
The Multiomics Analyses of Fecal Matrix and Its Significance to Coeliac Disease Gut Profiling

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Methods of Analysis
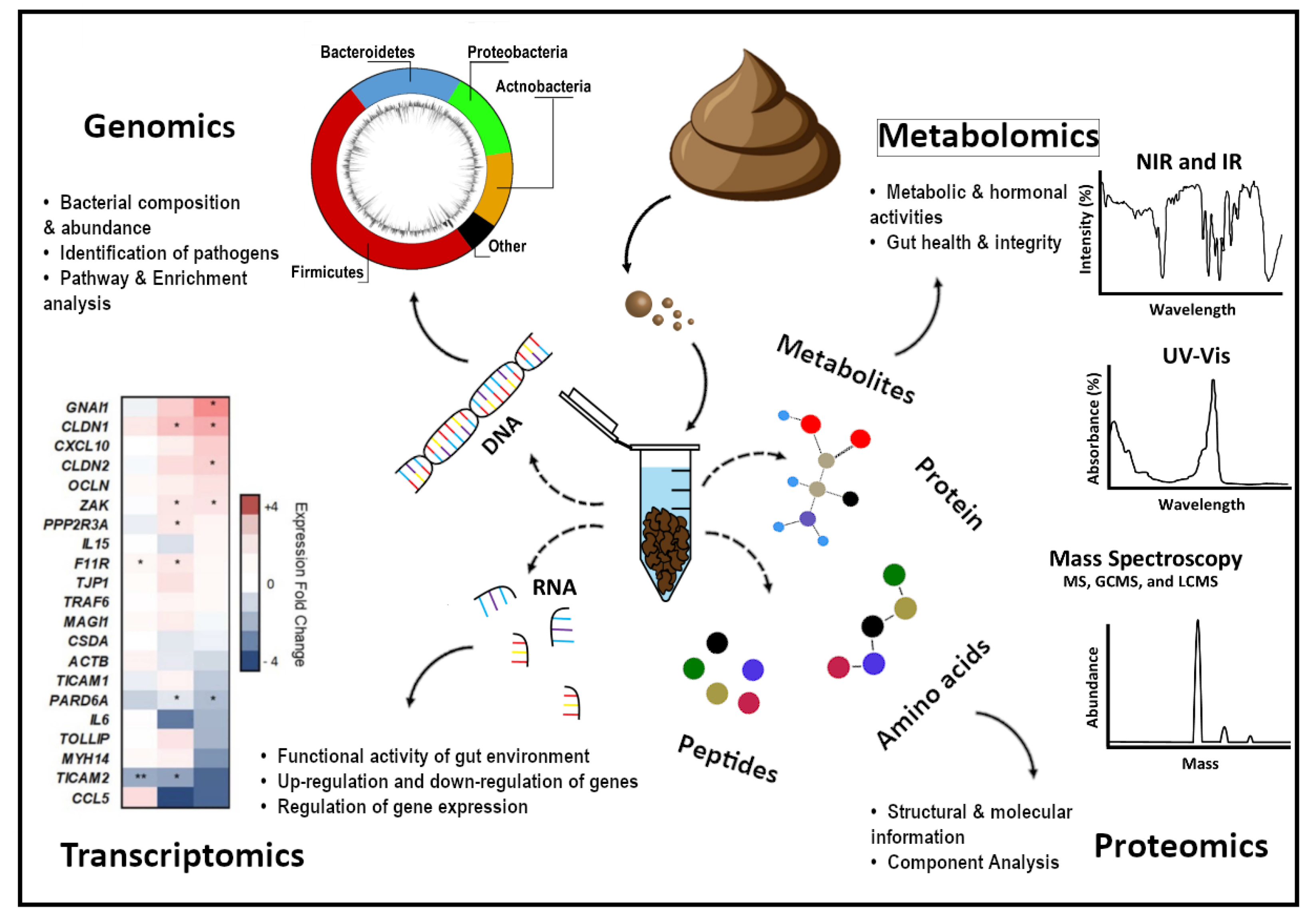
2.1. Metagenomics
2.1.1. Sample Collection and Storage
2.1.2. Lysis of Cells
2.1.3. Isolation and Extraction of DNA Sequences
2.1.4. Amplification and Sequencing
2.1.5. Bacterial Identification
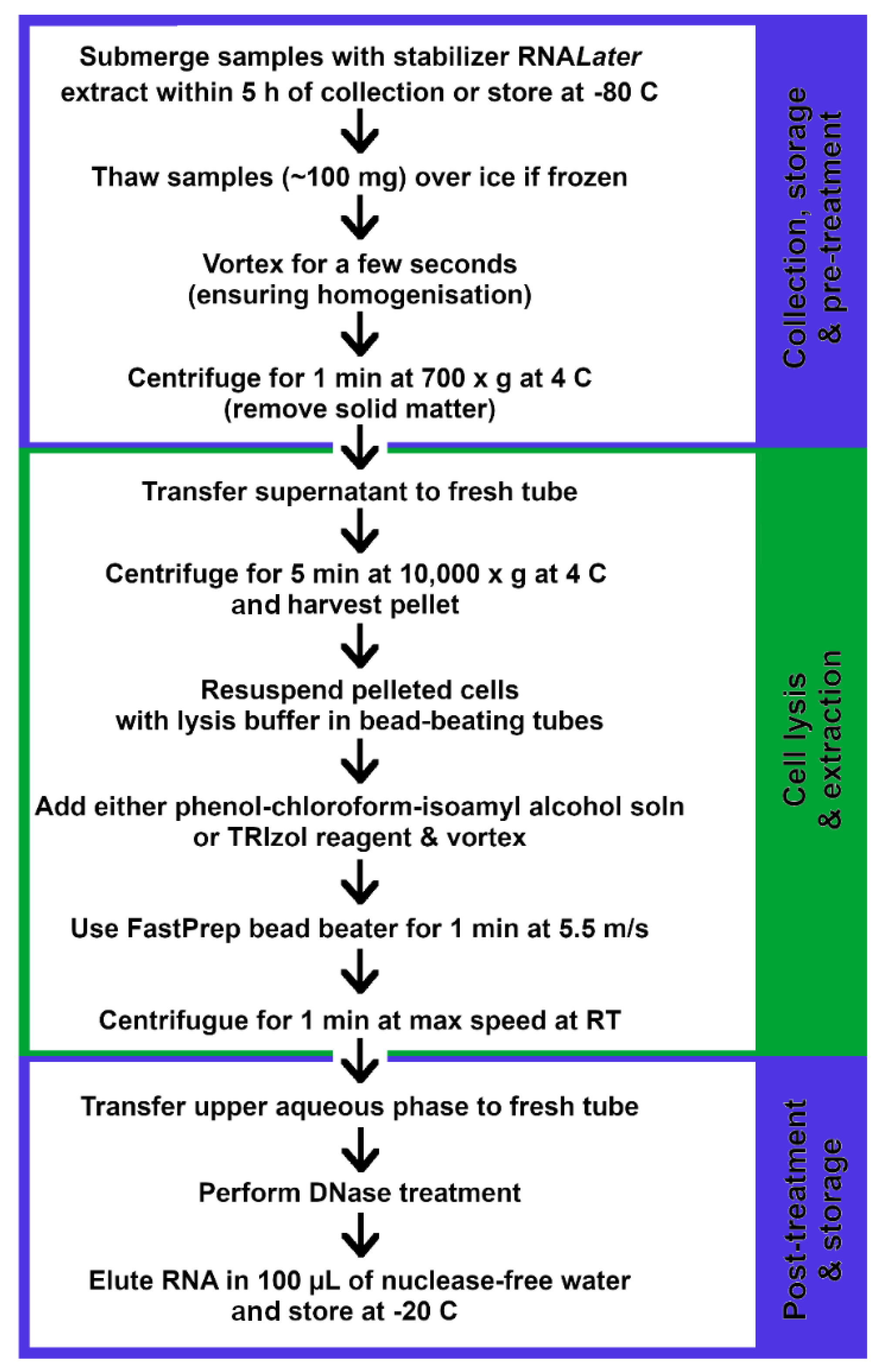
2.2. Transcriptomics
2.2.1. Sample Storage and Cell Lysis
2.2.2. Purification and Enrichment of mRNA Product
2.2.3. MicroRNA: A New Method for Shaping the Gut Microbiota
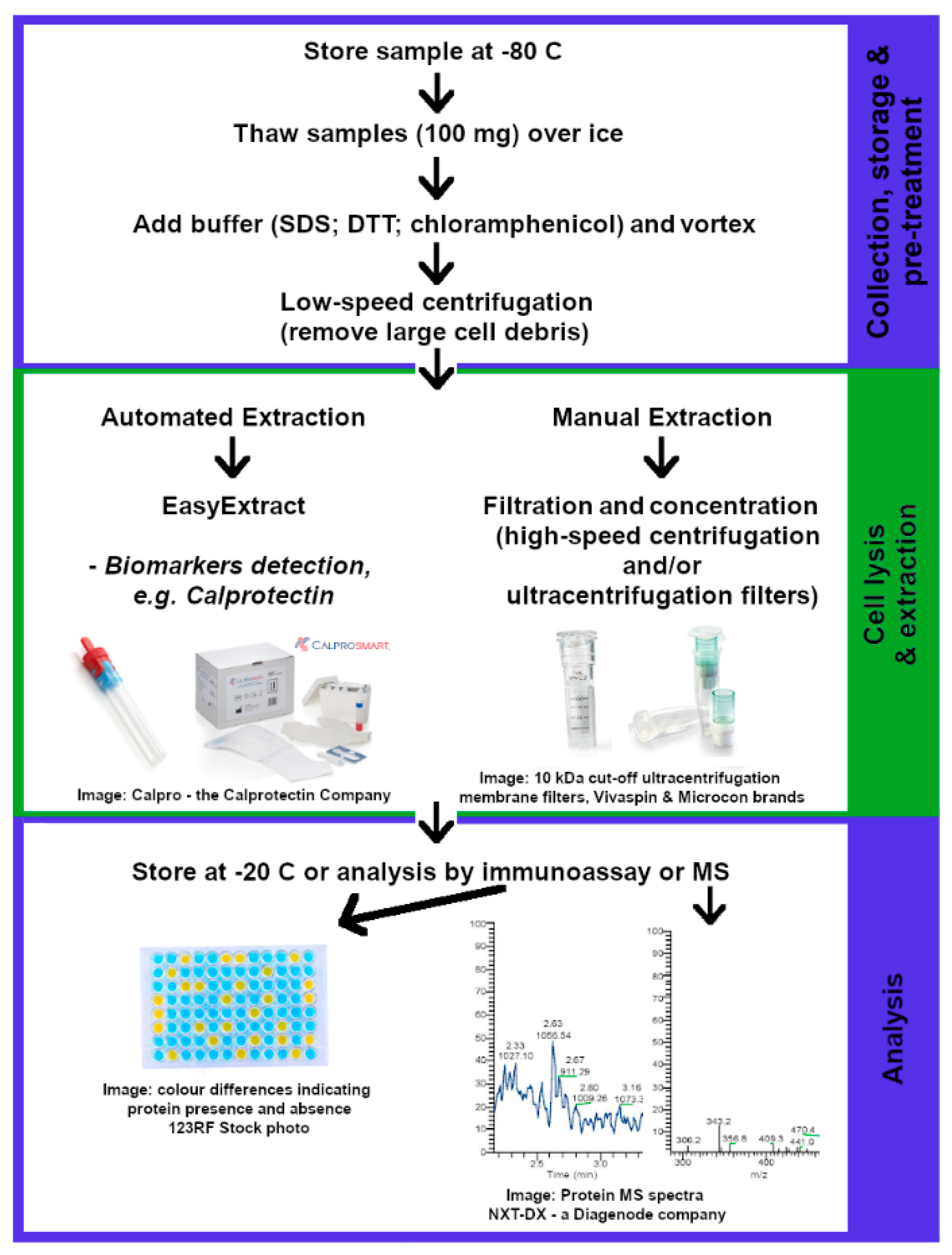
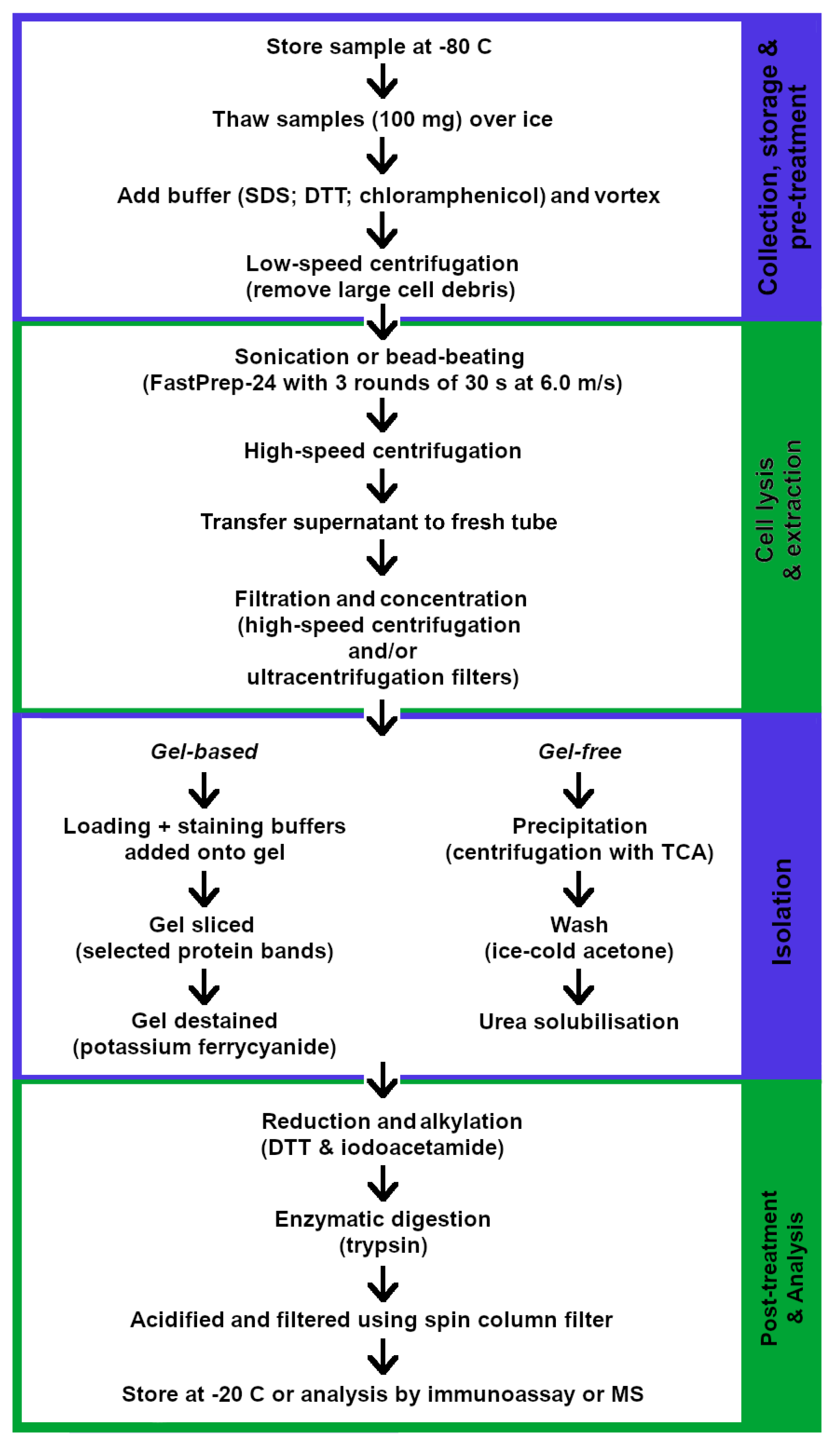
2.3. Proteomics
2.3.1. Sample Storage
2.3.2. Protein Extraction
2.3.3. Digestion and Peptide Isolation
2.3.4. Detection
2.4. Metabonomics
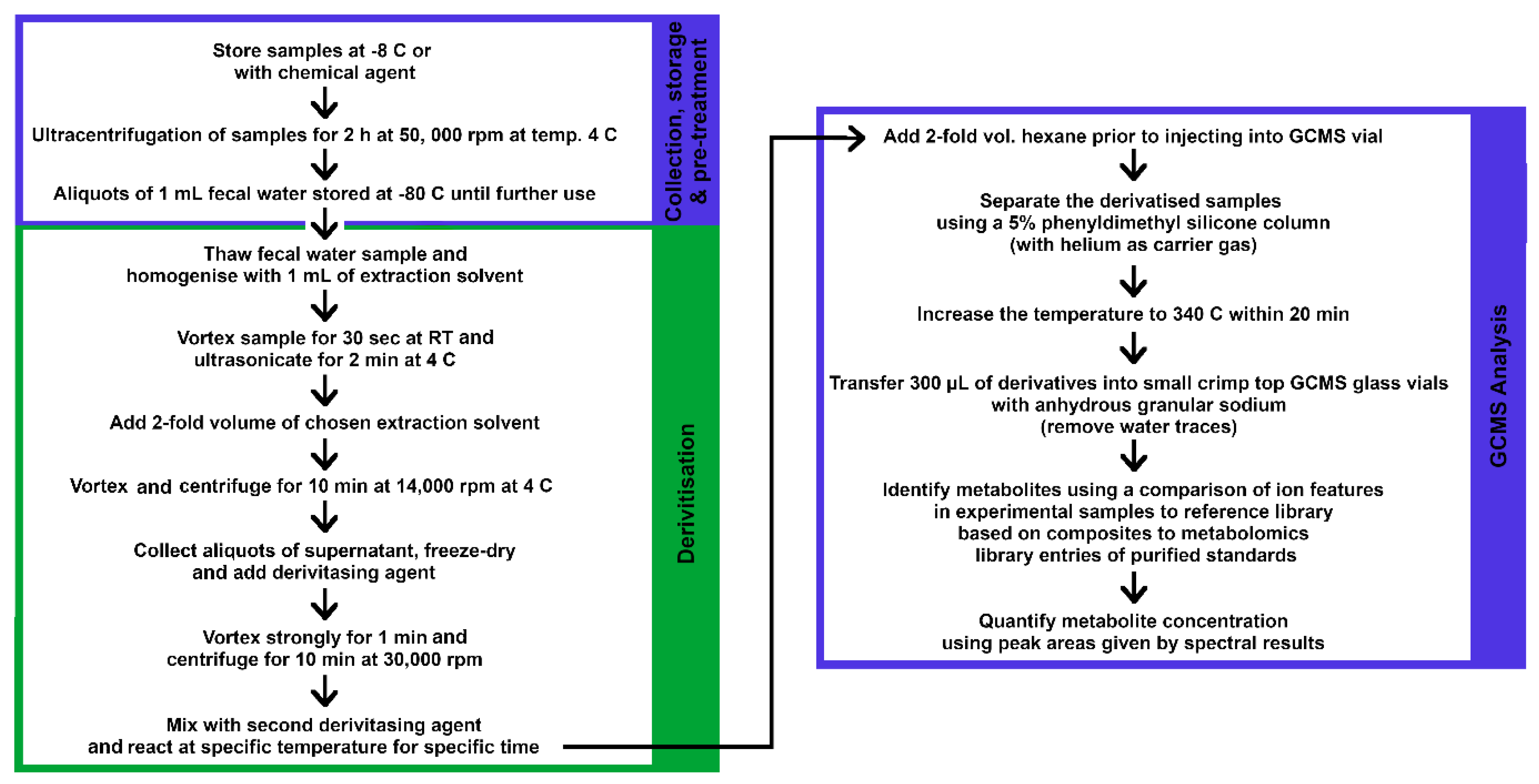
2.4.1. GC–MS Analysis
2.4.2. LC–MS Analysis for Metabolites
3. CeD Disease Gut Profiling
3.1. CeD Microbial Profiling
3.2. CeD mRNA and microRNA Profiling
3.3. CeD Protein Biomarkers Profiling
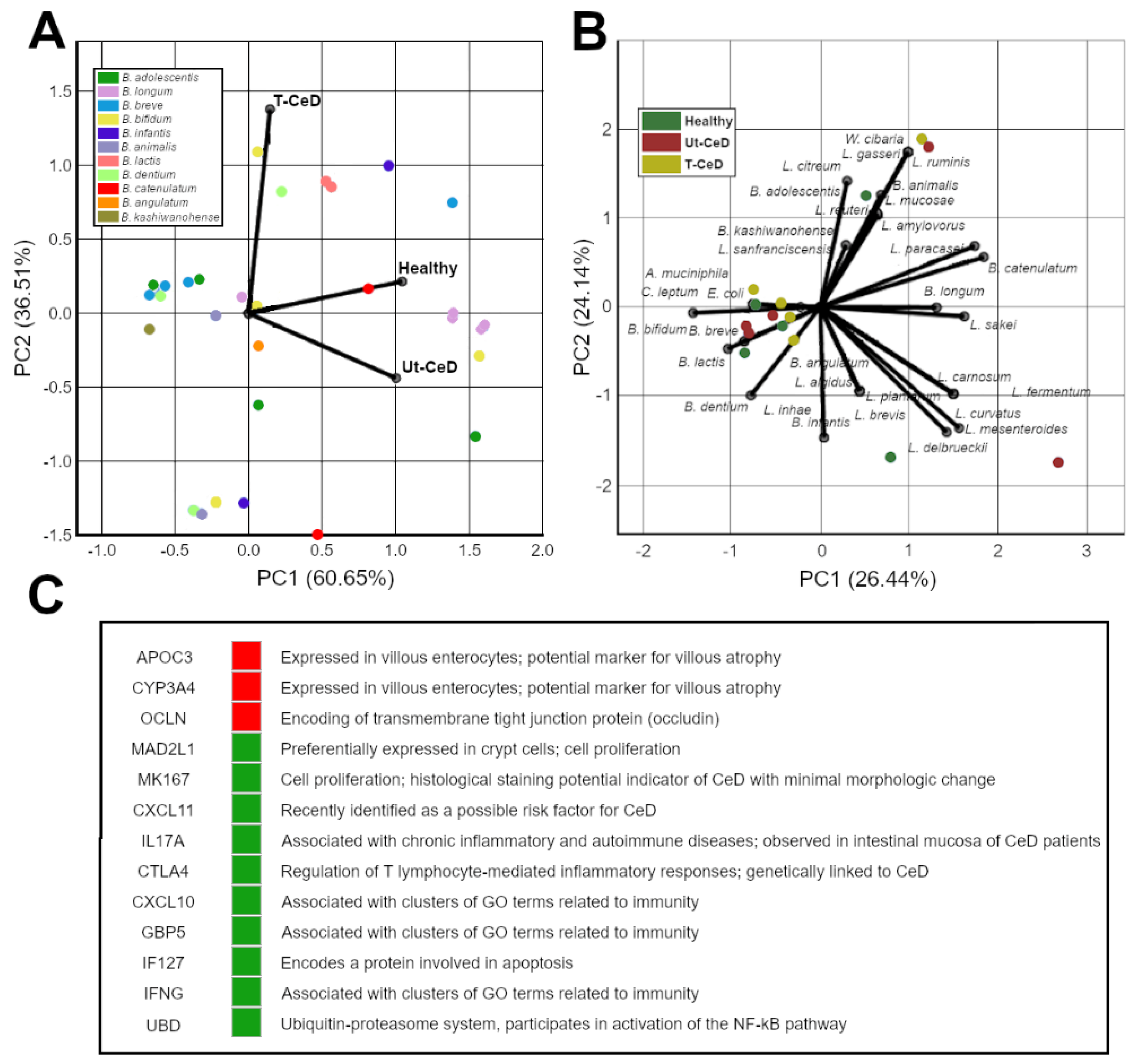
3.4. CeD Metabolic Profiling
3.5. Chemometrics and Machine Learning (ML)
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the Human Microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. 2009, 9, 313–324. [Google Scholar]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Whipps, J.M.; Lewis, K.; Cooke, R.C. Mycoparasitism and plant disease control. In Fungi in Biological Control Systems; Burge, M.N., Ed.; Manchester University Press: Manchester, UK, 1988; pp. 161–187. [Google Scholar]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghteling, P.D.; Walker, A.W. Why is initial bacterial colonization of the intestine important to the infant’s and child’s health? J. Pediatr. Gastroenterol. Nutr. 2015, 60, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O’Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Touhy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spor, A.; Koren, O.; Ley, R.E. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Jump, R.L.P.; Polinkovsky, A.; Hurless, K.; Sitzlar, B.; Eckart, K.; Tomas, M.; Deshpande, A.; Nerandzic, M.M.; Donskey, C.J. Metabolomics Analysis Identifies Intestinal MicrobiotaDerived Biomarkers of Colonization Resistance in Clindamycin-Treated Mice. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Gao, X.; Pujos-Guillot, E.; Martin, J.-F.; Galan, P.; Juste, C.; Jia, W.; Sebedio, J.-L. Metabolite analysis of human fecal water by gas chromatography/mass spectrometry with ethyl chloroformate derivatization. Anal. Biochem. 2009, 393, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Koboziev, I.; Webb, C.R.; Furr, K.L.; Grisham, M.B. Role of the Enteric Microbiota in Intestinal Homeostasis and Inflammation. Free Radic. Biol. Med. 2014, 68, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, M.A.; Bird, A.R. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2015, 7, 17–44. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadoo, S.; Dinev, I.; Chapman, J.; Hughes, R.J.; Van, T.T.H.; Moore, R.J.; Stanley, D. Selenium nanoparticles in poultry feed modify gut microbiota and increase abundance of Faecalibacterium prausnitzii. Appl. Microbiol. Biotechnol. 2018, 102, 1455–1466. [Google Scholar] [CrossRef]
- Prescott, S.L. History of medicine: Origin of the term microbiome and why it matters. Hum. Microbiome J. 2017, 4, 24–25. [Google Scholar] [CrossRef]
- Dubos, R.; Schaedler, R.W.; Costello, R.; Hoet, P. Indigenous, normal, and autochthonous flora of the gastrointestinal tract. J. Exp. Med. 1965, 122, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, K.Y.; Lee, M.-S. Gut microbiota and metabolic disorders. Diabetes Metab. 2015, 39, 198. [Google Scholar] [CrossRef] [Green Version]
- Perrier, C.; Corthesy, B. Gut permeability and food allergies. Clin. Exp. Allergy 2011, 41, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, M.L.; Bates, N.A.; Felix, K.M.; Wu, H.J.J. Impact of gut microbiota on gut-distal autoimmunity: A focus on T cells. Immunology 2019, 156, 305–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, G.; Keating, D.; Young, R.; Wong, M.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putignani, L.; Del Chierico, F.; Vernocchi, P.; Cicala, M.; Cucchiara, S.; Dallapiccola, B.; Group, D.S. Gut Microbiota dysbiosis as risk and premorbid factors of IBD and IBS along the childhood–Adulthood transition. Inflam. Bowel Dis. 2016, 22, 487–504. [Google Scholar] [CrossRef] [Green Version]
- Munyaka, P.M.; Khafipour, E.; Ghia, J.-E. External influence of early childhood establishment of gut microbiota and subsequent health implications. Front. Pediatr. 2014, 2, 109. [Google Scholar] [CrossRef] [Green Version]
- Kamada, N.; Kim, Y.-G.; Sham, H.P.; Vallance, B.A.; Puente, J.L.; Martens, E.C.; Núñez, G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 2012, 336, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Guttman, J.A.; Li, Y.; Wickham, M.E.; Deng, W.; Vogl, A.W.; Finlay, B.B. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell. Microbiol. 2006, 8, 634–645. [Google Scholar] [CrossRef]
- Council, N.R. The New Science of Metagenomics; The National Academies Press: Washington, DC, USA, 2007. [Google Scholar]
- Schaedler, R.W.; Dubos, R.; Costello, R. The development of the bacterial flora in the gastrointestinal tract of mice. J. Exp. Med. 1965, 122, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing Unculturable Bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Salvatore, F. Comparative Metagenomic Analysis of Human Gut Microbiome Composition Using Two Different Bioinformatic Pipelines. BioMed Res. Int. 2014, 2014, 325340. [Google Scholar] [CrossRef]
- Phua, L.C.; Koh, P.K.; Cheah, P.Y.; Ho, H.K.; Chan, E.C.Y. Global gas chromatography/time-of-flight mass spectrometry (GC/TOFMS)-based metabonomic profiling of lyophilized human feces. J. Chromatogr. B 2013, 937, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Green, P.H.; Cellier, C. Celiac disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D. Current concepts of celiac disease pathogenesis. Gastroenterology 2000, 119, 234–242. [Google Scholar] [CrossRef]
- Catassi, C.; Fasano, A. Celiac disease. In Gluten-Free Cereal Products and Beverages; Elsevier: Amsterdam, The Netherlands, 2008; pp. 1–27. [Google Scholar]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Grieb, A.; Bowers, R.M.; Oggerin, M.; Goudeau, D.; Lee, J.; Malmstrom, R.R.; Woyke, T.; Fuchs, B.M. A pipeline for targeted metagenomics of environmental bacteria. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef]
- Mandal, R.S.; Saha, S.; Das, S. Metagenomic surveys of gut microbiota. Genom. Proteom. Bioinform. 2015, 13, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Carroll, I.M.; Ringel-Kulka, T.; Siddle, J.P.; Klaenhammer, T.R.; Ringel, Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PLoS ONE 2012, 7, e46953. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Folsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Roesch, L.F.; Casella, G.; Simell, O.; Krischer, J.; Wasserfall, C.H.; Schatz, D.; Atkinson, M.A.; Neu, J.; Triplett, E.W. Influence of fecal sample storage on bacterial community diversity. Open Microbiol. J. 2009, 3, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, M.I.; Bergström, A.; Licht, T.R. Freezing fecal samples prior to DNA extraction affects the Firmicutes to Bacteroidetes ratio determined by downstream quantitative PCR analysis. FEMS Microbiol. Lett. 2012, 329, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Deane, J.; Rea, M.C.; O’Sullivan, Ó.; Ross, R.P.; O’Callaghan, G.; Plant, B.J.; Stanton, C. The effects of freezing on faecal microbiota as determined using MiSeq sequencing and culture-based investigations. PLoS ONE 2015, 10, e0119355. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.J.; Venema, K.; Keshavarzian, A.; Hamaker, B.R. Starch-entrapped microspheres show a beneficial fermentation profile and decrease in potentially harmful bacteria during in vitro fermentation in faecal microbiota obtained from patients with inflammatory bowel disease. Br. J. Nutr. 2010, 103, 1514–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlčková, K.; Mrázek, J.; Kopečný, J.; Petrželková, K.J. Evaluation of different storage methods to characterize the fecal bacterial communities of captive western lowland gorillas (Gorilla gorilla gorilla). J. Microbiol. Methods 2012, 91, 45–51. [Google Scholar] [CrossRef]
- Panek, M.; Paljetak, H.Č.; Barešić, A.; Perić, M.; Matijašić, M.; Lojkić, I.; Bender, D.V.; Krznarić, Ž.; Verbanac, D. Methodology challenges in studying human gut microbiota—Effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci. Rep. 2018, 8, 5143. [Google Scholar] [CrossRef] [Green Version]
- Roume, H.; Heintz-Buschart, A.; Muller, E.E.L.; Wilmes, P. Chapter Eleven—Sequential Isolation of Metabolites, RNA, DNA, and Proteins from the Same Unique Sample. Methods Enzymol. 2013, 531, 219–236. [Google Scholar]
- Nechvatal, J.M.; Ram, J.L.; Basson, M.D.; Namprachan, P.; Niec, S.R.; Badsha, K.Z.; Matherly, L.H.; Majumdar, A.P.N.; Kato, I. Fecal collection, ambient preservation, and DNA extraction for PCR amplification of bacterial and human markers from human feces. J. Microbiol. Methods 2008, 72, 124–132. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Yu, G.; Ma, B.; Ravel, J.; Goedert, J.J.; Sinha, R. Collection media and delayed freezing effects on microbial composition of human stool. Microbiome 2015, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Bleckmann, A.; Dresselhaus, T. Fluorescent whole-mount RNA in situ hybridization (F-WISH) in plant germ cells and the fertilized ovule. Methods 2016, 98, 66–73. [Google Scholar] [CrossRef]
- Hale, V.L.; Tan, C.L.; Knight, R.; Amato, K.R. Effect of preservation method on spider monkey (Ateles geoffroyi) fecal microbiota over 8 weeks. J. Microbiol. Methods 2015, 113, 16–26. [Google Scholar] [CrossRef]
- Dominianni, C.; Wu, J.; Hayes, R.B.; Ahn, J. Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiol. 2014, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuk, S.; Cetinkaya, U. Stool sample storage conditions for the preservation of Giardia intestinalis DNA. Memórias Inst. Oswaldo Cruz 2012, 107, 965–968. [Google Scholar] [CrossRef]
- Choo, J.M.; Leong, L.E.; Rogers, G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015, 5, 16350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariefdjohan, M.W.; Savaiano, D.A.; Nakatsu, C.H. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr. J. 2010, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.-H.; Peterson, C.M.; Raggio, A.; Keenan, M.J.; Martin, R.J.; Ravussin, E.; Marco, M.L. Impact of Different Fecal Processing Methods on Assessments of Bacterial Diversity in the Human Intestine. Front. Microbiol. 2016, 7, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salonen, A.; Nikkilä, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilić-Stojanović, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
- Smith, A.K.; Conneely, K.N.; Kilaru, V.; Mercer, K.B.; Weiss, T.E.; Bradley, B.; Tang, Y.; Gillespie, C.F.; Cubells, J.F.; Ressler, K.J. Differential Immune System DNA Methylation and Cytokine Regulation in Post-Traumatic Stress Disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156B, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrand, J.; Patron, K.; Legrand-Frossi, C.; Frippiat, J.-P.; Merlin, C.; Alauzet, C.; Lozniewski, A. Comparison of seven methods for extraction of bacterial DNA from fecal and cecal samples of mice. J. Microbiol. Methods 2014, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Alcon-Giner, C.; Caim, S.; Mitra, S.; Ketskemety, J.; Wegmann, U.; Wain, J.; Belteki, G.; Clarke, P.; Hall, L.J. Optimisation of 16S rRNA gut microbiota profiling of extremely low birth weight infants. BMC Genom. 2017, 18, 841. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.E.; Ruiz-Perez, F.; Jo, N.Y.; Vijayakumar, V.; Gong, M.Q.; Nataro, J.P. A Large Family of Antivirulence Regulators Modulates the Effects of Transcriptional Activators in Gram-negative Pathogenic Bacteria. PLoS Pathog. 2014, 10, e1004153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Martin, J.C.; Scott, P.; Parkhill, J.; Flint, H.J.; Scott, K.P. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 2015, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Maukonen, J.; Simões, C.; Saarela, M. The currently used commercial DNA-extraction methods give different results of clostridial and actinobacterial populations derived from human fecal samples. FEMS Microbiol. Ecol. 2012, 79, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–813. [Google Scholar] [CrossRef]
- Yu, M.; Liu, L.; Wang, S. Water-soluble dendritic-conjugated polyfluorenes: Synthesis, characterization, and interactions with DNA. J. Polym. Sci. Part A 2008, 46, 7462–7472. [Google Scholar] [CrossRef]
- Vandeventer, P.E.; Weigel, K.M.; Salazar, J.; Erwin, B.; Irvine, B.; Doebler, R.; Nadim, A.; Cangelosi, G.A.; Niemz, A. Mechanical Disruption of Lysis-Resistant Bacterial Cells by Use of a Miniature, Low-Power, Disposable Device. J. Clin. Microbiol. 2011, 49, 2533–2539. [Google Scholar] [CrossRef] [Green Version]
- Boer, R.; Petersb, R.; Gierveld, S.; Schuurman, T.; Kooistra-Smid, M.; Savelkoul, P. Improved detection of microbial DNA after bead-beating before DNA isolation. J. Microbiol. Methods 2010, 80, 209–211. [Google Scholar] [CrossRef]
- Kaser, M.; Ruf, M.-T.; Hauser, J.; Marsollier, L.; Pluschke, G. Optimized method for preparation of DNA from pathogenic and environmental mycobacteria. Appl. Environ. Microbiol. 2009, 75, 414–418. [Google Scholar] [CrossRef] [Green Version]
- Repetto, S.A.; AlbaSoto, C.D.; Cazorla, S.I.; Tayeldin, M.L.; Cuello, S.; Lasala, M.B.; Tekiel, V.S.; Cappa, S.M.G. An improved DNA isolation technique for PCR detection of Strongyloides stercoralis in stool samples. Acta Trop. 2013, 126, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. A structural taxonomy of DNA-binding domains. Nature 1991, 353, 715–719. [Google Scholar] [CrossRef]
- Bag, S.; Saha, B.; Mehta, O.; Anbumani, D.; Kumar, N.; Dayal, M.; Pant, A.; Kumar, P.; Saxena, S.; Allin, K.H.; et al. An Improved Method for High Quality Metagenomics DNA Extraction from Human and Environmental Samples. Sci. Rep. 2016, 6, 26775. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Cohen, D.B.; Ravel, J.; Abdo, Z.; Forney, L.J. Evaluation of Methods for the Extraction and Purification of DNA from the Human Microbiome. PLoS ONE 2012, 7, e33865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, K.; kiImachi, H.; Kawakami, S.; Nakamura, K.; Harada, H.; Ohashi, A. Evaluation of enzymatic cell treatments for application of CARD-FISH to methanogens. J. Microbiol. Methods 2008, 72, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-R.; Jiang, N. Extremely Rapid Extraction of DNA from Bacteria and Yeasts. Biotechnol. Lett. 2006, 28, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Structure and mechanism for DNA lesion recognition. Cell Res. 2008, 18, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryp, T.; Glorieux, G.; Joossens, M.; Vaneechoutte, M. Comparison of five assays for DNA extraction from bacterial cells in human faecal samples. J. Appl. Microbiol. 2020, 129, 378–388. [Google Scholar] [CrossRef]
- Davis, K.M.; Nakamura, S.; Weiser, J.N. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Investig. 2011, 121, 3666–3676. [Google Scholar] [CrossRef] [Green Version]
- Yokogawa, K.; Kawata, S.; Nishimura, S.; Ikeda, Y.; Yoshimura, Y. Mutanolysin, Bacteriolytic Agent for Cariogenic Streptococci: Partial Purification and Properties. Antimicrob. Agents Chaemother. 1974, 6, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.C.; Yiap, B.C. DNA, RNA, and Protein Extraction: The Past and The Present. J. Biomed. Biotechnol. 2009, 2009, 574398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ashram, S.; Nasr, I.A.; Suoa, X. Nucleic acid protocols: Extraction and optimization. Biotechnol. Rep. 2016, 12, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, K.; Kannamkumarath, S.S.; Wrobel, K.; Caruso, J.A. Hydrolysis of proteins with methanesulfonic acid for improved HPLC-ICP-MS determination of seleno-methionine in yeast and nuts. Anal. BioAnal. Chem. 2003, 375, 133–138. [Google Scholar] [CrossRef]
- Schindler, C.A.; Schuhardt, V.T. Lysostaphin: A new bacteriolytic agent for the staphylococcus. Proc. Natl. Acad. Sci. USA 1964, 51, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gründling, A.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus Mutants with Increased Lysostaphin Resistance. J. Bacteriol. 2006, 188, 6286–6297. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59. [Google Scholar] [CrossRef] [Green Version]
- Codling, C.; O’Mahony, L.; Shanahan, F.; Quigley, E.M.; Marchesi, J.R. A molecular analysis of fecal and mucosal bacterial communities in irritable bowel syndrome. Digest. Dis. Sci. 2010, 55, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Maeda, N.; Miron, I.M.; Yoh, M.; Izutsu, K.; Kataoka, C.; Honda, T.; Yasunaga, T.; Nakaya, T.; Kawai, J. Metagenomic diagnosis of bacterial infections. Emerg. Infect. Dis. 2008, 14, 1784. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Savard, P.; Lamarche, B.; Paradis, M.-E.; Thiboutot, H.; Laurin, É.; Roy, D. Impact of Bifidobacterium animalis subsp. lactis BB-12 and, Lactobacillus acidophilus LA-5-containing yoghurt, on fecal bacterial counts of healthy adults. Int. J. Food Microbiol. 2011, 149, 50–57. [Google Scholar] [CrossRef]
- Taipale, T.; Pienihäkkinen, K.; Isolauri, E.; Larsen, C.; Brockmann, E.; Alanen, P.; Jokela, J.; Söderling, E. Bifidobacterium animalis subsp. lactis BB-12 in reducing the risk of infections in infancy. Br. J. Nutr. 2011, 105, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniuchi, M.; Verweij, J.J.; Sethabutr, O.; Bodhidatta, L.; Garcia, L.; Maro, A.; Kumburu, H.; Gratz, J.; Kibiki, G.; Houpt, E.R. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagn. Microbiol. Infect. Dis. 2011, 71, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigsnaes, L.K.; Holck, J.; Meyer, A.S.; Licht, T.R. In vitro fermentation of sugar beet arabino-oligosaccharides by fecal microbiota obtained from patients with ulcerative colitis to selectively stimulate the growth of Bifidobacterium spp. and Lactobacillus spp. Appl. Environ. Microbiol. 2011, 77, 8336–8344. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Karlsson, C.; Olsson, C.; Adlerberth, I.; Wold, A.E.; Strachan, D.P.; Martricardi, P.M.; Åberg, N.; Perkin, M.R.; Tripodi, S. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 2008, 121, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, S.; Xie, Y.; Chen, X.; Zhao, Y.; He, D.; Li, J. Detection of methylated tissue factor pathway inhibitor 2 and human long DNA in fecal samples of patients with colorectal cancer in China. Cancer Epidemiol. 2012, 36, 73–77. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Weingarden, A.R.; Unno, T.; Khoruts, A.; Sadowsky, M.J. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 2013, 4, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoruts, A.; Dicksved, J.; Jansson, J.K.; Sadowsky, M.J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 2010, 44, 354–360. [Google Scholar] [CrossRef]
- Lauber, C.L.; Zhou, N.; Gordon, J.I.; Knight, R.; Fierer, N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol. Lett. 2010, 307, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claassen, S.; du Toit, E.; Kaba, M.; Moodley, C.; Zar, H.J.; Nicol, M.P. A comparison of the efficiency of five different commercial DNA extraction kits for extraction of DNA from faecal samples. J. Microbiol. Methods 2013, 94, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, N.A.; Walker, A.W.; Berry, S.H.; Duncan, S.H.; Farquarson, F.M.; Louis, P.; Thomson, J.M. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS ONE 2014, 9, e88982. [Google Scholar] [CrossRef]
- Nylund, L.; Heilig, H.G.; Salminen, S.; de Vos, W.M.; Satokari, R. Semi-automated extraction of microbial DNA from feces for qPCR and phylogenetic microarray analysis. J. Microbiol. Methods 2010, 83, 231–235. [Google Scholar] [CrossRef]
- Dridi, B.; Henry, M.; El Khechine, A.; Raoult, D.; Drancourt, M. High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS ONE 2009, 4, e7063. [Google Scholar] [CrossRef]
- Wu, G.D.; Lewis, J.D.; Hoffmann, C.; Chen, Y.-Y.; Knight, R.; Bittinger, K.; Hwang, J.; Chen, J.; Berkowsky, R.; Nessel, L. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Rangasamy, M.; Tan, S.Y.; Wang, H.; Siegfried, B.D. Evaluation of Five Methods for Total DNA Extraction from Western Corn Rootworm Beetles. PLoS ONE 2010, 5, e11963. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Engsbro, A.Ø.; Lukjancenko, O.; Kaas, R.S.; Lund, O.; Westh, H.; Aarestrup, F.M. Evaluating next-generation sequencing for direct clinical diagnostics in diarrhoeal disease. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, S.-H.; Kuang, Y.-S.; He, J.-R.; Lu, J.-H.; Luo, B.-J.; Jiang, F.-J.; Liu, Y.-Z.; Papasian, C.J.; Xia, H.-M. Effect of short-term room temperature storage on the microbial community in infant fecal samples. Sci. Rep. 2016, 6, 26648. [Google Scholar] [CrossRef] [PubMed]
- De Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef] [Green Version]
- Nistal, E.; Caminero, A.; Vivas, S.; de Morales, J.M.R.; de Miera, L.E.S.; Rodríguez-Aparicio, L.B.; Casqueiro, J. Differences in faecal bacteria populations and faecal bacteria metabolism in healthy adults and celiac disease patients. Biochimie 2012, 94, 1724–1729. [Google Scholar] [CrossRef]
- Ahlroos, T.; Tynkkynen, S. Quantitative strain-specific detection of Lactobacillus rhamnosus GG in human faecal samples by real-time PCR. J. Appl. Microbiol. 2009, 106, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksha, P.; Elbourne, A.; Gangadoo, S.; Brown, R.; Cozzolino, D.; Chapman, J. A review of methods for the detection of pathogenic microorganisms. Analyst 2019, 144, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Piterina, A.V.; Pembroke, J.T. Use of PCR-DGGE Based Molecular Methods to Analyse Microbial Community Diversity and Stability during the Thermophilic Stages of an ATAD Wastewater Sludge Treatment Process as an Aid to Performance Monitoring. ISRN Biotechnol. 2013, 2013, 162645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyzer, G.; Waal, E.C.d.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Ferris, M.J.; Muyzer, G.; Ward, D.M. Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 1996, 62, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Cardona, S.; Eck, A.; Cassellas, M.; Gallart, M.; Alastrue, C.; Dore, J.; Azpiroz, F.; Roca, J.; Guarner, F.; Manichanh, C. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol. 2012, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- De Palma, G.; Nadal, I.; Medina, M.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Van der Waaij, L.A.; Kroese, F.G.; Visser, A.; Nelis, G.F.; Westerveld, B.D.; Jansen, P.L.; Hunter, J.O. Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2004, 16, 669–674. [Google Scholar] [CrossRef]
- Nam, Y.-D.; Jung, M.-J.; Roh, S.W.; Kim, M.-S.; Bae, J.-W. Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS ONE 2011, 6, e22109. [Google Scholar] [CrossRef]
- Osman, M.A.; Neoh, H.-M.; Mutalib, N.S.B.; Chin, S.-F.; Jamal, R. 16S rRNA Gene Sequencing for Deciphering the Colorectal Cancer Gut Microbiome: Current Protocols and Workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Yap, G.C.; Hong, P.-Y.; Huang, C.-H.; Aw, M.M.; Shek, L.P.-C.; Liu, W.-T.; Lee, B.W. Immune-modulatory genomic properties differentiate gut microbiota of infants with and without eczema. PLoS ONE 2017, 12, e0184955. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Long, M.; Li, H.; Gatesoupe, F.-J.; Zhang, X.; Zhang, Q.; Feng, D.; Li, A. Multi-omics analysis reveals a correlation between the host phylogeny, gut microbiota and metabolite profiles in cyprinid fishes. Front. Microbiol. 2017, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Beckers, B.; De Beeck, M.O.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Hoang, H.T.; Le, D.H.; Le, T.T.H.; Nguyen, T.T.N.; Chu, H.H.; Nguyen, N.T.J.D. Metagenomic 16S rDNA amplicon data of microbial diversity of guts in Vietnamese humans with type 2 diabetes and nondiabetic adults. Data Brief 2021, 34, 106690. [Google Scholar] [CrossRef]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.J.C. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef]
- McKnight, D.T.; Huerlimann, R.; Bower, D.S.; Schwarzkopf, L.; Alford, R.A.; Zenger, K.R. Methods for normalizing microbiome data: An ecological perspective. Methods Ecol. Evol. 2018. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E.J.P.O. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Gosalbes, M.J.; Durbán, A.; Pignatelli, M.; Abellan, J.J.; Jiménez-Hernández, N.; Pérez-Cobas, A.E.; Latorre, A.; Moya, A. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS ONE 2011, 6, e17447. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of Metatranscriptomics in Microbiome Research. Bioinform. Biol. Insights 2016, 10, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.E.; Vos, P.; Ijames, S.; Lysle, D.T.; Allison, R.R.; Flake, G.; Sinar, D.R.; Naziri, W.; Marcuard, S.P.; Pennington, R. Transcriptomic Molecular Markers for Screening Human Colon Cancer in Stool and Tissue. Cancer Genom. Proteom. 2007, 4, 1–20. [Google Scholar]
- Reck, M.; Tomasch, J.; Deng, Z.; Jarek, M.; Husemann, P.; Wagner-Döbler, I. Stool metatranscriptomics: A technical guideline for mRNA stabilisation and isolation. BMC Genom. 2015, 16, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar]
- Booijink, C.C.G.M.; Boekhorst, J.; Zoetendal, E.G.; Smidt, H.; Kleerebezem, M.; de Vos, W.M. Metatranscriptome Analysis of the Human Fecal Microbiota Reveals Subject-Specific Expression Profiles, with Genes Encoding Proteins Involved in Carbohydrate Metabolism Being Dominantly Expressed. Appl. Environ. Microbiol. 2010, 76, 5533–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoukos, G.; Ciulla, D.M.; Huang, K.; Haas, B.J.; Izard, J.; Levin, J.Z.; Livny, J.; Earl, A.M.; Gevers, D.; Ward, D.V.; et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 2012, 13, R23. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.; Amstislavskiy, V.; Risch, T.; Schuette, M.; Dökel, S.; Ralser, M.; Balzereit, D.; Lehrach, H.; Yaspo, M.-L. Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genom. 2014, 15, 675. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; McSweeney, C.S. An Efficient RNA Extraction Method for Estimating Gut Microbial Diversity by Polymerase Chain Reaction. Curr. Microbiol. 2009, 58, 464–471. [Google Scholar] [CrossRef]
- Ilott, N.E.; Bollrath, J.; Danne, C.; Schiering, C.; Shale, M.; Adelmann, K.; Krausgruber, T.; Heger, A.; Sims, D.; Powrie, F. Defining the microbial transcriptional response to colitis through integrated host and microbiome profiling. ISME J. 2016, 10, 2389. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Booijink, C.C.; Klaassens, E.S.; Heilig, H.G.; Kleerebezem, M.; Smidt, H.; Vos, W.M.d. Isolation of RNA from bacterial samples of the human gastrointestinal tract. Nat. Protoc. 2006, 1, 954–959. [Google Scholar] [CrossRef]
- Shulman, L.M.; Hindiyeh, M.; Muhsen, K.; Cohen, D.; Mendelson, E.; Sofer, D. Evaluation of Four Different Systems for Extraction of RNA from Stool Suspensions Using MS-2 Coliphage as an Exogenous Control for RT-PCR Inhibition. PLoS ONE 2012, 7, e39455. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Rodrigo, B.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Cullen, B.R. Viruses and microRNAs. Nat. Genet. 2006, 38, S25. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Watanabe, Y. Small RNA world in plants. Tanpakushitsu Kakusan Koso 2006, 51, 2463–2470. [Google Scholar]
- Gottesman, S. Micros for microbes: Non-coding regulatory RNAs in bacteria. Trends Genet. 2005, 21, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Plasterk, R.H. Micro RNAs in animal development. Cell 2006, 124, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, F.E.; Jeffries, C.D.; Vos, P.W.; Flake, G.; Nuovo, G.J.; Sinar, D.R.; Naziri, W.; Marcuard, S.P. Diagnostic microRNA markers for screening sporadic human colon cancer and active ulcerative colitis in stool and tissue. Cancer Genom. Proteom. 2009, 6, 281–295. [Google Scholar]
- Link, A.; Becker, V.; Goel, A.; Wex, T.; Malfertheiner, P. Feasibility of fecal microRNAs as novel biomarkers for pancreatic cancer. PLoS ONE 2012, 7, e42933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhou, X.; Cai, W.; Shi, R.; Yang, G.; Yuan, L. Host intestinal epithelium derived mirnas shape the microbiota and its implication in cardiovascular diseases. J. Am. Coll. Cardiol. 2017, 69, 1075. [Google Scholar] [CrossRef]
- Peck, B.C.; Mah, A.T.; Pitman, W.A.; Ding, S.; Lund, P.K.; Sethupathy, P. Functional transcriptomics in diverse intestinal epithelial cell types reveals robust microRNA sensitivity in intestinal stem cells to microbial status. J. Biol. Chem. 2017, 292, 2586–2600. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Freudenberg, J.; Suh, Y.J.; Yang, Y. Using volcano plots and regularized-chi statistics in genetic association studies. Comput. Biol. Chem. 2014, 48, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, L.S.; Marchesi, J.R. Assessing the impact of long term frozen storage of faecal samples on protein concentration and protease activity. J. Microbiol. Methods 2016, 123, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhorst, J.; Boone, J.; Lauche, R.; Rueffer, A.; Dobos, G.J.J. Faecal lactoferrin, calprotectin, PMN-elastase, CRP, and white blood cell count as indicators for mucosal healing and clinical course of disease in patients with mild to moderate ulcerative colitis: Post hoc analysis of a prospective clinical trial. J. Crohn’s Colitis 2016, 10, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Lewis, S.J.; Creanor, S.; Ayling, R.M. Age-related faecal calprotectin, lactoferrin and tumour M2-PK concentrations in healthy volunteers. Ann. Clin. Biochem. 2010, 47, 259–263. [Google Scholar] [CrossRef]
- Ruiz, L.; Hidalgo, C.; Blanco-Míguez, A.; Lourenço, A.; Sánchez, B.; Margolles, A. Tackling probiotic and gut microbiota functionality through proteomics. J. Proteom. 2016, 147, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Dhaliwal, A.; Zeino, Z.; Tomkins, C.; Cheung, M.; Nwokolo, C.; Smith, S.; Harmston, C.; Arasaradnam, R. Utility of faecal calprotectin in inflammatory bowel disease (IBD): What cut-offs should we apply? Frontline Gastroenterol. 2015, 6, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, V.; Lauritzen, T.; Jelsness-JØrgensen, L.-P.; Moum, B. Validation of a new extraction device for measuring faecal calprotectin in inflammatory bowel disease, and comparison to established extraction methods. Scand. J. Clin. Lab. Investig. 2015, 75, 355–361. [Google Scholar] [CrossRef]
- Kolmeder, C.A.; Salojärvi, J.; Ritari, J.; De Been, M.; Raes, J.; Falony, G.; Vieira-Silva, S.; Kekkonen, R.A.; Corthals, G.L.; Palva, A. Faecal metaproteomic analysis reveals a personalized and stable functional microbiome and limited effects of a probiotic intervention in adults. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Young, J.C.; Pan, C.; Adams, R.M.; Brooks, B.; Banfield, J.F.; Morowitz, M.J.; Hettich, R.L. Metaproteomics Reveals Functional Shifts in Microbial and Human Proteins During Infant Gut Colonization Case. Proteomics 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Debyser, G.; Mesuere, B.; Clement, L.; Van de Weygaert, J.; Van Hecke, P.; Duytschaever, G.; Aerts, M.; Dawyndt, P.; De Boeck, K.; Vandamme, P. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, J.S.; Marcobal, A.; Sonnenburg, J.L.; Elias, J.E. Host-centric proteomics of stool: A novel strategy focused on intestinal responses to the gut microbiota. Mol. Cell. Proteom. 2013, 12, 3310–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, E.; Anselmo, M.; Calha, M.; Bottrill, A.; Duarte, I.; Andrew, P.W.; Faleiro, M.L. The intestinal proteome of diabetic and control children is enriched with different microbial and host proteins. Microbiology 2017, 163, 161–174. [Google Scholar] [CrossRef]
- Nowakowski, A.B.; Wobig, W.J.; Petering, D.H. Native SDS-PAGE: High resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. Metallomics 2014, 6, 1068–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Wang, K.; Huang, C.; Nice, E.C. Mining the fecal proteome: From biomarkers to personalised medicine. Exp. Rev. Proteom. 2017, 14, 445–459. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, F.; Chen, L.; Chen, K. Proteomics, metabolomics and metagenomics for type 2 diabetes and its complications. Life Sci. 2018, 212, 194–202. [Google Scholar] [CrossRef]
- Chauvin, A.; Boisvert, F.-M. Clinical proteomics in colorectal cancer, a promising tool for improving personalised medicine. Proteomes 2018, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, L.; Shan, G.; Sun, Z.; Zhang, F.; Xu, C.; Lou, X.; Li, S.; Du, H.; Chen, H.; Xu, G. Quantitative proteomic analysis reveals the deregulation of nicotinamide adenine dinucleotide metabolism and CD38 in inflammatory bowel disease. BioMed Res. Int. 2019, 2019, 3950628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarżyńska, E.; Kiersztyn, B.; Wilczyńska, P.; Jakimiuk, A.; Lisowska-Myjak, B. Total proteolytic activity and concentration of alpha-1 antitrypsin in meconium for assessment of the protease/antiprotease balance. Eur. J. Obstetr. Gynecol. Reproduct. Biol. 2018, 223, 133–138. [Google Scholar] [CrossRef]
- Maloupazoa Siawaya, A.; Kuissi Kamgaing, E.; Minto’o Rogombe, S.; Obiang, T.; Moungoyi Massala, E.; Magossou Mbadinga, M.; Leboueny, M.; Mvoundza Ndjindji, O.; Mveang-Nzoghe, A.; Ondo, J. HIV-exposed uninfected compared with unexposed infants show the presence of leucocytes, lower lactoferrin levels and antimicrobial-resistant micro-organisms in the stool. Paediatr. Int. Child Health 2019, 39, 249–258. [Google Scholar] [CrossRef]
- Annaházi, A.; Ábrahám, S.; Farkas, K.; Rosztóczy, A.; Inczefi, O.; Földesi, I.; Szűcs, M.; Rutka, M.; Theodorou, V.; Eutamene, H. A pilot study on faecal MMP-9: A new noninvasive diagnostic marker of colorectal cancer. Br. J. Cancer 2016, 114, 787–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widlak, M.; Thomas, C.; Thomas, M.; Tomkins, C.; Smith, S.; O’Connell, N.; Wurie, S.; Burns, L.; Harmston, C.; Evans, C. Diagnostic accuracy of faecal biomarkers in detecting colorectal cancer and adenoma in symptomatic patients. Aliment. Pharmacol. Ther. 2017, 45, 354–363. [Google Scholar] [CrossRef]
- Buisson, A.; Vazeille, E.; Minet-Quinard, R.; Goutte, M.; Bouvier, D.; Goutorbe, F.; Pereira, B.; Barnich, N.; Bommelaer, G. Faecal chitinase 3-like 1 is a reliable marker as accurate as faecal calprotectin in detecting endoscopic activity in adult patients with inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 43, 1069–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisowska-Myjak, B.; Muszyński, J.; Zborowska, H. Search for the Laboratory Parameters of Inflammation In the Serum and Faeces of Patients with Irritable Bowel Syndrome. Int. J. Gastroenterol. Res. Pract. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, F.S.; Burri, E.; Beglinger, C. The role and utility of faecal markers in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2015, 8, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Casavant, E.; Park, K.; Elias, J.E. Proteomic Discovery of Stool Protein Biomarkers for Distinguishing Pediatric Inflammatory Bowel Disease Flares. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Devaraj, S.; Hemarajata, P.; Versalovic, J. The human gut microbiome and body metabolism: Implications for obesity and diabetes. Clin. Chem. 2013, 59, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790. [Google Scholar] [CrossRef]
- Yu, M.; Jia, H.-M.; Zhou, C.; Yang, Y.; Sun, L.-L.; Zou, Z.-M. Urinary and fecal metabonomics study of the protective effect of Chaihu-Shu-Gan-San on antibiotic-induced gut microbiota dysbiosis in rats. Sci. Rep. 2017, 7, 46551. [Google Scholar] [CrossRef]
- Ng, J.S.Y.; Ryan, U.; Trengove, R.D.; Maker, G.L. Development of an untargeted metabolomics method for the analysis of human faecal samples using Cryptosporidium-infected samples. Mol. Biochem. Parasitol. 2012, 185, 145–150. [Google Scholar] [CrossRef]
- Hughes, R.; Magee, E.A.M.; Bingham, S. Protein Degradation in the Large Intestine: Relevance to Colorectal Cancer. Curr. Issues Intest. Microbiol. 2000, 1, 51–58. [Google Scholar]
- Rohloff, J. Analysis of Phenolic and Cyclic Compounds in Plants Using Derivatization Techniques in Combination with GC-MS-Based Metabolite Profiling. Molecules 2015, 20, 3431–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Zhang, H.; Aranibar, N.; Hanson, R.; Huang, Y.; Cheng, P.T.; Wu, S.; Bonacorsi, S.; Zhu, M.; Swaminathan, A.; et al. Structural elucidation of human oxidative metabolites of muraglitazar: Use of microbial bioreactors in the biosynthesis of metabolite standards. Drug Metab. Dispos. 2006, 34, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Soso, S.B.; Koziel, J.A.; Johnson, A.; Lee, Y.J.; Fairbanks, W.S. Analytical Methods for Chemical and Sensory Characterization of Scent-Markings in Large Wild Mammals: A Review. Sensors 2014, 14, 4428–4465. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: The combination of targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chapman, J.; Power, A.; Roberts, J.; Cozzolino, D. The Application of State-of-the-Art Analytic Tools (Biosensors and Spectroscopy) in Beverage and Food Fermentation Process Monitoring. Fermentation 2017, 3, 50. [Google Scholar] [CrossRef] [Green Version]
- Hough, R.; Archer, D.; Probert, C. A comparison of sample preparation methods for extracting volatile organic compounds (VOCs) from equine faeces using HS-SPME. Metabolomics 2018, 14, 19. [Google Scholar] [CrossRef] [Green Version]
- Deda, O.; Gika, H.G.; Wilson, I.D.; Theodoridis, G.A. An overview of fecal sample preparation for global metabolic profiling. J. Pharm. Biomed. Anal. 2015, 113, 137–150. [Google Scholar] [CrossRef]
- Poroyko, V.; Morowitz, M.; Bell, T.; Ulanov, A.; Wang, M.; Donovan, S.; Bao, N.; Gu, S.; Hong, L.; Alverdy, J.C.; et al. Diet creates metabolic niches in the “inmature gut” that shape microbial communities. Nutr. Hosp. 2011, 26, 1283–1295. [Google Scholar]
- Hublin, J.S.Y.N.; Ryan, U.; Trengove, R.; Maker, G. Metabolomic Profiling of Faecal Extracts from Cryptosporidium parvum Infection in Experimental Mouse Models. PLoS ONE 2013, 8, e77803. [Google Scholar] [CrossRef]
- Gao, X.; Pujos-Guillot, E.; Se’be’dio, J.-L. Development of a Quantitative Metabolomic Approach to Study Clinical Human Fecal Water Metabolome Based on Trimethylsilylation Derivatization and GC/MS Analysis. Anal. Chem. 2010, 82, 6447–6456. [Google Scholar] [CrossRef] [PubMed]
- Battistel, D.; Piazza, R.; Argiriadis, E.; Marchiori, E.; Radaelli, M.; Barbante, C. GC-MS method for determining faecal sterols as biomarkers of human and pastoral animal presence in freshwater sediments. Anal. Bioanal. Chem. 2015, 407, 8505–8514. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass Spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Strickland, J.R. Improved GC/MS method for quantitation of n-alkanes in plant and fecal material. J. Agricult. Food Chem. 2007, 55, 7301–7307. [Google Scholar] [CrossRef]
- Fang, M.; Ivanisevic, J.; Benton, H.P.; Johnson, C.H.; Patti, G.J.; Hoang, L.T.; Uritboonthai, W.; Kurczy, M.E.; Siuzdak, G. Thermal degradation of small molecules: A global metabolomic investigation. Anal. Chem. 2015, 87, 10935–10941. [Google Scholar] [CrossRef]
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot. 2004, 56, 219–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Huang, H.; Xu, W.; Chen, D.; Yu, J.; Li, J.; Li, L. Fecal metabolome profiling of liver cirrhosis and hepatocellular carcinoma patients by ultra performance liquid chromatography—Mass spectrometry. Anal. Chim. Acta 2011, 691, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Forcisi, S.; Moritz, F.; Lucio, M.; Lehmann, R.; Stefan, N.; Schmitt-Kopplin, P. Solutions for Low and High Accuracy Mass Spectrometric Data Matching: A Data-Driven Annotation Strategy in Nontargeted Metabolomics. Anal. Chem. 2015, 87, 8917−8924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernocchi, P.; Chierico, F.D.; Putignani, L. Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Macpherson, E.; Theodoridis, G.A.; Wilson, I.D. Evaluation of the repeatability of ultra-performance liquid chromatography–TOF-MS for global metabolic profiling of human urine samples. J. Chromatogr. B 2008, 87, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.; Zhang, A.-Y.; Cao, H.-C.; Lu, H.-F.; Wang, B.-H.; Xie, Q.; Xu, W.; Li, L.-J. Metabolomic analyses of faeces reveals malabsorption in cirrhotic patients. Digest. Liver Dis. 2013, 45, 677–682. [Google Scholar] [CrossRef]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability, and autoimmunity celiac disease and type 1 diabetes paradigms. Ann. N. Y. Acad. Sci. 2009, 1165, 195. [Google Scholar] [CrossRef] [Green Version]
- Verdu, E.F.; Schuppan, D.; Biology, M. The enemy within the gut: Bacterial pathogens in celiac autoimmunity. Nat. Struct. 2020, 27, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.Y.; Chen, Z.; McCluskey, J.; Anderson, R.P. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Primec, M.; Klemenak, M.; Di Gioia, D.; Aloisio, I.; Cionci, N.B.; Quagliariello, A.; Gorenjak, M.; Mičetić-Turk, D.; Langerholc, T. Clinical intervention using Bifidobacterium strains in celiac disease children reveals novel microbial modulators of TNF-α and short-chain fatty acids. Clin. Nutr. 2019, 38, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Serena, G.; Davies, C.; Cetinbas, M.; Sadreyev, R.I.; Fasano, A. Analysis of blood and fecal microbiome profile in patients with celiac disease. Hum. Microb. J. 2019, 11, 100049. [Google Scholar] [CrossRef]
- Sanz, Y.; Sánchez, E.; Marzotto, M.; Calabuig, M.; Torriani, S.; Dellaglio, F. Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol. Med. Microbiol. 2007, 51, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Bodkhe, R.; Shetty, S.A.; Dhotre, D.P.; Verma, A.K.; Bhatia, K.; Mishra, A.; Kaur, G.; Pande, P.; Bangarusamy, D.K.; Santosh, B.P. Comparison of small gut and whole gut microbiota of first-degree relatives with adult celiac disease patients and controls. Front. Microbiol. 2019, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares, M.; Walker, A.W.; Capilla, A.; Benítez-Páez, A.; Palau, F.; Parkhill, J.; Castillejo, G.; Sanz, Y. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome 2018, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drabińska, N.; Jarocka-Cyrta, E.; Markiewicz, L.H.; Krupa-Kozak, U. The effect of oligofructose-enriched inulin on faecal bacterial counts and microbiota-associated characteristics in celiac disease children following a gluten-free diet: Results of a randomized, placebo-controlled trial. Nutrients 2018, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harnett, J.; Myers, S.P.; Rolfe, M. Significantly higher faecal counts of the yeasts candida and saccharomyces identified in people with coeliac disease. Gut Pathog. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caminero, A.; Galipeau, H.J.; McCarville, J.L.; Johnston, C.W.; Bernier, S.P.; Russell, A.K.; Jury, J.; Herran, A.R.; Casqueiro, J.; Tye-Din, J.A. Duodenal bacteria from patients with celiac disease and healthy subjects distinctly affect gluten breakdown and immunogenicity. Gastroenterology 2016, 151, 670–683. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Yang, Y.; Tolstikov, V.; Kiebish, M.A.; Palm, N.W.; Ludvigsso, J.; Altindis, E. Children Developing Celiac Disease Have a Distinct and Proinflammatory Gut Microbiota in the First 5 Years of Life. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Sapone, A.; Lammers, K.M.; Casolaro, V.; Cammarota, M.; Giuliano, M.T.; De Rosa, M.; Stefanile, R.; Mazzarella, G.; Tolone, C.; Russo, M.I. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: Celiac disease and gluten sensitivity. BMC Med. 2011, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Bragde, H.; Jansson, U.; Jarlsfelt, I.; Söderman, J. Gene expression profiling of duodenal biopsies discriminates celiac disease mucosa from normal mucosa. Pediatr. Res. 2011, 69, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Bragde, H.; Jansson, U.; Fredrikson, M.; Grodzinsky, E.; Söderman, J.J.C. Celiac disease biomarkers identified by transcriptome analysis of small intestinal biopsies. Cell. Mol. Life Sci. 2018, 75, 4385–4401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauregi-Miguel, A.; Santin, I.; Garcia-Etxebarria, K.; Olazagoitia-Garmendia, A.; Romero-Garmendia, I.; Sebastian-delaCruz, M.; Irastorza, I.; Castellanos-Rubio, A.; Bilbao, J.R. MAGI2 gene region and celiac disease. Front. Nutr. 2019, 6, 187. [Google Scholar] [CrossRef]
- Cenit, M.C.; Olivares, M.; Codoñer-Franch, P.; Sanz, Y. Intestinal microbiota and celiac disease: Cause, consequence or co-evolution? Nutrients 2015, 7, 6900–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicerone, C.; Nenna, R.; Pontone, S. Th17, intestinal microbiota and the abnormal immune response in the pathogenesis of celiac disease. Gastroenterol. Hepatol. Bed Bench 2015, 8, 117. [Google Scholar]
- Barr, T.; Sureshchandra, S.; Ruegger, P.; Zhang, J.; Ma, W.; Borneman, J.; Grant, K.; Messaoudi, I. Concurrent gut transcriptome and microbiota profiling following chronic ethanol consumption in nonhuman primates. Gut Microb. 2018, 9, 338–356. [Google Scholar] [CrossRef] [Green Version]
- Schlaberg, R.; Barrett, A.; Edes, K.; Graves, M.; Paul, L.; Rychert, J.; Lopansri, B.K.; Leung, D.T. Fecal Host Transcriptomics for Non-invasive Human Mucosal Immune Profiling: Proof of Concept in Clostridium difficile Infection. Pathog. Immun. 2018, 3, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felli, C.; Baldassarre, A.; Masotti, A. Intestinal and circulating microRNAs in coeliac disease. Int. J. Mol. Sci. 2017, 18, 1907. [Google Scholar] [CrossRef] [Green Version]
- Ertekin, V.; Selimoglu, M.A.; Turgut, A.; Bakan, N. Fecal calprotectin concentration in celiac disease. J. Clin. Gastroenterol. 2010, 44, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Biskou, O.; Gardner-Medwin, J.; Mackinder, M.; Bertz, M.; Clark, C.; Svolos, V.; Russell, R.K.; Edwards, C.A.; McGrogan, P.; Gerasimidis, K. Faecal calprotectin in treated and untreated children with coeliac disease and juvenile idiopathic arthritis. J. Pediatr. Gastroenterol. Nutr. 2016, 63, e112–e115. [Google Scholar] [CrossRef] [Green Version]
- Rajani, S.; Huynh, H.Q.; Shirton, L.; Kluthe, C.; Spady, D.; Prosser, C.; Meddings, J.; Rempel, G.R.; Persad, R.; Turner, J.M. A Canadian study toward changing local practice in the diagnosis of pediatric celiac disease. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6234160. [Google Scholar] [CrossRef] [Green Version]
- Shahramian, I.; Bazi, A.; Shafie-Sabet, N.; Sargazi, A.; Aval, O.S.; Delaramnasab, M.; Behzadi, M.; Zaer-Sabet, Z. A Survey of Fecal Calprotectin in Children with Newly Diagnosed Celiac Disease with Villous Atrophy. Shiraz E-Med. J. 2019. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Pramanik, A.; Acharya, P.; Makharia, G.K. Non-Invasive Biomarkers for Celiac Disease. J. Clin. Med. 2019, 8, 885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tola, M.; Marino, M.; Casale, R.; Di Battista, V.; Borghini, R.; Picarelli, A. Extension of the celiac intestinal antibody (CIA) pattern through eight antibody assessments in fecal supernatants from patients with celiac disease. Immunobiology 2016, 221, 63–69. [Google Scholar] [CrossRef]
- Comino, I.; Fernández-Bañares, F.; Esteve, M.; Ortigosa, L.; Castillejo, G.; Fambuena, B.; Ribes-Koninckx, C.; Sierra, C.; Rodríguez-Herrera, A.; Salazar, J.C. Fecal gluten peptides reveal limitations of serological tests and food questionnaires for monitoring gluten-free diet in celiac disease patients. Am. J. Gastroenterol. 2016, 111, 1456. [Google Scholar] [CrossRef] [Green Version]
- Kappler, M.; Krauss-Etschmann, S.; Diehl, V.; Zeilhofer, H.; Koletzko, S. Detection of secretory IgA antibodies against gliadin and human tissue transglutaminase in stool to screen for coeliac disease in children: Validation study. BMJ 2006, 332, 213–214. [Google Scholar] [CrossRef] [Green Version]
- Caminero, A.; Nistal, E.; Arias, L.; Vivas, S.; Comino, I.; Real, A.; Sousa, C.; de Morales, J.M.R.; Ferrero, M.A.; Rodríguez-Aparicio, L.B. A gluten metabolism study in healthy individuals shows the presence of faecal glutenasic activity. Eur. J. Nutr. 2012, 51, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Roca, M.; Donat, E.; Masip, E.; Crespo Escobar, P.; Fornes-Ferrer, V.; Polo, B.; Ribes-Koninckx, C. Detection and quantification of gluten immunogenic peptides in feces of infants and their relationship with diet. Rev. Esp. Enferm. Dig. 2018, 111. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.D.L.; Rodríguez-Herrera, A.; Sousa, C.; Comino, I. Biomarkers to monitor gluten-free diet compliance in celiac patients. Nutrients 2017, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Comino, I.; Real, A.; Vivas, S.; Síglez, M.Á.; Caminero, A.; Nistal, E.; Casqueiro, J.; Rodríguez-Herrera, A.; Cebolla, A.; Sousa, C. Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer equivalent epitopes in feces. Am. J. Clin. Nutr. 2012, 95, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Gerasimidis, K.; Zafeiropoulou, K.; Mackinder, M.; Ijaz, U.Z.; Duncan, H.; Buchanan, E.; Cardigan, T.; Edwards, C.A.; McGrogan, P.; Russell, R.K. Comparison of clinical methods with the faecal gluten immunogenic peptide to assess gluten intake in coeliac disease. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, B.; Ferretti, F.; Cinci, F.; Biviano, I.; Santini, A.; Grande, E.; Quagliarella, F.; Terzuoli, L.; Bacarelli, M.R.; Bizzaro, N. Fecal gluten immunogenic peptides as indicators of dietary compliance in celiac patients. Minerva Gastroenterol. Dietol. 2020. [Google Scholar] [CrossRef]
- Stefanolo, J.P.; Tálamo, M.; Dodds, S.; de la Paz Temprano, M.; Costa, A.F.; Moreno, M.L.; Pinto-Sánchez, M.I.; Smecuol, E.; Vázquez, H.; Gonzalez, A. Real-world Gluten Exposure in Patients With Celiac Disease on Gluten-Free Diets, Determined From Gliadin Immunogenic Peptides in Urine and Fecal Samples. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Kim, J.-H.; An, H.J.; Garrido, D.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Proteomic analysis of Bifidobacterium longum subsp. infantis reveals the metabolic insight on consumption of prebiotics and host glycans. PLoS ONE 2013, 8, e57535. [Google Scholar] [CrossRef] [PubMed]
- Di Cagno, R.; Rizzello, C.G.; Gagliardi, F.; Ricciuti, P.; Ndagijimana, M.; Francavilla, R.; Guerzoni, M.E.; Crecchio, C.; Gobbetti, M.; De Angelis, M. Different fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl. Environ. Microbiol. 2009, 75, 3963–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cagno, R.; De Angelis, M.; De Pasquale, I.; Ndagijimana, M.; Vernocchi, P.; Ricciuti, P.; Gagliardi, F.; Laghi, L.; Crecchio, C.; Guerzoni, M.E. Duodenal and faecal microbiota of celiac children: Molecular, phenotype and metabolome characterization. BMC Microbiol. 2011, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Tjellström, B.; Stenhammar, L.; Högberg, L.; Fälth-Magnusson, K.; Magnusson, K.-E.; Midtvedt, T.; Sundqvist, T.; Norin, E. Gut microflora associated characteristics in children with celiac disease. Am. J. Gastroenterol. 2005, 100, 2784–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Andersen, D.; Roager, H.M.; Bahl, M.I.; Hansen, C.H.F.; Danneskiold-Samsøe, N.B.; Kristiansen, K.; Radulescu, I.D.; Sina, C.; Frandsen, H.L. Effects of gliadin consumption on the intestinal microbiota and metabolic homeostasis in mice fed a high-fat diet. Sci. Rep. 2017, 7, 44613. [Google Scholar] [CrossRef] [Green Version]
- Tjellström, B.; Högberg, L.; Stenhammar, L.; Fälth-Magnusson, K.; Magnusson, K.-E.; Norin, E.; Sundqvist, T.; Midtvedt, T. Faecal short-chain fatty acid pattern in childhood coeliac disease is normalised after more than one year’s gluten-free diet. Microb. Ecol. Health Dis. 2013, 24, 20905. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Ahern, A.; Carbone, C.; Temko, A.; Claesson, M.J.; Gasbarrini, A.; Tortora, G. Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 635–648. [Google Scholar] [CrossRef]
- Chapman, J.; Truong, V.K.; Elbourne, A.; Gangadoo, S.; Cheeseman, S.; Rajapaksha, P.; Latham, K.; Crawford, R.J.; Cozzolino, D. Combining Chemometrics and Sensors: Toward New Applications in Monitoring and Environmental Analysis. Chem. Rev. 2020. [Google Scholar] [CrossRef]
- Chapman, J.; Orrell-Trigg, R.; Kwoon, K.Y.; Truong, V.K.; Cozzolino, D. A High-Throughput and Machine Learning Resistance Monitoring System to Determine the Point of Resistance for Escherichia coli with tetracycline: Combining UV-Visible Spectrophotometry with Principal Component Analysis. Biotechnol. Bioeng. 2021. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Mode of Action |
|---|---|
| Lysozyme | Cleaves the bacterial cell wall [79] by catalyzing the hydrolysis of glycoside-linkages in the peptidoglycan layer [73]. |
| Mutanolysin | Rapidly solubilizes cell walls and removes reducing sugars and free amino groups. Greatly effective against some streptococci strains [80] and Gram-positive cell wall [73]. |
| Guanidine thiocyanate | A chaotropic agent, which disrupts the hydrogen bonding of a molecule and is used to inactivate DNase and RNase, enzymes that digest DNA and RNA, respectively [73,81]. |
| RNAse A | Degrades single-stranded RNA [82] |
| Proteinase K | Digests proteins by hydrolyzing peptide bonds [83] |
| Lysostaphin | Specifically breaks some Staphylococcus spp. [84] by cleaving the pentaglycine cross bridges of its cell wall [73,85] |
| Method | Concentration (μg/g) | Absorbance Ratio 260/280 | Absorbance Ratio 260/230 |
|---|---|---|---|
| Zoetendal (2006) [134] | >400 | <2.0 | <1.5 |
| Powersoil microbiome kit (MoBio) [134] | >200 | >2.0 | >2.25 |
| Bead beating + column [139] | 185.1 | 2.11 | 2.04 |
| TRIzol Max bacterial RNA kit (Invitrogen) [139] | 199.5 | 2.02 | 1.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gangadoo, S.; Rajapaksha Pathirannahalage, P.; Cheeseman, S.; Dang, Y.T.H.; Elbourne, A.; Cozzolino, D.; Latham, K.; Truong, V.K.; Chapman, J. The Multiomics Analyses of Fecal Matrix and Its Significance to Coeliac Disease Gut Profiling. Int. J. Mol. Sci. 2021, 22, 1965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041965
Gangadoo S, Rajapaksha Pathirannahalage P, Cheeseman S, Dang YTH, Elbourne A, Cozzolino D, Latham K, Truong VK, Chapman J. The Multiomics Analyses of Fecal Matrix and Its Significance to Coeliac Disease Gut Profiling. International Journal of Molecular Sciences. 2021; 22(4):1965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041965
Chicago/Turabian StyleGangadoo, Sheeana, Piumie Rajapaksha Pathirannahalage, Samuel Cheeseman, Yen Thi Hoang Dang, Aaron Elbourne, Daniel Cozzolino, Kay Latham, Vi Khanh Truong, and James Chapman. 2021. "The Multiomics Analyses of Fecal Matrix and Its Significance to Coeliac Disease Gut Profiling" International Journal of Molecular Sciences 22, no. 4: 1965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041965