antigen-1 |
| TIE2 | Tek Receptor Tyrosine Kinase |
| TYRO3 | Tyrosine-Protein Kinase Receptor 3 |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| WHO | World Health Organization |
| Wnt | Wingless-Related Integration Site |
| YAP | Yes-Associated Protein |
| ZEB1 | Zinc Finger E-Box Binding Homeobox 1 |
References
- European Association for the Study of the Liver. EASL clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Mak, L.-Y.; Cruz-Ramón, V.; Chinchilla-López, P.; Torres, H.A.; LoConte, N.K.; Rice, J.P.; Foxhall, L.E.; Sturgis, E.M.; Merrill, J.K.; Bailey, H.H.; et al. Global epidemiology, prevention, and management of hepatocellular carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 262–279. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.C.; Huang, S.F.; Chen, P.J.; Chen, C.L.; Chen, C.L.; Wu, C.C.; Tsai, C.C.; Lee, P.H.; Chen, M.F.; Lee, C.M.; et al. The hepatitis viral status in patients with hepatocellular carcinoma: A study of 3843 patients from taiwan liver cancer network. Medicine 2016, 95, e3284. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the global burden of disease study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- World-Health-Organization. Global Hepatitis Report, 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- World-Health-Organization. Global Health Sector Strategy on Viral Hepatitis 2016–2021: Towards Ending Viral Hepatitis; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the asia-pacific region with advanced hepatocellular carcinoma: A phase iii randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of osi-906: A selective and orally efficacious dual inhibitor of the igf-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef]
- Valaydon, Z.S.; Locarnini, S.A. The virological aspects of hepatitis b. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Colgrove, R.; Simon, G.; Ganem, D. Transcriptional activation of homologous and heterologous genes by the hepatitis b virus x gene product in cells permissive for viral replication. J. Virol. 1989, 63, 4019–4026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, H.; Ye, L. Effects of hepatitis b virus x protein on the development of liver cancer. J. Lab. Clin. Med. 2006, 147, 58–66. [Google Scholar] [CrossRef]
- Weil, R.; Sirma, H.; Giannini, C.; Kremsdorf, D.; Bessia, C.; Dargemont, C.; Bréchot, C.; Israël, A. Direct association and nuclear import of the hepatitis b virus x protein with the nf-κb inhibitor iκbα. Mol. Cell. Biol. 1999, 19, 6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgues, M.; Marrogi, A.J.; Spillare, E.A.; Wu, C.G.; Yang, Q.; Yoshida, M.; Wang, X.W. Interaction of the hepatitis b virus x protein with the crm1-dependent nuclear export pathway. J. Biol. Chem. 2001, 276, 22797–22803. [Google Scholar] [CrossRef] [Green Version]
- Kidd-Ljunggren, K.; Oberg, M.; Kidd, A.H. Hepatitis b virus x gene 1751 to 1764 mutations: Implications for hbeag status and disease. J. Gen. Virol. 1997, 78, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Duan, L.-X.; Guo, J.; Sun, B.; Woo, J.; Steensma, K.; Horiike, N.; Blumberg, B.S. X region deletion variants of hepatitis b virus in surface antigen-negative infections and non-a, non-b hepatitis. J. Infect. Dis. 1995, 172, 713–722. [Google Scholar] [CrossRef]
- Gearhart, T.L.; Bouchard, M.J. The hepatitis b virus x protein modulates hepatocyte proliferation pathways to stimulate viral replication. J. Virol. 2010, 84, 2675. [Google Scholar] [CrossRef] [Green Version]
- Ayub, A.; Ashfaq, U.A.; Haque, A. Hbv induced hcc: Major risk factors from genetic to molecular level. Biomed. Res. Int. 2013, 2013, 810461. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.-N.; Kuo, C.-F.; Ou, J.-H.J. Mechanisms of hepatitis b virus persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef]
- Umar, M.; Hamama Tul, B.; Umar, S.; Khan, H.A. Hbv perinatal transmission. Int. J. Hepatol. 2013, 2013, 875791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, B.J.; Alward, W.L.; Hall, D.B.; Heyward, W.L.; Bender, T.R.; Francis, D.P.; Maynard, J.E. Acute hepatitis b virus infection: Relation of age to the clinical expression of disease and subsequent development of the carrier state. J. Infect. Dis. 1985, 151, 599–603. [Google Scholar] [CrossRef]
- Tassopoulos, N.C.; Papaevangelou, G.J.; Sjogren, M.H.; Roumeliotou-Karayannis, A.; Gerin, J.L.; Purcell, R.H. Natural history of acute hepatitis b surface antigen-positive hepatitis in greek adults. Gastroenterology 1987, 92, 1844–1850. [Google Scholar] [CrossRef]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.-S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.-H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis c virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of hcv infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, A.; Cooke, G.S.; Nayagam, S.; Thursz, M.; Hallett, T.B. Scaling up prevention and treatment towards the elimination of hepatitis c: A global mathematical model. Lancet 2019, 393, 1319–1329. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular mechanisms of hepatitis c virus–induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Aydin, Y.; Widmer, K.E.; Nayak, L. Hepatocellular carcinoma mechanisms associated with chronic hcv infection and the impact of direct-acting antiviral treatment. J. Hepatocell. Carcinoma 2020, 7, 45–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Chu, Y.T.; Shau, W.Y.; Kuo, R.N.; Chen, P.J.; Lai, M.S. Treatment of patients with dual hepatitis c and b by peginterferon α and ribavirin reduced risk of hepatocellular carcinoma and mortality. Gut 2014, 63, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Marot, A.; Belaid, A.; Orlent, H.; Sersté, T.; Michielsen, P.; Colle, I.; Laleman, W.; de Galocsy, C.; Reynaert, H.; D’Heygere, F.; et al. Characteristics of patients with hepatitis b virus and hepatitis c virus dual infection in a western european country: Comparison with monoinfected patients. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.L.; Di Bisceglie, A.M.; Waggoner, J.G.; Waggoner, J.G.; Banks, S.M.; Hoofnagle, J.H. The significance of antibody to hepatitis c virus in patients with chronic hepatitis b. Hepatology 1991, 14, 64–67. [Google Scholar] [CrossRef]
- Al Karawi, M.A.; Mesa, G.A. Dual infection with hepatitis c and b viruses: Clinical and histological study in saudi patients. Hepatogastroenterology 1997, 44, 1404–1406. [Google Scholar]
- Thio, C.L.; Seaberg, E.C.; Skolasky, R.; Phair, J.; Visscher, B.; Muñoz, A.; Thomas, D.L. Hiv-1, hepatitis b virus, and risk of liver-related mortality in the multicenter cohort study (macs). Lancet 2002, 360, 1921–1926. [Google Scholar] [CrossRef]
- Soriano, V.; Vispo, E.; Labarga, P.; Medrano, J.; Barreiro, P. Viral hepatitis and hiv co-infection. Antiviral Res. 2010, 85, 303–315. [Google Scholar] [CrossRef]
- Maier, I.; Wu, G.Y. Hepatitis c and hiv co-infection: A review. World J. Gastroenterol. 2002, 8, 577–579. [Google Scholar] [CrossRef]
- Smith, K.B.; Smith, M.S. Obesity statistics. Prim. Care 2016, 43, 121–135, ix. [Google Scholar] [CrossRef]
- Rui, R.; Lou, J.; Zou, L.; Zhong, R.; Wang, J.; Xia, D.; Wang, Q.; Li, H.; Wu, J.; Lu, X.; et al. Excess body mass index and risk of liver cancer: A nonlinear dose-response meta-analysis of prospective studies. PLoS ONE 2012, 7, e44522. [Google Scholar] [CrossRef]
- Hashimoto, M.; Tashiro, H.; Kobayashi, T.; Kuroda, S.; Hamaoka, M.; Ohdan, H. Influence of higher bmi for hepatitis b- and c-related hepatocellular carcinomas. Langenbecks Arch. Surg. 2017, 402, 745–755. [Google Scholar] [CrossRef]
- Nugent, C.; Younossi, Z.M. Evaluation and management of obesity-related nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 432–441. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed nafld: Real-world study of 18 million patients in four european cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef]
- Kim, G.-A.; Lee, H.C.; Choe, J.; Kim, M.-J.; Lee, M.J.; Chang, H.-S.; Bae, I.Y.; Kim, H.-K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2018, 68, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Fartoux, L.; Goumard, C.; Scatton, O.; Wendum, D.; Rosmorduc, O.; Ratziu, V. Temporal trends, clinical patterns and outcomes of nafld-related hcc in patients undergoing liver resection over a 20-year period. Aliment. Pharmacol. Ther. 2017, 46, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.-L.; Ma, Y. Aflatoxin sufferer and p53 gene mutation in hepatocellular carcinoma. World J. Gastroenterol. 1998, 4, 28–29. [Google Scholar] [CrossRef]
- Macé, K.; Aguilar, F.; Wang, J.S.; Vautravers, P.; Gómez-Lechón, M.; Gonzalez, F.J.; Groopman, J.; Harris, C.C.; Pfeifer, A.M. Aflatoxin b1-induced DNA adduct formation and p53 mutations in cyp450-expressing human liver cell lines. Carcinogenesis 1997, 18, 1291–1297. [Google Scholar] [CrossRef] [Green Version]
- Kew, M.C. Synergistic interaction between aflatoxin and hepatitis b virus in hepatocarcinogenesis. Liver Int. 2013, 23, 405–409. [Google Scholar] [CrossRef]
- Stern, M.C.; Umbach, D.M.; Yu, M.C.; London, S.J.; Zhang, Z.-Q.; Taylor, J.A. Hepatitis b, aflatoxin b1, and p53 codon 249 mutation in hepatocellular carcinomas from guangxi, people’s republic of china, and a meta-analysis of existing studies. Cancer Epidemiol. Biomark. Prev. 2001, 10, 617. [Google Scholar]
- Chittmittrapap, S.; Chieochansin, T.; Chaiteerakij, R.; Treeprasertsuk, S.; Klaikaew, N.; Tangkijvanich, P.; Komolmit, P.; Poovorawan, Y. Prevalence of aflatoxin induced p53 mutation at codon 249 (r249s) in hepatocellular carcinoma patients with and without hepatitis b surface antigen (hbsag). Asian Pac. J. Cancer Prev. 2013, 14, 7675–7679. [Google Scholar] [CrossRef] [Green Version]
- Niemelä, O.; Parkkila, S.; Pasanen, M.; Iimuro, Y.; Bradford, B.; Thurman, R.G. Early alcoholic liver injury: Formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of cyp2e1 and cyp3a. Alcohol Clin. Exp. Res. 1998, 22, 2118–2124. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Horne, W.; Kamimura, S.; Niemelä, O.; Parkkila, S.; Ylä-Herttuala, S.; Brittenham, G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Investig. 1995, 96, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Ganne-Carrie, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Minicis, S.; Brenner, D.A. Oxidative stress in alcoholic liver disease: Role of nadph oxidase complex. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S98–S103. [Google Scholar] [CrossRef] [PubMed]
- Zago, A.; Moreira, F.P.; Jansen, K.; Lhullier, A.C.; da Silva, R.A.; de Oliveira, J.F.; Medeiros, J.R.C.; Colpo, G.B.; Portela, L.V.; Lara, D.R.; et al. Alcohol use disorder and inflammatory cytokines in a population sample of young adults. Drug Alcohol Depend. 2016, 4, 2. [Google Scholar] [CrossRef]
- Neupane, S.P.; Skulberg, A.; Skulberg, K.R.; Aass, H.C.D.; Bramness, J.G. Cytokine changes following acute ethanol intoxication in healthy men: A crossover study. Mediat. Inflamm. 2016, 2016, 3758590. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Galeone, C.; Rota, M.; Pelucchi, C.; Negri, E.; Bagnardi, V.; Corrao, G.; Boffetta, P.; La Vecchia, C. Alcohol and liver cancer: A systematic review and meta-analysis of prospective studies. Ann. Oncol. 2014, 25, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.; Ayer, T.; Bethea, E.D.; Tapper, E.B.; Chhatwal, J. Projected prevalence and mortality associated with alcohol-related liver disease in the USA, 2019–2040: A modelling study. Lancet Public Health 2020, 5, e316–e323. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. Hcc development is associated to peripheral insulin resistance in a mouse model of nash. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-like growth factor (igf) system in liver diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Hu, W.; Hu, J.; Wu, S.; Li, J.; Luo, Y.; Cao, M.; Zhou, H.; Jiang, X. Hepatitis b virus x protein promotes p3 transcript expression of the insulin-like growth factor 2 gene via inducing hypomethylation of p3 promoter in hepatocellular carcinoma. Liver Int. 2015, 35, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.L.; Kwon, T.; Bak, I.S.; Erikson, R.L.; Kim, B.Y.; Yu, D.Y. Igf-ii induced by hepatitis b virus x protein regulates emt via sumo mediated loss of e- cadherin in mice. Oncotarget 2016, 7, 56944–56957. [Google Scholar] [CrossRef]
- Kadakia, R.; Josefson, J. The relationship of insulin-like growth factor 2 to fetal growth and adiposity. Horm. Res. Paediatr. 2016, 85, 75–82. [Google Scholar] [CrossRef]
- Oberbauer, A.M. The regulation of igf-1 gene transcription and splicing during development and aging. Front. Oncol. 2013, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguay, S.J. Post-translational processing of insulin-like growth factors. Horm. Metab. Res. 1999, 31, 43–49. [Google Scholar] [CrossRef]
- Abdel-Wahab, R.; Shehata, S.; Hassan, M.M.; Habra, M.A.; Eskandari, G.; Tinkey, P.T.; Mitchell, J.; Lee, J.-S.; Amin, H.M.; Kaseb, A.O. Type i insulin-like growth factor as a liver reserve assessment tool in hepatocellular carcinoma. J. Hepatol. 2015, 2, 131–142. [Google Scholar]
- Weroha, S.J.; Haluska, P. The insulin-like growth factor system in cancer. Endocrinol. Metab. Clin. N. Am. 2012, 41, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Vyzantiadis, T.; Theodoridou, S.; Giouleme, O.; Harsoulis, P.; Evgenidis, N.; Vyzantiadis, A. Serum concentrations of insulin-like growth factor-i (igf-i) in patients with liver cirrhosis. Hepatogastroenterology 2003, 50, 814–816. [Google Scholar]
- Rosario, W.P. Normal values of serum igf-1 in adults: Results from a brazilian population. Arquivos Brasileiros de Endocrinologia Metabologia 2010, 54, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Huber, Y.; Bierling, F.; Labenz, C.; Koch, S.; Schmidtmann, I.; Kloeckner, R.; Schotten, S.; Huber, T.; Lang, H.; Woerns, M.A.; et al. Validation of insulin-like growth factor-1 as a prognostic parameter in patients with hepatocellular carcinoma in a european cohort. BMC Cancer 2018, 18, 774. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Luo, S.-M.; Liang, L.; Lai, J. Effects of parenteral nutrition without and with growth hormone on growth hormone/insulin-like growth factor-1 axis after hepatectomy in hepatocellular carcinoma with liver cirrhosis. J. Parenter. Enteral Nutr. 2007, 31, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Zapf, J.; Walter, H.; Froesch, E.R. Radioimmunological determination of insulin-like growth factors i and ii in normal subjects and in patients with growth disorders and extrapancreatic tumour hypoglycaemia. J. Clin. Investig. 1981, 68, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbel, R.E. Insulin-like growth factors i and ii. Eur. J. Biochem. 1990, 190, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.M.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Solé, M.; et al. Igf2 is up-regulated by epigenetic mechanisms in hepatocellular carcinomas and is an actionable oncogene product in experimental models. Gastroenterology 2016, 151, 1192–1205. [Google Scholar] [CrossRef]
- Fumihiko, H.; Shin-Ichiro, T. 40 years of igf1: Igf1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69. [Google Scholar]
- Wang, Z.; Ruan, Y.B.; Guan, Y.; Liu, S.H. Expression of igf-ii in early experimental hepatocellular carcinomas and its significance in early diagnosis. World J. Gastroenterol. 2003, 9, 267–270. [Google Scholar] [CrossRef]
- Philippou, A.; Maridaki, M.; Pneumaticos, S.; Koutsilieris, M. The complexity of the igf1 gene splicing, posttranslational modification and bioactivity. Mol. Med. 2014, 20, 202–214. [Google Scholar] [CrossRef]
- Lara-Diaz, V.J.; Castilla-Cortazar, I.; Martín-Estal, I.; García-Magariño, M.; Aguirre, G.A.; Puche, J.E.; de la Garza, R.G.; Morales, L.A.; Muñoz, U. Igf-1 modulates gene expression of proteins involved in inflammation, cytoskeleton, and liver architecture. J. Physiol. Biochem. 2017, 73, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Reiss, K.; Ferber, A.; Travali, S.; Porcu, P.; Phillips, P.D.; Baserga, R. The protooncogene c-myb increases the expression of insulin-like growth factor 1 and insulin-like growth factor 1 receptor messenger rnas by a transcriptional mechanism. Cancer Res. 1991, 51, 5997–6000. [Google Scholar] [PubMed]
- Werner, H.; Sarfstein, R.; LeRoith, D.; Bruchim, I. Insulin-like growth factor 1 signaling axis meets p53 genome protection pathways. Front. Oncol. 2016, 6, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shi, H.; Chen, H.; Gong, A.; Liu, Y.; Song, L.; Xu, X.; You, T.; Fan, X.; Wang, D.; et al. Dedifferentiation process driven by radiotherapy-induced hmgb1/tlr2/yap/hif-1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedemann, J.; Macaulay, V.M. Igf1r signalling and its inhibition. Endocr. Relat. Cancer 2006, 13, S33. [Google Scholar] [CrossRef]
- Vivian Hwa, Y.O.; Ron, G. Rosenfeld The insulin-like growth factor-binding protein (igfbp) superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar]
- Clemmons, D.R.; Busby, W.H.; Arai, T.; Nam, T.J.; Clarke, J.B.; Jones, J.I.; Ankrapp, D.K. Role of insulin-like growth factor binding proteins in the control of igf actions. Mol. Cell Endocrinol. 1998, 140, 19–24. [Google Scholar] [CrossRef]
- Yu, H.; Mistry, J.; Nicar, M.J.; Khosravi, M.J.; Diamandis, A.; van Doorn, J.; Juul, A. Insulin-like growth factors (igf-i, free igf-i, and igf-ii) and insulin-like growth factor binding proteins (igfbp-2, igfbp-3, igfbp-6, and als) in blood circulation. J. Clin. Lab. Anal. 1999, 13, 166–172. [Google Scholar] [CrossRef]
- Abdelhakeem, A.; Kaseb, A.O.; Hatia, R.; Abdel-Wahab, R.; Amin, H.M.; Hassan, M. Distribution of insulin growth factor-1 (igf-1) binding proteins in hepatocellular carcinoma with and without cirrhosis. J. Clin. Oncol. 2019, 37, 193. [Google Scholar] [CrossRef]
- Yumoto, E.; Nakatsukasa, H.; Hanafusa, T.; Yumoto, Y.; Nouso, K.; Matsumoto, E.; Onishi, T.; Takuma, Y.; Tanaka, H.; Fujikawa, T.; et al. Igfbp-3 expression in hepatocellular carcinoma involves abnormalities in tgf-beta and/or rb signaling pathways. Int. J. Oncol. 2005, 27, 1223–1230. [Google Scholar]
- Regel, I.; Eichenmüller, M.; Joppien, S.; Liebl, J.; Häberle, B.; Müller-Höcker, J.; Vollmar, A.; von Schweinitz, D.; Kappler, R. Igfbp3 impedes aggressive growth of pediatric liver cancer and is epigenetically silenced in vascular invasive and metastatic tumors. Mol. Cancer 2012, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Shen, F.; Weinfeld, M.; Sergi, C. Insulin growth factor binding protein 7 (igfbp7)-related cancer and igfbp3 and igfbp7 crosstalk. Front. Oncol. 2020, 10, 727. [Google Scholar] [CrossRef]
- Akiel, M.; Guo, C.; Li, X.; Rajasekaran, D.; Mendoza, R.G.; Robertson, C.L.; Jariwala, N.; Yuan, F.; Subler, M.A.; Windle, J.; et al. Igfbp7 deletion promotes hepatocellular carcinoma. Cancer Res. 2017, 77, 4014–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fürstenberger, G.; Senn, H.J. Insulin-like growth factors and cancer. Lancet Oncol. 2002, 3, 298–302. [Google Scholar] [CrossRef]
- Yan, J.; Yang, X.; Li, L.; Liu, P.; Wu, H.; Liu, Z.; Li, Q.; Liao, G.; Wang, X. Low expression levels of insulin-like growth factor binding protein-3 are correlated with poor prognosis for patients with hepatocellular carcinoma. Oncol. Lett. 2017, 13, 3395–3402. [Google Scholar] [CrossRef] [Green Version]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to egfr tyrosine kinase inhibitors in cancer cells is mediated by loss of igf-binding proteins. J. Clin. Investig. 2008, 11, 2609–2619. [Google Scholar] [CrossRef]
- Butt, A.J.; Firth, S.M.; King, M.A.; Baxter, R.C. Insulin-like growth factor-binding protein-3 modulates expression of bax and bcl-2 and potentiates p53-independent radiation-induced apoptosis in human breast cancer cells. J. Biol. Chem. 2000, 275, 39174–39181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleem, E.; Elshayeb, A.; Elhabachi, N.; Mansour, A.R.; Gowily, A.; Hela, A. Serum igfbp-3 is a more effective predictor than igf-1 and igf-2 for the development of hepatocellular carcinoma in patients with chronic hcv infection. Oncol. Lett. 2012, 3, 704–712. [Google Scholar] [CrossRef] [Green Version]
- El Tayebi, H.M.; Waly, A.A.; Assal, R.A.; Hosny, K.A.; Esmat, G.; Abdelaziz, A.I. Transcriptional activation of the igf-ii/igf-1r axis and inhibition of igfbp-3 by mir-155 in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 3206–3212. [Google Scholar] [CrossRef]
- Subramaniam, K.; Ooi, L.L.P.J.; Hui, K.M. Transcriptional down-regulation of igfbp-3 in human hepatocellular carcinoma cells is mediated by the binding of tia-1 to its at-rich element in the 3′-untranslated region. Cancer Lett. 2010, 297, 259–268. [Google Scholar] [CrossRef]
- Chen, D.; Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Bhutia, S.K.; Das, S.K.; Fuller, C.; Su, Z.Z.; Fisher, P.B.; Sarkar, D. Insulin-like growth factor-binding protein-7 functions as a potential tumor suppressor in hepatocellular carcinoma. Clin. Cancer Res. 2011, 17, 6693–6701. [Google Scholar] [CrossRef] [Green Version]
- Scharf, J.G.; Dombrowski, F.; Ramadori, G. The igf axis and hepatocarcinogenesis. Mol. Pathol. 2001, 54, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumminello, F.M.; Leto, G.; Pizzolanti, G.; Candiloro, V.; Crescimanno, M.; Crosta, L.; Flandina, C.; Montalto, G.; Soresi, M.; Carroccio, A.; et al. Cathepsin d, b and l circulating levels as prognostic markers of malignant progression. Anticancer Res. 1996, 16, 2315–2319. [Google Scholar] [PubMed]
- Rajah, R.; Katz, L.; Nunn, S.; Solberg, P.; Beers, T.; Cohen, P. Insulin-like growth factor binding protein (igfbp) proteases: Functional regulators of cell growth. Prog. Growth Factor Res. 1995, 6, 273–284. [Google Scholar] [CrossRef]
- Ryan, A.J.; Napoletano, S.; Fitzpatrick, P.A.; Currid, C.A.; O’Sullivan, N.C.; Harmey, J.H. Expression of a protease-resistant insulin-like growth factor-binding protein-4 inhibits tumour growth in a murine model of breast cancer. Br. J. Cancer 2009, 101, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Smith, E.P.; Kuroda, H.; Banach, W.; Chernausek, S.D.; Fagin, J.A. Targeted expression of a protease-resistant igfbp-4 mutant in smooth muscle of transgenic mice results in igfbp-4 stabilization and smooth muscle hypotrophy. J. Biol. Chem. 2002, 277, 21285–21290. [Google Scholar] [CrossRef] [Green Version]
- Brahmkhatri, V.P.; Prasanna, C.; Atreya, H.S. Insulin-like growth factor system in cancer: Novel targeted therapies. Biomed. Res. Int. 2015, 2015, 538019. [Google Scholar] [CrossRef] [Green Version]
- Bunn, R.C.; Fowlkes, J.L. Insulin-like growth factor binding protein proteolysis. Trends Endocrinol. Metab. 2003, 14, 176–181. [Google Scholar] [CrossRef]
- Bach, L.A.; Fu, P.; Yang, Z. Insulin-like growth factor-binding protein-6 and cancer. Clin. Sci. 2012, 124, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Tailor, P.D.; Kodeboyina, K.S.; Bai, S.; Patel, N.; Sharma, S.; Ratnani, A.; Copland, J.A.; She, J.X.; Sharma, A. Diagnostic and prognostic biomarker potential of kallikrein family genes in different cancer types. Oncotarget 2018, 9, 17876–17888. [Google Scholar] [CrossRef] [Green Version]
- Brouillet, J.P.; Hanslick, B.; Maudelonde, T.; Pivat, M.T.; Grenier, J.; Blanc, F.; Rochefort, H. Increased plasma cathepsin d concentration in hepatic carcinoma and cirrhosis but not in breast cancer. Clin. Biochem. 1991, 24, 491–496. [Google Scholar] [CrossRef]
- Yeh, H.C.; Lin, S.M.; Chen, M.F.; Pan, T.L.; Wang, P.W.; Yeh, C.T. Evaluation of serum matrix metalloproteinase (mmp)-9 to mmp-2 ratio as a biomarker in hepatocellular carcinoma. Hepatogastroenterology 2010, 57, 98–102. [Google Scholar]
- Kim, S.O.; Park, J.G.; Lee, Y.I. Increased expression of the insulin-like growth factor i (igf-i) receptor gene in hepatocellular carcinoma cell lines: Implications of igf-i receptor gene activation by hepatitis b virus x gene product. Cancer Res. 1996, 56, 3831–3836. [Google Scholar] [PubMed]
- Lee, Y.I.; Lee, S.; Lee, Y.; Bong, Y.S.; Hyun, S.W.; Yoo, Y.D.; Kim, S.J.; Kim, Y.W.; Poo, H.R. The human hepatitis b virus transactivator x gene product regulates sp1 mediated transcription of an insulin-like growth factor ii promoter 4. Oncogene 1998, 16, 2367–2380. [Google Scholar] [CrossRef] [Green Version]
- Sohda, T.; Kamimura, S.; Iwata, K.; Shijo, H.; Okumura, M. Immunohistochemical evidence of insulin-like growth factor ii in human small hepatocellular carcinoma with hepatitis c virus infection: Relationship to fatty change in carcinoma cells. J. Gastroenterol. Hepatol. 1997, 12, 224–228. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Candido, S.; Cervello, M.; Nicolosi, D.; Raiti, F.; Travali, S.; Spandidos, D.A.; Libra, M. The tumor microenvironment in hepatocellular carcinoma (review). Int. J. Oncol. 2012, 40, 1733–1747. [Google Scholar] [PubMed] [Green Version]
- Liu, Q.; Xu, Z.; Mao, S.; Chen, W.; Zeng, R.; Zhou, S.; Liu, J. Effect of hypoxia on hypoxia inducible factor-1α, insulin-like growth factor i and vascular endothelial growth factor expression in hepatocellular carcinoma hepg2 cells. Oncol. Lett. 2015, 9, 1142–1148. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Song, J.; Liu, K.; Ji, H.; Shen, H.; Hu, S.; Yang, G.; Du, Y.; Zou, X.; Jin, H.; et al. The expression of hypoxia-inducible factor-1α in hepatitis b virus-related hepatocellular carcinoma: Correlation with patients’ prognosis and hepatitis b virus x protein. Dig. Dis. Sci. 2008, 53, 3225–3233. [Google Scholar] [CrossRef]
- Yulyana, Y.; Ho, I.A.W.; Sia, K.C.; Newman, J.P.; Toh, X.Y.; Endaya, B.B.; Chan, J.K.Y.; Gnecchi, M.; Huynh, H.; Chung, A.Y.F.; et al. Paracrine factors of human fetal mscs inhibit liver cancer growth through reduced activation of igf-1r/pi3k/akt signaling. Mol. Ther. 2015, 23, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Rupp, C.; Scherzer, M.; Rudisch, A.; Unger, C.; Haslinger, C.; Schweifer, N.; Artaker, M.; Nivarthi, H.; Moriggl, R.; Hengstschläger, M.; et al. Igfbp7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor–stroma interaction. Oncogene 2015, 34, 815–825. [Google Scholar] [CrossRef]
- Sarfstein, R.; Werner, H. Minireview: Nuclear insulin and insulin-like growth factor-1 receptors: A novel paradigm in signal transduction. Endocrinology 2013, 154, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, L.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yan, X.; Ye, B.; Li, C.; Xia, P.; et al. The long noncoding rna lnctcf7 promotes self-renewal of human liver cancer stem cells through activation of wnt signaling. Cell Stem Cell 2015, 16, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamwal, G.; Singh, G.; Dar, M.S.; Singh, P.; Bano, N.; Syed, S.H.; Sandhu, P.; Akhter, Y.; Monga, S.P.; Dar, M.J. Identification of a unique loss-of-function mutation in igf1r and a crosstalk between igf1r and wnt/beta-catenin signaling pathways. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Bodzin, A.S.; Wei, Z.; Hurtt, R.; Gu, T.; Doria, C. Gefitinib resistance in hcc mahlavu cells: Upregulation of cd133 expression, activation of igf-1r signaling pathway, and enhancement of igf-1r nuclear translocation. J. Cell Physiol. 2012, 227, 2947–2952. [Google Scholar] [CrossRef]
- Shan, J.; Shen, J.; Liu, L.; Xia, F.; Xu, C.; Duan, G.; Xu, Y.; Ma, Q.; Yang, Z.; Zhang, Q.; et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology 2012, 56, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Wu, Y.C.; Chi, C.C.; Su, W.C.; Chang, P.J.; Lee, K.F.; Tung, T.H.; Wang, J.; Liu, J.J.; Tung, S.Y.; et al. Activation of il6/igfir confers poor prognosis of hbv-related hepatocellular carcinoma through induction of oct4/nanog expression. Clin. Cancer Res. 2015, 21, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.J.H.; Thng, D.K.H.; Lim, J.J.; Toh, T.B. Jak/stat signaling in hepatocellular carcinoma. Hepat. Oncol. 2020, 7, HEP18. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting stat family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Mao, J.; Yang, H.; Cui, T.; Pan, P.; Kabir, N.; Chen, D.; Ma, J.; Chen, X.; Chen, Y.; Yang, Y. Combined treatment with sorafenib and silibinin synergistically targets both hcc cells and cancer stem cells by enhanced inhibition of the phosphorylation of stat3/erk/akt. Eur. J. Pharmacol. 2018, 832, 39–49. [Google Scholar] [CrossRef]
- Chang, T.S.; Chen, C.L.; Wu, Y.C.; Liu, J.J.; Kuo, Y.C.; Lee, K.F.; Lin, S.Y.; Lin, S.E.; Tung, S.Y.; Kuo, L.M.; et al. Inflammation promotes expression of stemness-related properties in hbv-related hepatocellular carcinoma. PLoS ONE 2016, 11, e0149897. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.C.; Su, Y.T.; Chi, C.C.; Kuo, Y.C.; Lee, K.F.; Wu, Y.C.; Lan, P.C.; Yang, M.H.; Chang, T.S.; Huang, Y.A.-O. Dnmt3b/oct4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through il-6/stat3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Youness, R.A.; El-Tayebi, H.M.; Assal, R.A.; Hosny, K.; Esmat, G.; Abdelaziz, A.I. Microrna-486-5p enhances hepatocellular carcinoma tumor suppression through repression of igf-1r and its downstream mtor, stat3 and c-myc. Oncol. Lett. 2016, 12, 2567–2573. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wang, Q.; Wang, B.; Sun, Q.; He, Z.; Hong, J.; Kuehn, F.; Liu, E.; Zhang, Z. Igf-1 induces the epithelial-mesenchymal transition via stat5 in hepatocellular carcinoma. Oncotarget 2017, 8, 111922–111930. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, C.; Zhang, Q.; Shen, J.; Zhang, H.; Shan, J.; Duan, G.; Guo, D.; Chen, X.; Cheng, J.; et al. Sirt1-mediated transcriptional regulation of sox2 is important for self-renewal of liver cancer stem cells. Hepatology 2016, 64, 814–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in hcc cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Mizushima, T.; Yokoyama, Y.; Hirose, H.; Wu, X.; Qian, Y.; Ikehata, K.; Miyoshi, N.; Takahashi, H.; Haraguchi, N.; et al. Sox2 is associated with cancer stem-like properties in colorectal cancer. Sci. Rep. 2018, 8, 17639. [Google Scholar] [CrossRef]
- Apostolou, P.; Toloudi, M.; Chatziioannou, M.; Ioannou, E.; Papasotiriou, I. Cancer stem cells stemness transcription factors expression correlates with breast cancer disease stage. Curr. Stem Cell Res. Ther. 2012, 7, 415–419. [Google Scholar] [CrossRef]
- Müller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, M.Y.; Guo, Q.; Liu, J.; Yang, R.; Bai, J.; Wang, W.; Cai, Y.; Han, R.; Lv, Y.Q.; Ding, L.; et al. An fgfr/akt/sox2 signaling axis controls pancreatic cancer stemness. Front. Cell Dev. Biol. 2020, 8, 287. [Google Scholar] [CrossRef]
- Bu, Y.; Jia, Q.A.; Ren, Z.G.; Zhang, J.B.; Jiang, X.M.; Liang, L.; Xue, T.C.; Zhang, Q.B.; Wang, Y.H.; Zhang, L.; et al. Maintenance of stemness in oxaliplatin-resistant hepatocellular carcinoma is associated with increased autocrine of igf1. PLoS ONE 2014, 9, e89686. [Google Scholar] [CrossRef]
- Xia, Q.; Han, T.; Yang, P.; Wang, R.; Li, H.; Zhang, J.; Zhou, X. Microrna-28-5p regulates liver cancer stem cell expansion via igf-1 pathway. Stem Cells Int. 2019, 2019, 8734362. [Google Scholar] [CrossRef]
- Chen, P.C.; Kuo, Y.C.; Chuong, C.M.; Huang, Y.H. Niche modulation of igf-1r signaling: Its role in stem cell pluripotency, cancer reprogramming, and therapeutic applications. Front. Cell Dev. Biol. 2021, 8, 1780. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The hippo-yap pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Zhou, Q.; Pignoni, F. Yki/yap, sd/tead and hth/meis control tissue specification in the drosophila eye disc epithelium. PLoS ONE 2011, 6, e22278. [Google Scholar] [CrossRef]
- Chan, S.W.; Lim, C.J.; Loo, L.S.; Chong, Y.F.; Huang, C.; Hong, W. Teads mediate nuclear retention of taz to promote oncogenic transformation. J. Biol. Chem. 2009, 284, 14347–14358. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, W.M., Jr.; Hong, J.-H.; Yaffe, M.B.; Farrance, I.K.G. The transcriptional co-activator taz interacts differentially with transcriptional enhancer factor-1 (tef-1) family members. Biochem. J. 2005, 225, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. Tead/tef transcription factors utilize the activation domain of yap65, a src/yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. Tead mediates yap-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, C.B.; Cooper, L.F.; Yang, X.; Karsenty, G.; Aukhil, I. Transcriptional coactivation of bone-specific transcription factor cbfa1 by taz. Mol. Cell Biol. 2003, 23, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Lin, S.J.; Chen, Y.; Voon, D.-c.; Zhu, F.; Chuang, L.S.H.; Wang, T.; Tan, P.; Lee, S.C.; Yeoh, K.G.; et al. Runx3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene 2015, 35, 1–11. [Google Scholar] [CrossRef]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A ww domain protein taz is a critical coactivator for tbx5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [Green Version]
- Strassburger, K.; Tiebe, M.; Pinna, F.; Breuhahn, K.; Teleman, A.A. Insulin/igf signaling drives cell proliferation in part via yorkie/yap. Dev. Biol. 2012, 367, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Borreguero-Muñoz, N.; Fletcher, G.C.; Aguilar-Aragon, M.; Elbediwy, A.; Vincent-Mistiaen, Z.I.; Thompson, B.J. The hippo pathway integrates pi3k-akt signals with mechanical and polarity cues to control tissue growth. PLoS Biol. 2019, 17, e3000509. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.D.; Yuan, T.; Yan, F.J.; Zeng, C.M.; Dai, X.Y.; Chen, Z.b.; Chen, Y.; Zhou, T.; Fan, G.H.; et al. Multikinase inhibitor ct-707 targets liver cancer by interrupting the hypoxia-activated igf-1r–yap axis. Cancer Res. 2018, 78, 3995–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.L.; Chen, H.A.; Bamodu, O.A.; Lee, K.F.; Tzeng, Y.M.; Lee, W.H.; Tsai, J.T. Ovatodiolide suppresses yes-associated protein 1-modulated cancer stem cell phenotypes in highly malignant hepatocellular carcinoma and sensitizes cancer cells to chemotherapy in vitro. Toxicol. In Vitro 2018, 51, 74–82. [Google Scholar] [CrossRef]
- Van Haele, M.; Moya, I.M.; Karaman, R.; Rens, G.; Snoeck, J.; Govaere, O.; Nevens, F.; Verslype, C.; Topal, B.; Monbaliu, D.; et al. Yap and taz heterogeneity in primary liver cancer: An analysis of its prognostic and diagnostic role. Int. J. Mol. Sci. 2019, 20, 638. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Wang, Y.; Wu, J.; Huang, G.; Liu, B.; Ye, B.; Du, Y.; Gao, G.; Tian, Y.; He, L.; et al. Lncbrm initiates yap1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 2016, 7, 13608. [Google Scholar] [CrossRef] [Green Version]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.J.; Kim, H.; Park, Y.N. Increased expression of yes-associated protein 1 in hepatocellular carcinoma with stemness and combined hepatocellular-cholangiocarcinoma. PLoS ONE 2013, 8, e75449. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.G.; Li, H.L.; Fu, S.R.; Chen, X.F.; Lu, L.G. Surface markers of liver cancer stem cells and innovative targeted-therapy strategies for hcc (review). Oncol. Lett. 2018, 15, 2039–2048. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.H.; Luo, Q.; Liu, L.L.; Song, G.B. Liver cancer stem cell markers: Progression and therapeutic implications. World J. Gastroenterol. 2016, 22, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Schulte, L.A.; Lopez-Gil, J.C.; Sainz, B., Jr.; Hermann, P.C. The cancer stem cell in hepatocellular carcinoma. Cancers 2020, 12, 684. [Google Scholar] [CrossRef] [Green Version]
- Benabou, E.; Salame, Z.; Wendum, D.; Lequoy, M.; Tahraoui, S.; Merabtene, F.; Chretien, Y.; Scatton, O.; Rosmorduc, O.; Fouassier, L.; et al. Insulin receptor isoform a favors tumor progression in human hepatocellular carcinoma by increasing stem/progenitor cell features. Cancer Lett. 2019, 450, 155–168. [Google Scholar] [CrossRef]
- Ye, Y.; Guo, J.; Xiao, P.; Ning, J.; Zhang, R.; Liu, P.; Yu, W.; Xu, L.; Zhao, Y.; Yu, J. Macrophages-induced long noncoding rna h19 up-regulation triggers and activates the mir-193b/mapk1 axis and promotes cell aggressiveness in hepatocellular carcinoma. Cancer Lett. 2020, 469, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, C.; Yu, K.; Shi, S.; Chen, H.; Qian, Y.; Mei, Z. Aquaporin-9, mediated by igf2, suppresses liver cancer stem cell properties via augmenting ros/beta-catenin/foxo3a signaling. Mol. Cancer Res. 2020, 18, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xiong, L.; Li, Q.; Lin, L.; Miao, X.; Yan, S.; Hong, Z.; Yang, L.; Wen, Y.; Deng, X. 3d modeling of cancer stem cell niche. Oncotarget 2017, 9, 1326–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumor Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendall, S.C.; Stewart, M.H.; Menendez, P.; George, D.; Vijayaragavan, K.; Werbowetski-Ogilvie, T.; Ramos-Mejia, V.; Rouleau, A.; Yang, J.; Bossé, M.; et al. Igf and fgf cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 2007, 448, 1015–1021. [Google Scholar] [CrossRef]
- Youssef, A.; Aboalola, D.; Han, V.K. The roles of insulin-like growth factors in mesenchymal stem cell niche. Stem Cells Int. 2017, 2017, 9453108. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting igf-1 receptor-insulin receptor substrate and stat3 signaling. Oncogene 2016, 35, 2634–2644. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.; Shaked, Y.; Tsai, K.K. Targeting the interplay between cancer fibroblasts, mesenchymal stem cells, and cancer stem cells in desmoplastic cancers. Front. Oncol. 2019, 9, 688. [Google Scholar] [CrossRef] [Green Version]
- Beasley, R.P.; Hwang, L.Y.; Lin, C.C.; Chien, C.S. Hepatocellular carcinoma and hepatitis b virus. A prospective study of 22 707 men in taiwan. Lancet 1981, 2, 1129–1133. [Google Scholar] [CrossRef]
- Ng, K.Y.; Chai, S.; Tong, M.; Guan, X.Y.; Lin, C.H.; Ching, Y.P.; Xie, D.; Cheng, A.S.; Ma, S. C-terminal truncated hepatitis b virus x protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016, 7, 24005–24017. [Google Scholar] [CrossRef] [PubMed]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. Hbv-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arville, C.N.; Nouri-Aria, K.T.; Johnson, P.; Williams, R. Regulation of insulin-like growth factor ii gene expression by hepatitis b virus in hepaocellular carcinoma. Hepatology 1991, 13, 310–315. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, Z.; Chen, H.; Zhang, L.; Zhuo, F.; Yang, Q. Serum from chronic hepatitis b patients promotes growth and proliferation via the igf-ii/igf-ir/mek/erk signaling pathway in hepatocellular carcinoma cells. Cell Physiol. Biochem. 2018, 47, 39–53. [Google Scholar] [CrossRef]
- Nielsen, K.O.; Mirza, A.H.; Kaur, S.; Jacobsen, K.S.; Winther, T.N.; Glebe, D.; Pociot, F.; Hogh, B.; Størling, J. Hepatitis b virus suppresses the secretion of insulin-like growth factor binding protein 1 to facilitate anti-apoptotic igf-1 effects in hepg2 cells. Exp. Cell Res. 2018, 370, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.; Zhang, H.; Diab, A.; Pascuzzi, P.E.; Lefrancois, L.; Fares, N.; Bancel, B.; Merle, P.; Andrisani, O. Epcam-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis b virus-infected hepatocytes. J. Hepatol. 2016, 65, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Ching, R.H.H.; Sze, K.M.F.; Lau, E.Y.T.; Chiu, Y.T.; Lee, J.M.F.; Ng, I.O.L.; Lee, T.K.W. C-terminal truncated hepatitis b virus x protein regulates tumorigenicity, self-renewal and drug resistance via stat3/nanog signaling pathway. Oncotarget 2017, 8, 23507–23516. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yang, X.; Wang, L.; Zhang, C. Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol. Lett. 2018, 16, 679–686. [Google Scholar] [CrossRef]
- Wang, X.; Sun, W.; Shen, W.; Xia, M.; Chen, C.; Xiang, D.; Ning, B.; Cui, X.; Li, H.; Li, X.; et al. Long non-coding rna dilc regulates liver cancer stem cells via il-6/stat3 axis. J. Hepatol. 2016, 64, 1283–1294. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, Y.; Tan, X.; Ke, K.; Zheng, X.; Wang, F.; Lan, S.; Liao, N.; Cai, Z.; Shi, Y.; et al. Inflammatory micro-environment contributes to stemness properties and metastatic potential of hcc via the nf-kappab/mir-497/sall4 axis. Mol. Ther. Oncolytics 2019, 15, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tan, W.Y.; Chen, Q.R.; Chen, X.P.; Fu, K.; Zhao, Y.Y.; Chen, Z.W. Daintain/aif-1 promotes breast cancer proliferation via activation of the nf-kappab/cyclin d1 pathway and facilitates tumor growth. Cancer Sci. 2008, 99, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Feng, Z.; Jia, S.; Wang, W.; Du, Z.; Chen, N.; Chen, Z. Daintain/aif-1 promotes breast cancer cell migration by up-regulated tnf-alpha via activate p38 mapk signaling pathway. Breast Cancer Res. Treat. 2012, 131, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, D.M.; Brandy, N.Z.D.; da Silva, R.L.L.; Haddock, N.L.; Kacsinta, A.D.; de Moura, T.R.; Lipscomb, M.W. Allograft inflammatory factor-1 governs hematopoietic stem cell differentiation into cdc1 and monocyte-derived dendritic cells through irf8 and relb in vitro. Front. Immunol. 2019, 10, 173. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Du, Z.; Jiang, H.; Huang, X.; Chen, Z.; Chen, N. Daintain/aif-1 accelerates the activation of insulin-like growth factor-1 receptor signaling pathway in hepg2 cells. Oncol. Rep. 2015, 34, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Yu, D.-Y.; Yang, Z.-F.; Liu, D.-Y.; Cao, H.-Q.; Liao, X.-W. Igfbp2 upregulates zeb1 expression and promotes hepatocellular carcinoma progression through nf-κb signaling pathway. Dig. Liver Dis. 2020, 52, 573–581. [Google Scholar] [CrossRef]
- Liu, F.; Sun, Y.; Liu, B.; Lu, J.; Li, H.; Zhu, H.; Gao, H.; Zhou, X.; Chang, H. Insulin-like growth factor-1 induces epithelial-mesenchymal transition in hepatocellular carcinoma by activating survivin. Oncol. Rep. 2018, 40, 952–958. [Google Scholar] [CrossRef] [Green Version]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (bay 43-9006, nexavar®), a dual-action inhibitor that targets raf/mek/erk pathway in tumor cells and tyrosine kinases vegfr/pdgfr in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar]
- Keating, G.M.; Santoro, A. Sorafenib. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Carneiro, B.A.; Chandra, S.; Pai, S.G.; Chae, Y.K.; Kaplan, J.B.; Garrett, H.B.; Agulnik, M.; Kopp, P.A.; Giles, F.J. Spotlight on lenvatinib in the treatment of thyroid cancer: Patient selection and perspectives. Drug Des. Devel. Ther. 2016, 10, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Zschäbitz, S.; Grüllich, C. Lenvantinib: A tyrosine kinase inhibitor of vegfr 1-3, fgfr 1-4, pdgfrα, kit and ret. Recent Results Cancer Res. 2018, 211, 187–198. [Google Scholar] [PubMed]
- Nakazawa, Y.; Kawano, S.; Matsui, J.; Funahashi, Y.; Tohyama, O.; Muto, H.; Nakagawa, T.; Matsushima, T. Multitargeting strategy using lenvatinib and golvatinib: Maximizing anti-angiogenesis activity in a preclinical cancer model. Cancer Sci. 2015, 106, 201–207. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (resorce): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Duensing, A.; Boichuk, S.; Rausch, J. New developments in management of gastrointestinal stromal tumors: Regorafenib, the new player in the team. Gastrointest. Cancer Targets Ther. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.T.; Chu, P.Y.; Shiau, C.W.; Chen, Y.L.; Li, Y.S.; Hung, M.H.; Chen, L.J.; Chen, P.L.; Su, J.C.; Lin, P.Y.; et al. Stat3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5768–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; González-Gallego, J.; Mauriz, J.L. Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S. Review of regorafenib for the treatment of hepatocellular carcinoma. Gastroenterol. Hepatol. 2017, 13, 491–495. [Google Scholar]
- Deeks, E.D. Cabozantinib: A review in advanced hepatocellular carcinoma. Target. Oncol. 2019, 14, 107–113. [Google Scholar] [CrossRef]
- Trojan, J. Cabozantinib for the treatment of advanced hepatocellular carcinoma: Current data and future perspectives. Drugs 2020, 80, 1203–1210. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- De Luca, E.; Marino, D.; Di Maio, M. Ramucirumab, a second-line option for patients with hepatocellular carcinoma: A review of the evidence. Cancer Manag. Res. 2020, 12, 3721–3729. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (reach-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Yen, C.J.; Kudo, M.; Lim, H.Y.; Hsu, C.H.; Vogel, A.; Brandi, G.; Cheng, R.; Nitu, I.S.; Abada, P.; Hsu, Y.; et al. Efficacy and safety of ramucirumab in asian and non-asian patients with advanced hepatocellular carcinoma and elevated alpha-fetoprotein: Pooled individual data analysis of two randomized studies. Liver Cancer 2020, 9, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, T.; Ishigami, M.; Ito, T.; Ishizu, Y.; Honda, T.; Ishikawa, T.; Fujishiro, M. Initial experience of ramucirumab treatment after lenvatinib failure for patients with advanced hepatocellular carcinoma. Anticancer Res. 2020, 40, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. Lba38_pr - checkmate 459: A randomized, multi-center phase iii study of nivolumab (nivo) vs sorafenib (sor) as first-line (1l) treatment in patients (pts) with advanced hepatocellular carcinoma (ahcc). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (keynote-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in keynote-240: A randomized, double-blind, phase iii trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Colagrande, S.; Inghilesi, A.L.; Aburas, S.; Taliani, G.G.; Nardi, C.; Marra, F. Challenges of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7645–7659. [Google Scholar] [CrossRef]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase iii failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal. Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Hwang, S.; Keam, B.; Yu, Y.S.; Kim, J.H.; Kim, D.S.; Bae, S.H.; Kim, G.D.; Lee, J.K.; Seo, Y.B.; et al. Gene signature for sorafenib susceptibility in hepatocellular carcinoma: Different approach with a predictive biomarker. Liver Cancer 2020, 9, 182–192. [Google Scholar] [CrossRef]
- Lin, X.; Li, A.-m.; Li, Y.-H.; Luo, R.-C.; Zou, Y.-J.; Liu, Y.-Y.; Liu, C.; Xie, Y.-Y.; Zuo, S.; Liu, Z.; et al. Silencing myh9 blocks hbx-induced gsk3β ubiquitination and degradation to inhibit tumor stemness in hepatocellular carcinoma. Signal. Transduct. Target. Ther. 2020, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Witt-Kehati, D.; Fridkin, A.; Alaluf, M.B.; Zemel, R.; Shlomai, A. Inhibition of pmapk14 overcomes resistance to sorafenib in hepatoma cells with hepatitis b virus. Transl. Oncol. 2018, 11, 511–517. [Google Scholar] [CrossRef]
- Lu, S.-N.; Wang, J.-H.; Su, C.-W.; Wang, T.-E.; Dai, C.-Y.; Chen, C.-H.; Chen, R.-C.; Yang, S.-S.; Hung, C.-F.; Huang, S.-F.; et al. Management consensus guideline for hepatocellular carcinoma: 2016 updated by the Taiwan liver cancer association and the gastroenterological society of Taiwan. J. Formosan Med. Assoc. 2018, 117, 381–403. [Google Scholar] [CrossRef]
- Wang, F.; Bank, T.; Malnassy, G.; Arteaga, M.; Shang, N.; Dalheim, A.; Ding, X.; Cotler, S.J.; Denning, M.F.; Nishimura, M.I.; et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol. Commun. 2018, 2, 732–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeannot, V.; Busser, B.; Vanwonterghem, L.; Michallet, S.; Ferroudj, S.; Cokol, M.; Coll, J.L.; Ozturk, M.; Hurbin, A. Synergistic activity of vorinostat combined with gefitinib but not with sorafenib in mutant kras human non-small cell lung cancers and hepatocarcinoma. Onco Targets Ther. 2016, 9, 6843–6855. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M. Picropodophyllin and sorafenib synergistically suppress the proliferation and motility of hepatocellular carcinoma cells. Oncol. Lett. 2014, 8, 2023–2026. [Google Scholar] [CrossRef]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and igf/fgf signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.-A.; Murphy, P.R. Microrna: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutay, H.; Bai, S.; Datta, J.; Motiwala, T.; Pogribny, I.; Frankel, W.; Jacob, S.T.; Ghoshal, K. Downregulation of mir-122 in the rodent and human hepatocellular carcinomas. J. Cell Biochem. 2006, 99, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. Microrna-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting igf-1r to regulate ras/raf/erk signaling pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, J.; Sun, F.; Qi, M.; Su, P.; Liu, H.; Gao, L.; Jiao, M.; Wu, Z.; Xiang, L.; et al. Enhancer of zeste 2 polycomb repressive complex 2 subunit promotes sorafenib resistance of hepatocellular carcinoma though insulin-like growth factor 1 receptor. Anticancer Drugs 2019, 30, e0746. [Google Scholar] [CrossRef]
- Lin, Z.; Xia, S.; Liang, Y.; Ji, L.; Pan, Y.; Jiang, S.; Wan, Z.; Tao, L.; Chen, J.; Lin, C.; et al. Lxr activation potentiates sorafenib sensitivity in hcc by activating microrna-378a transcription. Theranostics 2020, 10, 8834–8850. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. Igf-binding proteins: Why do they exist and why are there so many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Mazerbourg, S.; Monget, P. Insulin-like growth factor binding proteins and igfbp proteases: A dynamic system regulating the ovarian folliculogenesis. Front. Endocrinol. 2018, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Wu, J.; Lv, Z.; Peng, C.; Chen, J.; Zhang, C.; He, B.; Tong, R.; Hu, W.; Ding, C.; et al. Targeting mybbp1a suppresses hcc progression via inhibiting igf1/akt pathway by cpg islands hypo-methylation dependent promotion of igfbp5. EBioMedicine 2019, 44, 225–236. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zuo, Q.; Hu, B.; Jin, H.; Wang, C.; Cheng, Z.; Deng, X.; Yang, C.; Ruan, H.; Yu, C.; et al. A novel, liver-specific long noncoding rna linc01093 suppresses hcc progression by interaction with igf2bp1 to facilitate decay of gli1 mrna. Cancer Lett. 2019, 450, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-Y.; Mok, M.T.; Kang, W.; Yang, W.; Tang, W.; Wu, F.; Xu, L.; Yan, M.; Yu, Z.; Lee, S.-D.; et al. Loss of tumor suppressor igfbp4 drives epigenetic reprogramming in hepatic carcinogenesis. Nucleic Acids Res. 2018, 46, 8832–8847. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, X.; Li, N.; Du, C.; Yang, B.; Qin, W.; Fu, J.; Markowitz, G.J.; Wang, H.; Ma, J.; et al. Ccl22 signaling contributes to sorafenib resistance in hepatitis b virus-associated hepatocellular carcinoma. Pharmacol. Res. 2020, 157, 104800. [Google Scholar] [CrossRef]
- Saile, B.; DiRocco, P.; Dudas, J.; El-Armouche, H.; Sebb, H.; Eisenbach, C.; Neubauer, K.; Ramadori, G. Igf-i induces DNA synthesis and apoptosis in rat liver hepatic stellate cells (hsc) but DNA synthesis and proliferation in rat liver myofibroblasts (rmf). Lab. Investig. 2004, 84, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Lee, Y.K.; Kim, H.J.; Park, O.J.; Kim, Y.M. Ampk interacts with β-catenin in the regulation of hepatocellular carcinoma cell proliferation and survival with selenium treatment. Oncol. Rep. 2016, 35, 1566–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippolis, C.; Refolo, M.G.; D’Alessandro, R.; Carella, N.; Messa, C.; Cavallini, A.; Carr, B.I. Resistance to multikinase inhibitor actions mediated by insulin like growth factor-1. J. Exp. Clin. Cancer Res. 2015, 34, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suemura, S.; Kodama, T.; Myojin, Y.; Yamada, R.; Shigekawa, M.; Hikita, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. Crispr loss-of-function screen identifies the hippo signaling pathway as the mediator of regorafenib efficacy in hepatocellular carcinoma. Cancers 2019, 11, 1362. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Zhang, S.; Ma, D.; Yan, D.; Zhang, G.; Cao, Y.; Wang, Z.; Wu, J.; Jiang, C. Targeting sphk2 reverses acquired resistance of regorafenib in hepatocellular carcinoma. Front. Oncol. 2020, 10, 694. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.W.; Zhou, D.M.; Zhang, Z.Y.; Zhang, J.K.; Zhu, H.J.; Lin, N.M.; Zhang, C. Suppression of lsd1 enhances the cytotoxic and apoptotic effects of regorafenib in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 512, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating β-catenin/c-myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46. [Google Scholar] [CrossRef]
- Refolo, M.G.; Lippolis, C.; Carella, N.; Cavallini, A.; Messa, C.; D’Alessandro, R. Chlorogenic acid improves the regorafenib effects in human hepatocellular carcinoma cells. Int. J. Mol. Sci. 2018, 19, 1518. [Google Scholar] [CrossRef] [Green Version]
- Desbois-Mouthon, C.; Cacheux, W.; Blivet-Van Eggelpoel, M.J.; Barbu, V.; Fartoux, L.; Poupon, R.; Housset, C.; Rosmorduc, O. Impact of igf-1r/egfr cross-talks on hepatoma cell sensitivity to gefitinib. Int. J. Cancer 2006, 119, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Ezzoukhry, Z.; Louandre, C.; Trecherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.C.; Maziere, J.C.; Galmiche, A. Egfr activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Jiang, S.; Li, J.; Chen, H.; Zhang, X. Activation of the hgf/c-met axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-met expression. Med. Oncol. 2020, 37, 24. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Gansukh, B.; Shia, J.; Kalin, M.; Katz, S.; Abad, L.; et al. A phase ii study of cixutumumab (imc-a12, nsc742460) in advanced hepatocellular carcinoma. J. Hepatol. 2014, 60, 319–324. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; O’Donnell, R.; Semrad, T.J.; Mack, P.; Blanchard, S.; Bahary, N.; Jiang, Y.; Yen, Y.; Wright, J.; Chen, H.; et al. A phase i trial of escalating doses of cixutumumab (imc-a12) and sorafenib in the treatment of advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2018, 81, 957–963. [Google Scholar] [CrossRef]
- Wu, J.; Zhu, A.X. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J. Hematol. Oncol. 2011, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.J.; Fartoux, L.; Bouattour, M.; Bumsel, F.; Dreyer, C.; Raymond, E.; Rosmorduc, O. A phase i study of ave1642, a human monoclonal antibody–blocking insulin-like growth factor-1 receptor (igf-1r), given as a single agent and in combination with sorafenib as first-line therapy in patients with advanced hepatocellular carcinoma (hcc). J. Clin. Oncol. 2011, 29, 270. [Google Scholar] [CrossRef]
- McKian, K.P.; Haluska, P. Cixutumumab. Expert Opin. Investig. Drugs 2009, 18, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Gansukh, B.; Chou, J.F.; Shia, J.; Capanu, M.; Kalin, M.; Chen, H.X.; Zojwalla, N.J.; Katz, S.; Reidy, D.L.; et al. Phase ii study of cixutumumab (imc-a12, nsc742460; c) in hepatocellular carcinoma (hcc). J. Clin. Oncol. 2011, 29, 4043. [Google Scholar] [CrossRef]
- Prenner, S.; Kulik, L. 46-hepatocellular carcinoma. In Zakim and Boyer’s Hepatology, 7th ed.; Sanyal, A.J., Boyer, T.D., Lindor, K.D., Terrault, N.A., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 668–692. [Google Scholar]
- Rieder, S.; Michalski, C.W.; Friess, H.; Kleeff, J. Insulin-like growth factor signaling as a therapeutic target in pancreatic cancer. Anticancer Agents Med. Chem. 2011, 11, 427–433. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
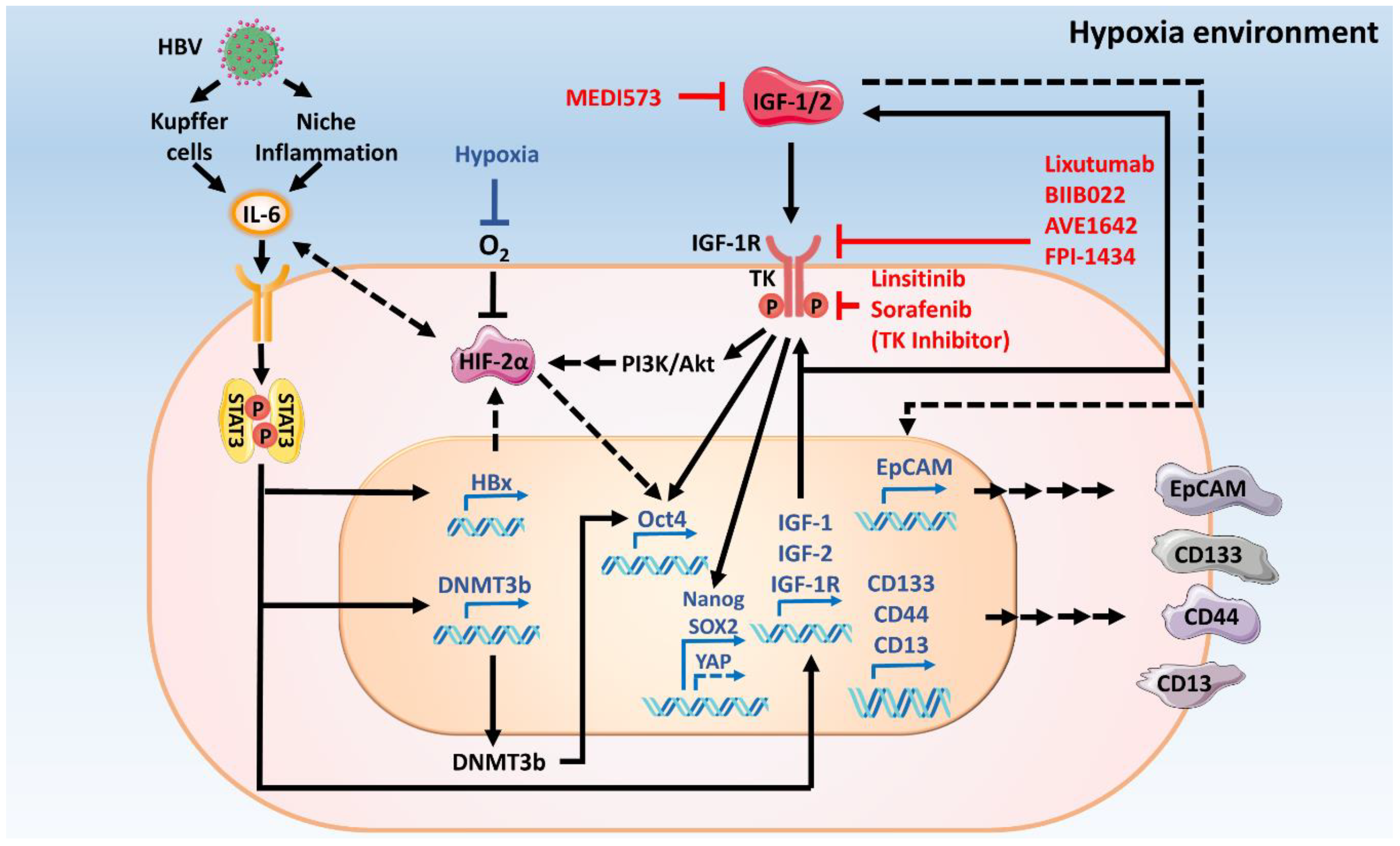{kind=link}
| Compound | Locations of Clinical Trial | Mechanism of Action | Phase | Combination Treatment | No. of Participants (n) | HCC Etiology of Participants | Conclusion | Clinical Trial No. or Ref. | Year of Clinical Trial |
|---|---|---|---|---|---|---|---|---|---|
| Monoclonal antibodies | |||||||||
| MEDI-573 | United States | Anti IGFI and IGFII | Phase 1b | Combination with sorafenib | 6 | Unspecific | Safety population included all participants who received any amount of study treatment. | NCT01498952 | 2012–2013 |
| Cixutumumab (IMC-A12) | United States | Anti IGF-1R | Phase II | Single | 24 | Unspecific | Cixutumumab alone did not show the clinical meaningful activity in recruited patients with HCC. | NCT00639509 [259] | 2008–2011 |
| Cixutumumab (IMC-A12) | United States | Anti IGF-1R | Phase I | Combination with sorafenib | 21 | Unspecific | Combination of cixutumumab and sorafenib showed limited clinical efficacy in patients with HCC. | NCT01008566 [260] | 2009–2016 |
| BIIB022 | United States Singapore Taiwan United Kingdom | Anti IGF-1R | Phase Ib | Combination with sorafenib | 40 | Unspecific | The dose of BIIB022 was established for clinical trial phase II. | NCT00956436 [261] | 2009–2011 |
| AVE1642 | France | Anti IGF-1R | Phase I | Single and Combination with sorafenib | 13 | Unspecific | AVE1642 can be safely combined with active doses of sorafenib. | NCT00791544 [262] | 2008–2009 |
| FPI-1434 | United States Australia Canada | Anti IGF-1R | Phase I | Single | 38 | Unspecific | Recruiting. | NCT03746431 | 2019–2022 |
| Inhibitors | |||||||||
| Linsitinib (OSI-906) | United States Belgium France Germany Hong Kong Italy Korea Singapore Spain Taiwan | IGF-1R phosphorylation inhibitor | Phase II | Single | 23 | Unspecific | Terminated due to safety issue observed. | NCT01101906 | 2011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, M.-H.T.; Jeng, H.-Y.; Kuo, Y.-C.; Diony Nanda, J.; Brahmadhi, A.; Ling, T.-Y.; Chang, T.-S.; Huang, Y.-H. The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance. Int. J. Mol. Sci. 2021, 22, 1931. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041931
Ngo M-HT, Jeng H-Y, Kuo Y-C, Diony Nanda J, Brahmadhi A, Ling T-Y, Chang T-S, Huang Y-H. The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance. International Journal of Molecular Sciences. 2021; 22(4):1931. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041931
Chicago/Turabian StyleNgo, Mai-Huong Thi, Han-Yin Jeng, Yung-Che Kuo, Josephine Diony Nanda, Ageng Brahmadhi, Thai-Yen Ling, Te-Sheng Chang, and Yen-Hua Huang. 2021. "The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance" International Journal of Molecular Sciences 22, no. 4: 1931. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041931