
An Evidence-Based Systematic Review of Human Knee Post-Traumatic Osteoarthritis (PTOA): Timeline of Clinical Presentation and Disease Markers, Comparison of Knee Joint PTOA Models and Early Disease Implications


Abstract
:1. Introduction
2. Methodology
3. Results of the Clinical Studies That Measured Inflammatory and Other Key Biochemical Biomarkers in Patients Who Have Had Knee Trauma
3.1. Continuous Localized Inflammation from Months to Years after Knee Trauma
3.2. Activation of the Complement System Correlates with Inflammation Months to Years after Knee Trauma
3.3. Release of MMPs, ECM Components and Damage to the Cartilage Collagen Network Months to Years after Knee Trauma
3.4. Systemic Measurable Effects after Knee Trauma
3.5. Local and Circulating Inflammatory Cytokines and Other Biochemical Biomarkers Once PTOA Is Diagnosed
3.6. The Role of Inflammation in the Pathogenesis of Knee PTOA—Summary of Clinical Proof
4. Results of the In Vivo, Ex Vivo and In Vitro Models That Simulate Inflammation and/or Injury of the Knee Joint
4.1. In Vivo Knee Joint Inflammatory, Injury and PTOA Animal Models
4.2. Considerations in Using In Vitro Chondrocyte and Ex Vivo Articular Cartilage Models
4.3. In Vitro Chondrocyte and Ex Vivo Articular Cartilage Explant Injury and/or Inflammatory Models
4.4. Co-Culture Studies
5. Clinical Findings vs. Experimental Evidence
5.1. Comparing the Concentrations of Inflammatory Cytokines Used in Laboratory Models vs. the Clinical Presentation
5.2. Comparing the Results of In Vivo, Ex Vivo and In Vitro Laboratory Models vs. the Clinical Presentation
5.3. Assessing Clinical and Experimental Results to Determine if IL-1β, TNF-α, IL-6 and IL-17 Are Possible Causal Factors in Inducing Progression towards Knee PTOA
6. Summary, Outlook and Early Disease Considerations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACAN | Aggrecan |
| ACI | Autologous chondrocyte implantation |
| ACL | Anterior cruciate ligament |
| ACLT | Anterior cruciate ligament transection |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| AI | Artificial intelligence |
| ARGS | Alanine–Arginine–Glycine–Serine |
| BMI | Body mass index |
| C1,2C | Type I/II collagen degradation |
| C2C | Type II collagen cleavage product |
| C2M | MMP-mediated type II collagen |
| CCL2 | CC-chemokine ligand 2, also known as MCP-1 |
| CH | Chondrocyte |
| CHI3L1 | Chitinase 3-like 1 |
| CLU | Clusterin |
| COLI | Collagen I |
| COLII | Collagen II |
| COMP | Cartilage oligomeric matrix protein |
| CPII | Procollagen II c-propeptide |
| CRP | C-reactive protein |
| CS | Chondroitin sulphate |
| CTX-I | C-terminal crosslinked telopeptide type I collagen |
| CTX-II | C-terminal crosslinked telopeptide type II collagen |
| CXCL | Chemokine ligand |
| DAMPs | Damage-associated molecular patterns |
| ECM | Extracellular matrix |
| FB | Factor B |
| FRI | Fluorescence reflectance imaging |
| gp 130 | Glycol–protein 130 |
| HMGB1 | High mobility group box 1 |
| IFN-γ | Interferon gamma |
| IL | Interleukin |
| IL-1ra | IL-1 receptor antagonist |
| IL-6R | IL-6 receptor |
| iNOS | Inducible NO synthase |
| KOOS4 | Knee Injury and Osteoarthritis Outcome Score 4 |
| LIF | Leukemia inhibitor factor |
| LPS | Lipopolysaccharide |
| MASP | MBL-associated serine proteases |
| MBL | Mannose binding lectin |
| MCL | Medial collateral ligament |
| MSC | Mesenchymal stem cells |
| MCP-1 | Monocyte chemoattractant protein-1, also known as CCL2 |
| MIP-1β | Macrophage inflammatory protein-1 beta, also known as CCL4 |
| MIP-3α | Macrophage inflammatory protein-3 alpha, also known as CCL20 |
| MMPs | Matrix metalloproteinases |
| MMX | Partial medial meniscectomy |
| MNC | Mononuclear cells |
| MRI | Magnetic resonance imaging |
| NLRP3 | Nucleotide-binding oligomerization domain-like receptor pyrin domain-containing-3 |
| NO | Nitric oxide |
| NTX-1 | N-terminal telopeptides of type I collagen |
| OA | Osteoarthritis |
| OCL | Osteocalcin |
| OPN | Osteopontin |
| PG | Proteoglycan |
| PLGA | Poly (lactic-co-glycolic acid) |
| PPA | Pyrophosphate arthritis, also known as pseudogout |
| PRP | Platelet rich plasma |
| PRR | Pattern-recognition receptors |
| PTOA | Post-traumatic osteoarthritis |
| RA | Rheumatoid arthritis |
| SASP | Senescence-associated secretory phenotype |
| SCSO | Superficial zone chondrocytes |
| SF | Synovial fluid |
| sGAG | Sulfated glycosaminoglycan |
| SPARC | Secreted protein acidic and rich in cysteine, also known as osteonectin |
| sTCC | Soluble terminal complement complex |
| SnCs | Senescent cells |
| TIMP | Tissue inhibitor of metalloproteinases |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor alpha |
| TSG-6 | Product of tumor necrosis factor alpha-stimulated gene 6 |
| Th17 | T helper 17 |
| VEGF | Vascular endothelial growth factor |
References
- Brown, T.D.; Johnston, R.C.; Saltzman, C.L.; Marsh, J.L.; Buckwalter, J.A. Posttraumatic osteoarthritis: A first estimate of incidence, prevalence, and burden of disease. J. Orthop. Trauma 2006, 20, 739–744. [Google Scholar] [CrossRef]
- Schenker, M.L.; Mauck, R.L.; Ahn, J.; Mehta, S. Pathogenesis and prevention of posttraumatic osteoarthritis after intra-articular fracture. J. Am. Acad. Orthop. Surg. 2014, 22, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Lohmander, L.S.; Englund, P.M.; Dahl, L.L.; Roos, E.M. The long-term consequence of anterior cruciate ligament and meniscus injuries: Osteoarthritis. Am. J. Sports Med. 2007, 35, 1756–1769. [Google Scholar] [CrossRef] [Green Version]
- Gelber, A.C.; Hochberg, M.C.; Mead, L.A.; Wang, N.Y.; Wigley, F.M.; Klag, M.J. Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann. Intern. Med. 2000, 133, 321–328. [Google Scholar] [CrossRef]
- Gillquist, J.; Messner, K. Anterior cruciate ligament reconstruction and the long-term incidence of gonarthrosis. Sports Med. 1999, 27, 143–156. [Google Scholar] [CrossRef]
- Roos, H.; Lauren, M.; Adalberth, T.; Roos, E.M.; Jonsson, K.; Lohmander, L.S. Knee osteoarthritis after meniscectomy: Prevalence of radiographic changes after twenty-one years, compared with matched controls. Arthritis Rheumatol. 1998, 41, 687–693. [Google Scholar] [CrossRef]
- Laird, A.; Keating, J.F. Acetabular fractures: A 16-year prospective epidemiological study. J. Bone Jt. Surg. Br. 2005, 87, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.P.; Marsh, J.L. High-energy fractures of the tibial plateau. Knee function after longer follow-up. J. Bone Jt. Surg. 2002, 84, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Zhang, Y. An update on the epidemiology of knee and hip osteoarthritis with a view to prevention. Arthritis Rheumatol. 1998, 41, 1343–1355. [Google Scholar] [CrossRef]
- Muthuri, S.G.; McWilliams, D.F.; Doherty, M.; Zhang, W. History of knee injuries and knee osteoarthritis: A meta-analysis of observational studies. Osteoarthr. Cartil. 2011, 19, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Olson, S.A.; Horne, P.; Furman, B.; Huebner, J.; Al-Rashid, M.; Kraus, V.B.; Guilak, F. The role of cytokines in posttraumatic arthritis. J. Am. Acad. Orthop. Surg. 2014, 22, 29–37. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [Green Version]
- Boehme, K.A.; Rolauffs, B. Onset and progression of human osteoarthritis-can growth factors, inflammatory cytokines, or differential mirna expression concomitantly induce proliferation, ecm degradation, and inflammation in articular cartilage? Int. J. Mol. Sci. 2018, 19, 2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chubinskaya, S.; Wimmer, M.A. Key pathways to prevent posttraumatic arthritis for future molecule-based therapy. Cartilage 2013, 4, 13S–21S. [Google Scholar] [CrossRef] [Green Version]
- Punzi, L.; Galozzi, P.; Luisetto, R.; Favero, M.; Ramonda, R.; Oliviero, F.; Scanu, A. Post-traumatic arthritis: Overview on pathogenic mechanisms and role of inflammation. RMD Open 2016, 2, e000279. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, J.; Sambamurthy, N.; Scanzello, C.R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Fedak, K.M.; Bernal, A.; Capshaw, Z.A.; Gross, S. Applying the bradford hill criteria in the 21st century: How data integration has changed causal inference in molecular epidemiology. Emerg. Themes Epidemiol. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.N.; Patel, V.; McCormack, R. The approach to the painful joint. 2019. Available online: https://emedicine.medscape.com/article/336054-overview (accessed on 14 November 2020).
- Haller, J.M.; McFadden, M.; Kubiak, E.N.; Higgins, T.F. Inflammatory cytokine response following acute tibial plateau fracture. J. Bone Jt. Surg. 2015, 97, 478–483. [Google Scholar] [CrossRef]
- Haller, J.M.; Swearingen, C.A.; Partridge, D.; McFadden, M.; Thirunavukkarasu, K.; Higgins, T.F. Intraarticular matrix metalloproteinases and aggrecan degradation are elevated after articular fracture. Clin. Orthop. Relat. Res. 2015, 473, 3280–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sward, P.; Frobell, R.; Englund, M.; Roos, H.; Struglics, A. Cartilage and bone markers and inflammatory cytokines are increased in synovial fluid in the acute phase of knee injury (hemarthrosis)—A cross-sectional analysis. Osteoarthr. Cartil. 2012, 20, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Bigoni, M.; Sacerdote, P.; Turati, M.; Franchi, S.; Gandolla, M.; Gaddi, D.; Moretti, S.; Munegato, D.; Augusti, C.A.; Bresciani, E.; et al. Acute and late changes in intraarticular cytokine levels following anterior cruciate ligament injury. J. Orthop. Res. 2013, 31, 315–321. [Google Scholar] [CrossRef]
- Irie, K.; Uchiyama, E.; Iwaso, H. Intraarticular inflammatory cytokines in acute anterior cruciate ligament injured knee. Knee 2003, 10, 93–96. [Google Scholar] [CrossRef]
- Elsaid, K.A.; Fleming, B.C.; Oksendahl, H.L.; Machan, J.T.; Fadale, P.D.; Hulstyn, M.J.; Shalvoy, R.; Jay, G.D. Decreased lubricin concentrations and markers of joint inflammation in the synovial fluid of patients with anterior cruciate ligament injury. Arthritis Rheumatol. 2008, 58, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, J.B.; Stabler, T.V.; Flannery, C.R.; Kraus, V.B. Changes in serum and synovial fluid biomarkers after acute injury (nct00332254). Arthritis Res. Ther. 2010, 12, R229. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.E.; Paterson, E.; Freidin, A.; Kenny, M.; Judge, A.; Saklatvala, J.; Williams, A.; Vincent, T.L. Acute molecular changes in synovial fluid following human knee injury: Association with early clinical outcomes. Arthritis Rheumatol. 2016, 68, 2129–2140. [Google Scholar] [CrossRef]
- Lattermann, C.; Jacobs, C.A.; Bunnell, M.P.; Jochimsen, K.N.; Abt, J.P.; Reinke, E.K.; Gammon, L.G.; Huebner, J.L.; Kraus, V.B.; Spindler, K.P. Logistical challenges and design considerations for studies using acute anterior cruciate ligament injury as a potential model for early posttraumatic osteoarthritis. J. Orthop. Res. 2017, 35, 641–650. [Google Scholar] [CrossRef]
- Dahlberg, L.; Roos, H.; Saxne, T.; Heinegard, D.; Lark, M.W.; Hoerrner, L.A.; Lohmander, L.S. Cartilage metabolism in the injured and uninjured knee of the same patient. Ann. Rheum. Dis. 1994, 53, 823–827. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, H.; Shirakura, K.; Kimura, M.; Terauchi, M.; Shinozaki, T.; Watanabe, H.; Takagishi, K. Changes in biochemical parameters after anterior cruciate ligament injury. Int. Orthop. 2006, 30, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Struglics, A.; Larsson, S.; Kumahashi, N.; Frobell, R.; Lohmander, L.S. Changes in cytokines and aggrecan args neoepitope in synovial fluid and serum and in c-terminal crosslinking telopeptide of type ii collagen and n-terminal crosslinking telopeptide of type i collagen in urine over five years after anterior cruciate ligament rupture: An exploratory analysis in the knee anterior cruciate ligament, nonsurgical versus surgical treatment trial. Arthritis Rheumatol. 2015, 67, 1816–1825. [Google Scholar]
- Sarafan, N.; Fakoor, M.; Mehdinasab, A.; Bahadoram, M.; Ashtary-Larky, D.; Mahdavi, H.; Javanmardi, F. Post-traumatic arthritis: The role of cytokine levels in serum and synovial fluid. Glob. J. Health Sci. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Lohmander, L.S.; Atley, L.M.; Pietka, T.A.; Eyre, D.R. The release of crosslinked peptides from type ii collagen into human synovial fluid is increased soon after joint injury and in osteoarthritis. Arthritis Rheumatol. 2003, 48, 3130–3139. [Google Scholar] [CrossRef] [PubMed]
- Lohmander, L.S.; Ionescu, M.; Jugessur, H.; Poole, A.R. Changes in joint cartilage aggrecan after knee injury and in osteoarthritis. Arthritis Rheumatol. 1999, 42, 534–544. [Google Scholar] [CrossRef]
- Struglics, A.; Okroj, M.; Sward, P.; Frobell, R.; Saxne, T.; Lohmander, L.S.; Blom, A.M. The complement system is activated in synovial fluid from subjects with knee injury and from patients with osteoarthritis. Arthritis Res. Ther. 2016, 18, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struglics, A.; Larsson, S.; Pramhed, A.; Frobell, R.; Sward, P. Changes in synovial fluid and serum concentrations of cartilage oligomeric matrix protein over 5 years after anterior cruciate ligament rupture: An exploratory analysis in the kanon trial. Osteoarthr. Cartil. 2018, 26, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.; Lohmander, L.S.; Struglics, A. Synovial fluid level of aggrecan args fragments is a more sensitive marker of joint disease than glycosaminoglycan or aggrecan levels: A cross-sectional study. Arthritis Res. Ther. 2009, 11, R92. [Google Scholar] [CrossRef] [Green Version]
- Panina, S.B.; Krolevets, I.V.; Milyutina, N.P.; Sagakyants, A.B.; Kornienko, I.V.; Ananyan, A.A.; Zabrodin, M.A.; Plotnikov, A.A.; Vnukov, V.V. Circulating levels of proinflammatory mediators as potential biomarkers of post-traumatic knee osteoarthritis development. J. Orthop. Traumatol. 2017, 18, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.; Watt, F.; Sawaji, Y.; Hermansson, M.; Saklatvala, J. Activin a is an anticatabolic autocrine cytokine in articular cartilage whose production is controlled by fibroblast growth factor 2 and nf-kappab. Arthritis Rheumatol. 2007, 56, 3715–3725. [Google Scholar] [CrossRef]
- Chou, C.H.; Attarian, D.E.; Wisniewski, H.G.; Band, P.A.; Kraus, V.B. Tsg-6—A double-edged sword for osteoarthritis (oa). Osteoarthr. Cartil. 2018, 26, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, H.G.; Colon, E.; Liublinska, V.; Karia, R.J.; Stabler, T.V.; Attur, M.; Abramson, S.B.; Band, P.A.; Kraus, V.B. Tsg-6 activity as a novel biomarker of progression in knee osteoarthritis. Osteoarthr. Cartil. 2014, 22, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Liu, J.; Lu, H. Correlation of concentrations of activin a with occurrence and severity of knee osteoarthritis. J. Musculoskelet. Neuronal. Interact. 2018, 18, 320–322. [Google Scholar] [PubMed]
- Hart, M.L.; Walsh, M.C.; Stahl, G.L. Initiation of complement activation following oxidative stress. In vitro and in vivo observations. Mol. Immunol. 2004, 41, 165–171. [Google Scholar] [CrossRef]
- John, T.; Stahel, P.F.; Morgan, S.J.; Schulze-Tanzil, G. Impact of the complement cascade on posttraumatic cartilage inflammation and degradation. Histol. Histopathol. 2007, 22, 781–790. [Google Scholar]
- Silawal, S.; Triebel, J.; Bertsch, T.; Schulze-Tanzil, G. Osteoarthritis and the complement cascade. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2018, 11, 1179544117751430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobezie, R.; Kho, A.; Krastins, B.; Sarracino, D.A.; Thornhill, T.S.; Chase, M.; Millett, P.J.; Lee, D.M. High abundance synovial fluid proteome: Distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 2007, 9, R36. [Google Scholar] [CrossRef] [Green Version]
- Corvetta, A.; Pomponio, G.; Rinaldi, N.; Luchetti, M.M.; Di Loreto, C.; Stramazzotti, D. Terminal complement complex in synovial tissue from patients affected by rheumatoid arthritis, osteoarthritis and acute joint trauma. Clin. Exp. Rheumatol. 1992, 10, 433–438. [Google Scholar] [PubMed]
- Ritter, S.Y.; Subbaiah, R.; Bebek, G.; Crish, J.; Scanzello, C.R.; Krastins, B.; Sarracino, D.; Lopez, M.F.; Crow, M.K.; Aigner, T.; et al. Proteomic analysis of synovial fluid from the osteoarthritic knee: Comparison with transcriptome analyses of joint tissues. Arthritis Rheumatol. 2013, 65, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Bollmann, M.; Colombo, F.; Marco, P.L.D.M.; Brandstaedter, K.; Lohmann, C.H.; Bertrand, J. Inhibition of the complement system component c5 as possible treatment in oa. Osteoarthr. Cartil. 2018, 26, S108. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [Green Version]
- Riegger, J.; Huber-Lang, M.; Brenner, R.E. Crucial role of the terminal complement complex in chondrocyte death and hypertrophy after cartilage trauma. Osteoarthr. Cartil. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Puente, P.; Mateos, J.; Fernandez-Costa, C.; Oreiro, N.; Fernandez-Lopez, C.; Ruiz-Romero, C.; Blanco, F.J. Identification of a panel of novel serum osteoarthritis biomarkers. J. Proteome Res. 2011, 10, 5095–5101. [Google Scholar] [CrossRef]
- Ammitzboll, C.G.; Thiel, S.; Ellingsen, T.; Deleuran, B.; Jorgensen, A.; Jensenius, J.C.; Stengaard-Pedersen, K. Levels of lectin pathway proteins in plasma and synovial fluid of rheumatoid arthritis and osteoarthritis. Rheumatol. Int. 2012, 32, 1457–1463. [Google Scholar] [CrossRef]
- Teunis, T.; Beekhuizen, M.; Van Osch, G.V.; Schuurman, A.H.; Creemers, L.B.; van Minnen, L.P. Soluble mediators in posttraumatic wrist and primary knee osteoarthritis. Arch. Bone Jt. Surg. 2014, 2, 146–150. [Google Scholar]
- Zhu, Z.; Otahal, P.; Wang, B.; Jin, X.; Laslett, L.L.; Wluka, A.E.; Antony, B.; Han, W.; Wang, X.; Winzenberg, T.; et al. Cross-sectional and longitudinal associations between serum inflammatory cytokines and knee bone marrow lesions in patients with knee osteoarthritis. Osteoarthr. Cartil. 2017, 25, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Xu, J.; Cai, J.; Zheng, S.; Yang, X.; Ding, C. Serum levels of resistin and interleukin-17 are associated with increased cartilage defects and bone marrow lesions in patients with knee osteoarthritis. Mod. Rheumatol. 2017, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, H.; Meng, Z.; Wei, M. Correlation of il-17 level in synovia and severity of knee osteoarthritis. Med. Sci. Monit. 2015, 21, 1732–1736. [Google Scholar] [PubMed] [Green Version]
- Bai, Y.; Gao, S.; Liu, Y.; Jin, S.; Zhang, H.; Su, K. Correlation between interleukin-17 gene polymorphism and osteoarthritis susceptibility in han chinese population. BMC Med. Genet. 2019, 20, 20. [Google Scholar] [CrossRef] [Green Version]
- Snelling, S.J.; Bas, S.; Puskas, G.J.; Dakin, S.G.; Suva, D.; Finckh, A.; Gabay, C.; Hoffmeyer, P.; Carr, A.J.; Lubbeke, A. Presence of il-17 in synovial fluid identifies a potential inflammatory osteoarthritic phenotype. PLoS ONE 2017, 12, e0175109. [Google Scholar] [CrossRef] [Green Version]
- Pengas, I.; Eldridge, S.; Assiotis, A.; McNicholas, M.; Mendes, J.E.; Laver, L. Mmp-3 in the peripheral serum as a biomarker of knee osteoarthritis, 40 years after open total knee meniscectomy. J. Exp. Orthop. 2018, 5, 21. [Google Scholar] [CrossRef]
- Bao, J.P.; Chen, W.P.; Feng, J.; Hu, P.F.; Shi, Z.L.; Wu, L.D. Leptin plays a catabolic role on articular cartilage. Mol. Biol. Rep. 2010, 37, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Beekhuizen, M.; Gierman, L.M.; van Spil, W.E.; Van Osch, G.J.; Huizinga, T.W.; Saris, D.B.; Creemers, L.B.; Zuurmond, A.M. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 2013, 21, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Imamura, M.; Ezquerro, F.; Marcon Alfieri, F.; Vilas Boas, L.; Tozetto-Mendoza, T.R.; Chen, J.; Ozcakar, L.; Arendt-Nielsen, L.; Rizzo Battistella, L. Serum levels of proinflammatory cytokines in painful knee osteoarthritis and sensitization. Int. J. Inflam. 2015, 2015, 329792. [Google Scholar] [CrossRef] [PubMed]
- Ersoy, Y.; Ozerol, E.; Baysal, O.; Temel, I.; MacWalter, R.S.; Meral, U.; Altay, Z.E. Serum nitrate and nitrite levels in patients with rheumatoid arthritis, ankylosing spondylitis, and osteoarthritis. Ann. Rheum. Dis. 2002, 61, 76–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnokutsky, S.; Oshinsky, C.; Attur, M.; Ma, S.; Zhou, H.; Zheng, F.; Chen, M.; Patel, J.; Samuels, J.; Pike, V.C.; et al. Serum urate levels predict joint space narrowing in non-gout patients with medial knee osteoarthritis. Arthritis Rheumatol. 2017, 69, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Denoble, A.E.; Huffman, K.M.; Stabler, T.V.; Kelly, S.J.; Hershfield, M.S.; McDaniel, G.E.; Coleman, R.E.; Kraus, V.B. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc. Natl. Acad. Sci. USA 2011, 108, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Olsson, O.; Isacsson, A.; Englund, M.; Frobell, R.B. Epidemiology of intra- and peri-articular structural injuries in traumatic knee joint hemarthrosis—Data from 1145 consecutive knees with subacute mri. Osteoarthr. Cartil. 2016, 24, 1890–1897. [Google Scholar] [CrossRef] [Green Version]
- Pacquelet, S.; Presle, N.; Boileau, C.; Dumond, H.; Netter, P.; Martel-Pelletier, J.; Pelletier, J.P.; Terlain, B.; Jouzeau, J.Y. Interleukin 17, a nitric oxide-producing cytokine with a peroxynitrite-independent inhibitory effect on proteoglycan synthesis. J. Rheumatol. 2002, 29, 2602–2610. [Google Scholar]
- Wang, Z.; Zheng, C.; Zhong, Y.; He, J.; Cao, X.; Xia, H.; Ba, H.; Li, P.; Wu, S.; Peng, C. Interleukin-17 can induce osteoarthritis in rabbit knee joints similar to hulth’s method. BioMed Res. Int. 2017, 2017, 2091325. [Google Scholar] [CrossRef] [Green Version]
- Malfait, A.M.; Tortorella, M.; Thompson, J.; Hills, R.; Meyer, D.M.; Jaffee, B.D.; Chinn, K.; Ghoreishi-Haack, N.; Markosyan, S.; Arner, E.C. Intra-articular injection of tumor necrosis factor-alpha in the rat: An acute and reversible in vivo model of cartilage proteoglycan degradation. Osteoarthr. Cartil. 2009, 17, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meegeren, M.E.; Roosendaal, G.; Jansen, N.W.; Lafeber, F.P.; Mastbergen, S.C. Blood-induced joint damage: The devastating effects of acute joint bleeds versus micro-bleeds. Cartilage 2013, 4, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.L.; Brandt, K.D.; O’Connor, B.L.; Visco, D.M.; Albrecht, M.E. Synovitis and osteoarthritic changes in canine articular cartilage after anterior cruciate ligament transection. Effect of surgical hemostasis. Arthritis Rheumatol. 1990, 33, 1406–1415. [Google Scholar] [CrossRef]
- Jansen, N.W.; Roosendaal, G.; Bijlsma, J.W.; Degroot, J.; Lafeber, F.P. Exposure of human cartilage tissue to low concentrations of blood for a short period of time leads to prolonged cartilage damage: An in vitro study. Arthritis Rheumatol. 2007, 56, 199–207. [Google Scholar] [CrossRef]
- Van Vulpen, L.F.D.; Popov-Celeketic, J.; van Meegeren, M.E.R.; Coeleveld, K.; van Laar, J.M.; Hack, C.E.; Schutgens, R.E.G.; Mastbergen, S.C.; Lafeber, F. A fusion protein of interleukin-4 and interleukin-10 protects against blood-induced cartilage damage in vitro and in vivo. J. Thromb. Haemost. 2017, 15, 1788–1798. [Google Scholar] [CrossRef] [Green Version]
- van Meegeren, M.E.; Roosendaal, G.; Jansen, N.W.; Wenting, M.J.; van Wesel, A.C.; van Roon, J.A.; Lafeber, F.P. Il-4 alone and in combination with il-10 protects against blood-induced cartilage damage. Osteoarthr. Cartil. 2012, 20, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, N.W.; Roosendaal, G.; Hooiveld, M.J.; Bijlsma, J.W.; van Roon, J.A.; Theobald, M.; Lafeber, F.P. Interleukin-10 protects against blood-induced joint damage. Br. J. Haematol. 2008, 142, 953–961. [Google Scholar] [CrossRef]
- Melin Furst, C.; Ahrman, E.; Bratteby, K.; Waldemarson, S.; Malmstrom, J.; Blom, A.M. Quantitative mass spectrometry to study inflammatory cartilage degradation and resulting interactions with the complement system. J. Immunol. 2016, 197, 3415–3424. [Google Scholar] [CrossRef]
- Sohn, D.H.; Sokolove, J.; Sharpe, O.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Lindstrom, T.M.; Hwang, I.; Boyer, K.A.; Andriacchi, T.P.; et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via toll-like receptor 4. Arthritis Res. Ther. 2012, 14, R7. [Google Scholar] [CrossRef] [Green Version]
- Colten, H.R.; Ooi, Y.M.; Edelson, P.J. Synthesis and secretion of complement proteins by macrophages. Ann. N. Y. Acad. Sci. 1979, 332, 482–490. [Google Scholar] [CrossRef]
- Lubbers, R.; van Schaarenburg, R.A.; Kwekkeboom, J.C.; Levarht, E.W.N.; Bakker, A.M.; Mahdad, R.; Monteagudo, S.; Cherifi, C.; Lories, R.J.; Toes, R.E.M.; et al. Complement component c1q is produced by isolated articular chondrocytes. Osteoarthr. Cartil. 2020, 28, 675–684. [Google Scholar] [CrossRef]
- Bradley, K.; North, J.; Saunders, D.; Schwaeble, W.; Jeziorska, M.; Woolley, D.E.; Whaley, K. Synthesis of classical pathway complement components by chondrocytes. Immunology 1996, 88, 648–656. [Google Scholar]
- Schulze-Tanzil, G.; Kohl, B.; El Sayed, K.; Arens, S.; Ertel, W.; Stolzel, K.; John, T. Anaphylatoxin receptors and complement regulatory proteins in human articular and non-articular chondrocytes: Interrelation with cytokines. Cell Tissue Res. 2012, 350, 465–475. [Google Scholar] [CrossRef]
- Ignatius, A.; Schoengraf, P.; Kreja, L.; Liedert, A.; Recknagel, S.; Kandert, S.; Brenner, R.E.; Schneider, M.; Lambris, J.D.; Huber-Lang, M. Complement c3a and c5a modulate osteoclast formation and inflammatory response of osteoblasts in synergism with il-1beta. J. Cell. Biochem. 2011, 112, 2594–2605. [Google Scholar] [CrossRef] [Green Version]
- Assirelli, E.; Dolzani, P.; Pulsatelli, L.; Addimanda, O.; Lisignoli, G.; Mariani, E.; Meliconi, R. Complement factor expression in osteoarthritis joint compartments. Osteoarthr. Cartil. 2016, 24, S383–S384. [Google Scholar] [CrossRef]
- Furman, B.D.; Strand, J.; Hembree, W.C.; Ward, B.D.; Guilak, F.; Olson, S.A. Joint degeneration following closed intraarticular fracture in the mouse knee: A model of posttraumatic arthritis. J. Orthop. Res. 2007, 25, 578–592. [Google Scholar] [CrossRef]
- Furman, B.D.; Mangiapani, D.S.; Zeitler, E.; Bailey, K.N.; Horne, P.H.; Huebner, J.L.; Kraus, V.B.; Guilak, F.; Olson, S.A. Targeting pro-inflammatory cytokines following joint injury: Acute intra-articular inhibition of interleukin-1 following knee injury prevents post-traumatic arthritis. Arthritis Res. Ther. 2014, 16, R134. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Hembree, W.C.; Furman, B.D.; Tippets, L.; Cattel, D.; Huebner, J.L.; Little, D.; DeFrate, L.E.; Kraus, V.B.; Guilak, F.; et al. Acute joint pathology and synovial inflammation is associated with increased intra-articular fracture severity in the mouse knee. Osteoarthr. Cartil. 2011, 19, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Sandell, L.J.; Chinzei, N.; Holguin, N.; Silva, M.J.; Schiavinato, A.; Rai, M.F. Therapeutic efficacy of intra-articular hyaluronan derivative and platelet-rich plasma in mice following axial tibial loading. PLoS ONE 2017, 12, e0175682. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Guidolin, D. Potential mechanism of action of intra-articular hyaluronan therapy in osteoarthritis: Are the effects molecular weight dependent? Semin. Arthritis Rheumatol. 2002, 32, 10–37. [Google Scholar] [CrossRef]
- Christiansen, B.A.; Guilak, F.; Lockwood, K.A.; Olson, S.A.; Pitsillides, A.A.; Sandell, L.J.; Silva, M.J.; van der Meulen, M.C.; Haudenschild, D.R. Non-invasive mouse models of post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Rolauffs, B.; Kurz, B.; Felka, T.; Rothdiener, M.; Uynuk-Ool, T.; Aurich, M.; Frank, E.; Bahrs, C.; Badke, A.; Stockle, U.; et al. Stress-vs-time signals allow the prediction of structurally catastrophic events during fracturing of immature cartilage and predetermine the biomechanical, biochemical, and structural impairment. J. Struct. Biol. 2013, 183, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Rolauffs, B.; Muehleman, C.; Li, J.; Kurz, B.; Kuettner, K.E.; Frank, E.; Grodzinsky, A.J. Vulnerability of the superficial zone of immature articular cartilage to compressive injury. Arthritis Rheumatol. 2010, 62, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- DiMicco, M.A.; Patwari, P.; Siparsky, P.N.; Kumar, S.; Pratta, M.A.; Lark, M.W.; Kim, Y.J.; Grodzinsky, A.J. Mechanisms and kinetics of glycosaminoglycan release following in vitro cartilage injury. Arthritis Rheumatol. 2004, 50, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, K.A.; Chu, B.T.; Anderson, M.J.; Haudenschild, D.R.; Christiansen, B.A. Comparison of loading rate-dependent injury modes in a murine model of post-traumatic osteoarthritis. J. Orthop. Res. 2014, 32, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Satkunananthan, P.B.; Anderson, M.J.; De Jesus, N.M.; Haudenschild, D.R.; Ripplinger, C.M.; Christiansen, B.A. In vivo fluorescence reflectance imaging of protease activity in a mouse model of post-traumatic osteoarthritis. Osteoarthr. Cartil. 2014, 22, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Holguin, N.; Silva, M.J.; Fu, M.; Liao, W.; Sandell, L.J. Early response of mouse joint tissue to noninvasive knee injury suggests treatment targets. Arthritis Rheumatol. 2014, 66, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Bajpayee, A.G.; De la Vega, R.E.; Scheu, M.; Varady, N.H.; Yannatos, I.A.; Brown, L.A.; Krishnan, Y.; Fitzsimons, T.J.; Bhattacharya, P.; Frank, E.H.; et al. Sustained intra-cartilage delivery of low dose dexamethasone using a cationic carrier for treatment of post traumatic osteoarthritis. Eur. Cells Mater. 2017, 34, 341–364. [Google Scholar]
- Pickarski, M.; Hayami, T.; Zhuo, Y.; Duong, L.T. Molecular changes in articular cartilage and subchondral bone in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. BMC Musculoskelet. Disord. 2011, 12, 197. [Google Scholar] [CrossRef] [Green Version]
- Teeple, E.; Jay, G.D.; Elsaid, K.A.; Fleming, B.C. Animal models of osteoarthritis: Challenges of model selection and analysis. AAPS J. 2013, 15, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Kuettner, K.E.; Cole, A.A. Cartilage degeneration in different human joints. Osteoarthr. Cartil. 2005, 13, 93–103. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.M. Animal models of osteoarthritis: Comparisons and key considerations. Vet. Pathol. 2015, 52, 803–818. [Google Scholar] [CrossRef]
- Fitzgerald, J. Enhanced cartilage repair in ’healer’ mice-new leads in the search for better clinical options for cartilage repair. Semin. Cell Dev. Biol. 2017, 62, 78–85. [Google Scholar]
- Rai, M.F.; Sandell, L.J. Regeneration of articular cartilage in healer and non-healer mice. Matrix Biol. 2014, 39, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Malda, J.; Benders, K.E.; Klein, T.J.; de Grauw, J.C.; Kik, M.J.; Hutmacher, D.W.; Saris, D.B.; van Weeren, P.R.; Dhert, W.J. Comparative study of depth-dependent characteristics of equine and human osteochondral tissue from the medial and lateral femoral condyles. Osteoarthr. Cartil. 2012, 20, 1147–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aicher, W.K.; Rolauffs, B. The spatial organisation of joint surface chondrocytes: Review of its potential roles in tissue functioning, disease and early, preclinical diagnosis of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 645–653. [Google Scholar] [CrossRef]
- Rolauffs, B.; Rothdiener, M.; Bahrs, C.; Badke, A.; Weise, K.; Kuettner, K.E.; Kurz, B.; Aurich, M.; Grodzinsky, A.J.; Aicher, W.K. Onset of preclinical osteoarthritis: The angular spatial organization permits early diagnosis. Arthritis Rheumatol. 2011, 63, 1637–1647. [Google Scholar] [CrossRef]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative remodeling of the spatial organization of human superficial chondrocytes distant from focal early osteoarthritis. Arthritis Rheumatol. 2010, 62, 489–498. [Google Scholar]
- Rolauffs, B.; Williams, J.M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Distinct horizontal patterns in the spatial organization of superficial zone chondrocytes of human joints. J. Struct. Biol. 2008, 162, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, B.D.; Furman, B.D.; Huebner, J.L.; Kraus, V.B.; Guilak, F.; Olson, S.A. Absence of posttraumatic arthritis following intraarticular fracture in the mrl/mpj mouse. Arthritis Rheumatol. 2008, 58, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.A.; Furman, B.D.; Kraus, V.B.; Huebner, J.L.; Guilak, F. Therapeutic opportunities to prevent post-traumatic arthritis: Lessons from the natural history of arthritis after articular fracture. J. Orthop. Res. 2015, 33, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Conde, J.; Scotece, M.; Abella, V.; Lopez, V.; Pino, J.; Gomez, R.; Gomez-Reino, J.J.; Gualillo, O. Choosing the right chondrocyte cell line: Focus on nitric oxide. J. Orthop. Res. 2015, 33, 1784–1788. [Google Scholar] [CrossRef]
- Park, E.; Hart, M.L.; Rolauffs, B.; Stegemann, J.P.; Ramkumar, T.A. Bioresponsive microspheres for on-demand delivery of anti-inflammatory cytokines for articular cartilage repair. J. Biomed. Mater. Res. A 2020, 108, 722–733. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Lago, F.; Reino, J.J.; Gualillo, O. Signalling pathway involved in nitric oxide synthase type ii activation in chondrocytes: Synergistic effect of leptin with interleukin-1. Arthritis Res. Ther. 2005, 7, R581–R591. [Google Scholar] [CrossRef] [Green Version]
- Otero, M.; Gomez Reino, J.J.; Gualillo, O. Synergistic induction of nitric oxide synthase type ii: In vitro effect of leptin and interferon-gamma in human chondrocytes and atdc5 chondrogenic cells. Arthritis Rheumatol. 2003, 48, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Mayhewy, M.; Kevorkian, L.; Swingler, T.; Bevan, D.; Stubberfield, C.; Moore, A.; Gavrilovi, J. A modified and enhanced atdc-5 chondrogenesis model produces an articular-like. Osteoarthr. Cartil. 2014, 22. [Google Scholar]
- Hubka, K.M.; Dahlin, R.L.; Meretoja, V.V.; Kasper, F.K.; Mikos, A.G. Enhancing chondrogenic phenotype for cartilage tissue engineering: Monoculture and coculture of articular chondrocytes and mesenchymal stem cells. Tissue Eng. Part B Rev. 2014, 20, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Benya, P.D.; Padilla, S.R.; Nimni, M.E. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978, 15, 1313–1321. [Google Scholar] [CrossRef]
- Benya, P.D.; Shaffer, J.D. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell 1982, 30, 215–224. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Coolsen, M.M.; Voss, L.; Surtel, D.A.; Cremers, A.; van Rhijn, L.W.; Welting, T.J. Redifferentiation of dedifferentiated human articular chondrocytes: Comparison of 2d and 3d cultures. Osteoarthr. Cartil. 2012, 20, 1170–1178. [Google Scholar] [CrossRef] [Green Version]
- Aurich, M.; Hofmann, G.O.; Gras, F.; Rolauffs, B. Human osteochondritis dissecans fragment-derived chondrocyte characteristics ex vivo, after monolayer expansion-induced de-differentiation, and after re-differentiation in alginate bead culture. BMC Musculoskelet. Disord. 2018, 19, 168. [Google Scholar] [CrossRef] [PubMed]
- Hsieh-Bonassera, N.D.; Wu, I.; Lin, J.K.; Schumacher, B.L.; Chen, A.C.; Masuda, K.; Bugbee, W.D.; Sah, R.L. Expansion and redifferentiation of chondrocytes from osteoarthritic cartilage: Cells for human cartilage tissue engineering. Tissue Eng. Part A 2009, 15, 3513–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babur, B.K.; Ghanavi, P.; Levett, P.; Lott, W.B.; Klein, T.; Cooper-White, J.J.; Crawford, R.; Doran, M.R. The interplay between chondrocyte redifferentiation pellet size and oxygen concentration. PLoS ONE 2013, 8, e58865. [Google Scholar] [CrossRef] [Green Version]
- Meretoja, V.V.; Dahlin, R.L.; Wright, S.; Kasper, F.K.; Mikos, A.G. Articular chondrocyte redifferentiation in 3d co-cultures with mesenchymal stem cells. Tissue Eng. Part C Methods 2014, 20, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, E.; Hofmann, S.; Stok, K.; Notbohm, H.; Muller, R.; Rotter, N. Chondrocyte redifferentiation in 3d: The effect of adhesion site density and substrate elasticity. J. Biomed. Mater. Res. A 2012, 100, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Levett, P.A.; Melchels, F.P.; Schrobback, K.; Hutmacher, D.W.; Malda, J.; Klein, T.J. Chondrocyte redifferentiation and construct mechanical property development in single-component photocrosslinkable hydrogels. J. Biomed. Mater. Res. A 2014, 102, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, G.; Lopez-Ruiz, E.; Kwiatkowski, W.; Montanez, E.; Arrebola, F.; Carrillo, E.; Gray, P.C.; Izpisua Belmonte, J.C.; Choe, S.; Peran, M.; et al. Activin a/bmp2 chimera ab235 drives efficient redifferentiation of long term cultured autologous chondrocytes. Sci. Rep. 2015, 5, 16400. [Google Scholar] [CrossRef] [Green Version]
- Rothdiener, M.; Uynuk-Ool, T.; Sudkamp, N.; Aurich, M.; Grodzinsky, A.J.; Kurz, B.; Rolauffs, B. Human osteoarthritic chondrons outnumber patient- and joint-matched chondrocytes in hydrogel culture-future application in autologous cell-based oa cartilage repair? J. Tissue Eng. Regen. Med. 2018, 12, e1206–e1220. [Google Scholar] [CrossRef] [PubMed]
- Aurich, M.; Hofmann, G.O.; Best, N.; Rolauffs, B. Induced redifferentiation of human chondrocytes from articular cartilage lesion in alginate bead culture after monolayer dedifferentiation: An alternative cell source for cell-based therapies? Tissue Eng. Part A 2018, 24, 275–286. [Google Scholar] [CrossRef]
- Aurich, M.; Eger, W.; Rolauffs, B.; Margulis, A.; Kuettner, K.E.; Mollenhauer, J.A.; Cole, A.A. Ankle chondrocytes are more resistant to interleukin-1 than chondrocytes derived from the knee. Orthopade 2006, 35, 784–790. [Google Scholar] [CrossRef]
- Aurich, M.; Squires, G.R.; Reiner, A.; Mollenhauer, J.A.; Kuettner, K.E.; Poole, A.R.; Cole, A.A. Differential matrix degradation and turnover in early cartilage lesions of human knee and ankle joints. Arthritis Rheumatol. 2005, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Eger, W.; Aurich, M.; Schumacher, B.L.; Mollenhauer, J.; Kuettner, K.E.; Cole, A.A. Differences in metabolism of chondrocytes of the knee and ankle joint. Z. Orthop. Grenzgeb. 2003, 141, 18–20. [Google Scholar] [PubMed]
- Aurich, M.; Hofmann, G.O.; Rolauffs, B. Differences in type ii collagen turnover of osteoarthritic human knee and ankle joints. Int. Orthop. 2017. [Google Scholar] [CrossRef]
- Aurich, M.; Hofmann, G.O.; Muckley, T.; Mollenhauer, J.; Rolauffs, B. In vitro phenotypic modulation of chondrocytes from knees of patients with osteochondritis dissecans: Implications for chondrocyte implantation procedures. J. Bone Jt. Surg. Br. 2012, 94, 62–67. [Google Scholar] [CrossRef]
- Felka, T.; Rothdiener, M.; Bast, S.; Uynuk-Ool, T.; Zouhair, S.; Ochs, B.G.; De Zwart, P.; Stoeckle, U.; Aicher, W.K.; Hart, M.L.; et al. Loss of spatial organization and destruction of the pericellular matrix in early osteoarthritis in vivo and in a novel in vitro methodology. Osteoarthr. Cartil. 2016, 24, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, A.I.; Beekhuizen, M.; Hart, M.C.; Radstake, T.R.; Dhert, W.J.; Saris, D.B.; van Osch, G.J.; Creemers, L.B. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 2014, 16, 441. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Fitzgerald, J.B.; Xu, J.; Willers, C.; Wood, D.; Grodzinsky, A.J.; Zheng, M.H. Gene expression profiles of human chondrocytes during passaged monolayer cultivation. J. Orthop. Res. 2008, 26, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Kurz, B.; Lemke, A.K.; Fay, J.; Pufe, T.; Grodzinsky, A.J.; Schunke, M. Pathomechanisms of cartilage destruction by mechanical injury. Ann. Anat. 2005, 187, 473–485. [Google Scholar] [CrossRef]
- Nickien, M.; Heuijerjans, A.; Ito, K.; van Donkelaar, C.C. Comparison between in vitro and in vivo cartilage overloading studies based on a systematic literature review. J. Orthop. Res. 2018. [Google Scholar] [CrossRef]
- Behrendt, P.; Preusse-Prange, A.; Kluter, T.; Haake, M.; Rolauffs, B.; Grodzinsky, A.J.; Lippross, S.; Kurz, B. Il-10 reduces apoptosis and extracellular matrix degradation after injurious compression of mature articular cartilage. Osteoarthr. Cartil. 2016, 24, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of il-10 in mechanically injured cartilage and cellularized collagen aci grafts. Osteoarthr. Cartil. 2018, 26, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, Y.; Khabut, A.; Chubinskaya, S.; Grodzinsky, A.J.; Onnerfjord, P. Quantitative proteomics analysis of cartilage response to mechanical injury and cytokine treatment. Matrix Biol. 2017, 63, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Cook, M.N.; DiMicco, M.A.; Blake, S.M.; James, I.E.; Kumar, S.; Cole, A.A.; Lark, M.W.; Grodzinsky, A.J. Proteoglycan degradation after injurious compression of bovine and human articular cartilage in vitro: Interaction with exogenous cytokines. Arthritis Rheumatol. 2003, 48, 1292–1301. [Google Scholar] [CrossRef]
- Lu, Y.C.; Evans, C.H.; Grodzinsky, A.J. Effects of short-term glucocorticoid treatment on changes in cartilage matrix degradation and chondrocyte gene expression induced by mechanical injury and inflammatory cytokines. Arthritis Res. Ther. 2011, 13, R142. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Lee, J.H.; DiMicco, M.A.; Vanderploeg, E.J.; Blake, S.M.; Hung, H.H.; Plaas, A.H.; James, I.E.; Song, X.Y.; Lark, M.W.; et al. Mechanical injury potentiates proteoglycan catabolism induced by interleukin-6 with soluble interleukin-6 receptor and tumor necrosis factor alpha in immature bovine and adult human articular cartilage. Arthritis Rheumatol. 2009, 60, 2985–2996. [Google Scholar] [CrossRef]
- Stevens, A.L.; Wishnok, J.S.; White, F.M.; Grodzinsky, A.J.; Tannenbaum, S.R. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Mol. Cell Proteomics 2009, 8, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Torzilli, P.A.; Bhargava, M.; Chen, C.T. Mechanical loading of articular cartilage reduces il-1-induced enzyme expression. Cartilage 2011, 2, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Bajpayee, A.G.; Quadir, M.A.; Hammond, P.T.; Grodzinsky, A.J. Charge based intra-cartilage delivery of single dose dexamethasone using avidin nano-carriers suppresses cytokine-induced catabolism long term. Osteoarthr. Cartil. 2016, 24, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkeviciute, D.; Aspberg, A.; He, Y.; Bay-Jensen, A.C.; Onnerfjord, P. Characterization of the interleukin-17 effect on articular cartilage in a translational model: An explorative study. BMC Rheumatol. 2020, 4, 30. [Google Scholar] [CrossRef]
- Clutterbuck, A.L.; Smith, J.R.; Allaway, D.; Harris, P.; Liddell, S.; Mobasheri, A. High throughput proteomic analysis of the secretome in an explant model of articular cartilage inflammation. J. Proteom. 2011, 74, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Kongdang, P.; Chokchaitaweesuk, C.; Tangyuenyong, S.; Ongchai, S. Proinflammatory effects of il-1beta combined with il-17a promoted cartilage degradation and suppressed genes associated with cartilage matrix synthesis in vitro. Molecules 2019, 24, 3682. [Google Scholar] [CrossRef] [Green Version]
- Lubberts, E.; Joosten, L.A.; van de Loo, F.A.; van den Gersselaar, L.A.; van den Berg, W.B. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheumatol. 2000, 43, 1300–1306. [Google Scholar] [CrossRef]
- Li, Y.; Frank, E.H.; Wang, Y.; Chubinskaya, S.; Huang, H.H.; Grodzinsky, A.J. Moderate dynamic compression inhibits pro-catabolic response of cartilage to mechanical injury, tumor necrosis factor-alpha and interleukin-6, but accentuates degradation above a strain threshold. Osteoarthr. Cartil. 2013, 21, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.T.; Moradi, B.; Smith, M.M.; Jackson, C.J.; Little, C.B. Activation of matrix metalloproteinases 2, 9, and 13 by activated protein c in human osteoarthritic cartilage chondrocytes. Arthritis Rheumatol. 2014, 66, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Chubinskaya, S.; Schoeberl, B.; Florine, E.; Kopesky, P.; Grodzinsky, A.J. Effects of insulin-like growth factor-1 and dexamethasone on cytokine-challenged cartilage: Relevance to post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; Van Offel, J.F.; Bridts, C.H.; De Clerck, L.S. Influence of pro-inflammatory (il-1 alpha, il-6, tnf-alpha, ifn-gamma) and anti-inflammatory (il-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; de Andres, M.C.; Hashimoto, K.; Itoi, E.; Oreffo, R.O. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Qureshi, H.Y.; El Mabrouk, M.; Sylvester, J.; Ahmad, M.; Zafarullah, M. Inhibition of interleukin 1-induced matrix metalloproteinase 13 expression in human chondrocytes by interferon gamma. Ann. Rheum. Dis. 2007, 66, 782–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrotin, Y.E.; De Groote, D.D.; Labasse, A.H.; Gaspar, S.E.; Zheng, S.X.; Geenen, V.G.; Reginster, J.Y. Effects of exogenous il-1 beta, tnf alpha, il-6, il-8 and lif on cytokine production by human articular chondrocytes. Osteoarthr. Cartil. 1996, 4, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Roman-Blas, J.A.; Stokes, D.G.; Jimenez, S.A. Modulation of tgf-beta signaling by proinflammatory cytokines in articular chondrocytes. Osteoarthr. Cartil. 2007, 15, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.; Andriamanalijaona, R.; Mathy-Hartert, M.; Henrotin, Y.; Pujol, J.P. Comparative effects of il-1beta and hydrogen peroxide (h2o2) on catabolic and anabolic gene expression in juvenile bovine chondrocytes. Osteoarthr. Cartil. 2005, 13, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, T.T.; Bader, D.L.; Lee, D.A. Anti-inflammatory effects of il-4 and dynamic compression in il-1beta stimulated chondrocytes. Biochem. Biophys. Res. Commun. 2006, 339, 241–247. [Google Scholar] [CrossRef]
- Chen, X.Y.; Hao, Y.R.; Wang, Z.; Zhou, J.L.; Jia, Q.X.; Qiu, B. The effect of vascular endothelial growth factor on aggrecan and type ii collagen expression in rat articular chondrocytes. Rheumatol. Int. 2012, 32, 3359–3364. [Google Scholar] [CrossRef]
- Bougault, C.; Gosset, M.; Houard, X.; Salvat, C.; Godmann, L.; Pap, T.; Jacques, C.; Berenbaum, F. Stress-induced cartilage degradation does not depend on the nlrp3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheumatol. 2012, 64, 3972–3981. [Google Scholar] [CrossRef]
- Muller, R.D.; John, T.; Kohl, B.; Oberholzer, A.; Gust, T.; Hostmann, A.; Hellmuth, M.; Laface, D.; Hutchins, B.; Laube, G.; et al. Il-10 overexpression differentially affects cartilage matrix gene expression in response to tnf-alpha in human articular chondrocytes in vitro. Cytokine 2008, 44, 377–385. [Google Scholar] [CrossRef] [PubMed]
- John, T.; Muller, R.D.; Oberholzer, A.; Zreiqat, H.; Kohl, B.; Ertel, W.; Hostmann, A.; Tschoeke, S.K.; Schulze-Tanzil, G. Interleukin-10 modulates pro-apoptotic effects of tnf-alpha in human articular chondrocytes in vitro. Cytokine 2007, 40, 226–234. [Google Scholar] [CrossRef]
- Scotece, M.; Conde, J.; Abella, V.; Lopez, V.; Lago, F.; Pino, J.; Gomez-Reino, J.J.; Gualillo, O. Nucb2/nesfatin-1: A new adipokine expressed in human and murine chondrocytes with pro-inflammatory properties, an in vitro study. J. Orthop. Res. 2014, 32, 653–660. [Google Scholar] [CrossRef]
- LeGrand, A.; Fermor, B.; Fink, C.; Pisetsky, D.S.; Weinberg, J.B.; Vail, T.P.; Guilak, F. Interleukin-1, tumor necrosis factor alpha, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin e2 production in explants of human osteoarthritic knee menisci. Arthritis Rheumatol. 2001, 44, 2078–2083. [Google Scholar] [CrossRef]
- Na, H.S.; Park, J.S.; Cho, K.H.; Kwon, J.Y.; Choi, J.; Jhun, J.; Kim, S.J.; Park, S.H.; Cho, M.L. Interleukin-1-interleukin-17 signaling axis induces cartilage destruction and promotes experimental osteoarthritis. Front. Immunol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Attur, M.G.; Patel, R.N.; Abramson, S.B.; Amin, A.R. Interleukin-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheumatol. 1997, 40, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Barak, T.; Quach, J.; Lotz, M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and nf-kappab. J. Biol. Chem. 1998, 273, 27467–27473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honorati, M.C.; Bovara, M.; Cattini, L.; Piacentini, A.; Facchini, A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthr. Cartil. 2002, 10, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Mimpen, J.Y.; Snelling, S.J.B. Chondroprotective factors in osteoarthritis: A joint affair. Curr. Rheumatol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanigawa, S.; Aida, Y.; Kawato, T.; Honda, K.; Nakayama, G.; Motohashi, M.; Suzuki, N.; Ochiai, K.; Matsumura, H.; Maeno, M. Interleukin-17f affects cartilage matrix turnover by increasing the expression of collagenases and stromelysin-1 and by decreasing the expression of their inhibitors and extracellular matrix components in chondrocytes. Cytokine 2011, 56, 376–386. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Mineau, F.; Jovanovic, D.; Di Battista, J.A.; Pelletier, J.P. Mitogen-activated protein kinase and nuclear factor kappab together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: Possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (mapkapk). Arthritis Rheumatol. 1999, 42, 2399–2409. [Google Scholar]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Fossiez, F.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A t cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheumatol. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Katz, Y.; Nadiv, O.; Beer, Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: A possible role as a "fine-tuning cytokine" in inflammation processes. Arthritis Rheumatol. 2001, 44, 2176–2184. [Google Scholar] [CrossRef]
- Kondo, M.; Yamaoka, K.; Sonomoto, K.; Fukuyo, S.; Oshita, K.; Okada, Y.; Tanaka, Y. Il-17 inhibits chondrogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e79463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, P.; Albrecht, D.; Andereya, S.; Angele, P.; Ateschrang, A.; Aurich, M.; Baumann, M.; Bosch, U.; Erggelet, C.; Fickert, S.; et al. Autologous chondrocyte implantation (ACI) for cartilage defects of the knee: A guideline by the working group "clinical tissue regeneration" of the German Society of Orthopaedics and Trauma (DGOU). Knee 2016. [Google Scholar] [CrossRef] [Green Version]
- Aurich, M.; Bedi, H.S.; Smith, P.J.; Rolauffs, B.; Muckley, T.; Clayton, J.; Blackney, M. Arthroscopic treatment of osteochondral lesions of the ankle with matrix-associated chondrocyte implantation: Early clinical and magnetic resonance imaging results. Am. J. Sports Med. 2011, 39, 311–319. [Google Scholar] [CrossRef]
- Ochs, B.G.; Muller-Horvat, C.; Albrecht, D.; Schewe, B.; Weise, K.; Aicher, W.K.; Rolauffs, B. Remodeling of articular cartilage and subchondral bone after bone grafting and matrix-associated autologous chondrocyte implantation for osteochondritis dissecans of the knee. Am. J. Sports Med. 2011, 39, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Selig, M.; Lauer, J.C.; Hart, M.L.; Rolauffs, B. Mechanotransduction and stiffness-sensing: Mechanisms and opportunities to control multiple molecular aspects of cell phenotype as a design cornerstone of cell-instructive biomaterials for articular cartilage repair. Int. J. Mol. Sci. 2020, 21, 5399. [Google Scholar] [CrossRef]
- Lubrano, E.; Perrotta, F.M. Secukinumab for ankylosing spondylitis and psoriatic arthritis. Ther. Clin. Risk Manag. 2016, 12, 1587–1592. [Google Scholar] [CrossRef] [Green Version]
- Sandy, J.D.; Chan, D.D.; Trevino, R.L.; Wimmer, M.A.; Plaas, A. Human genome-wide expression analysis reorients the study of inflammatory mediators and biomechanics in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Ruiz de Morales, J.M.G.; Puig, L.; Dauden, E.; Canete, J.D.; Pablos, J.L.; Martin, A.O.; Juanatey, C.G.; Adan, A.; Montalban, X.; Borruel, N.; et al. Critical role of interleukin (il)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting il-17 and th17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Faust, H.J.; Zhang, H.; Han, J.; Wolf, M.T.; Jeon, O.H.; Sadtler, K.; Pena, A.N.; Chung, L.; Maestas, D.R., Jr.; Tam, A.J.; et al. Il-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Investig. 2020, 130, 5493–5507. [Google Scholar] [CrossRef]
- Akeson, G.; Malemud, C.J. A role for soluble il-6 receptor in osteoarthritis. J. Funct. Morphol. Kinesiol. 2017, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Wiegertjes, R.; van de Loo, F.A.J.; Blaney Davidson, E.N. A roadmap to target interleukin-6 in osteoarthritis. Rheumatology (Oxford) 2020, 59, 2681–2694. [Google Scholar] [CrossRef]
- Ahrman, E.; Lorenzo, P.; Holmgren, K.; Grodzinsky, A.J.; Dahlberg, L.E.; Saxne, T.; Heinegard, D.; Onnerfjord, P. Novel cartilage oligomeric matrix protein (comp) neoepitopes identified in synovial fluids from patients with joint diseases using affinity chromatography and mass spectrometry. J. Biol. Chem. 2014, 289, 20908–20916. [Google Scholar] [CrossRef] [Green Version]
- Flannery, C.R.; Little, C.B.; Hughes, C.E.; Curtis, C.L.; Caterson, B.; Jones, S.A. Il-6 and its soluble receptor augment aggrecanase-mediated proteoglycan catabolism in articular cartilage. Matrix Biol. 2000, 19, 549–553. [Google Scholar] [CrossRef]
- Pasquali Ronchetti, I.; Guerra, D.; Taparelli, F.; Boraldi, F.; Bergamini, G.; Mori, G.; Zizzi, F.; Frizziero, L. Morphological analysis of knee synovial membrane biopsies from a randomized controlled clinical study comparing the effects of sodium hyaluronate (hyalgan) and methylprednisolone acetate (depomedrol) in osteoarthritis. Rheumatology (Oxford) 2001, 40, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daghestani, H.N.; Pieper, C.F.; Kraus, V.B. Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee osteoarthritis. Arthritis Rheumatol. 2015, 67, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.J.; Leckenby, A.; Reynolds, G.; Spiering, R.; Pratt, A.G.; Rankin, K.S.; Isaacs, J.D.; Haniffa, M.A.; Milling, S.; Hilkens, C.M. Macrophage proliferation distinguishes 2 subgroups of knee osteoarthritis patients. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Aristizabal, A.; Gandhi, R.; Mahomed, N.N.; Marshall, K.W.; Viswanathan, S. Synovial fluid monocyte/macrophage subsets and their correlation to patient-reported outcomes in osteoarthritic patients: A cohort study. Arthritis Res. Ther. 2019, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Sward, P.; Wang, Y.; Hansson, M.; Lohmander, L.S.; Grodzinsky, A.J.; Struglics, A. Coculture of bovine cartilage with synovium and fibrous joint capsule increases aggrecanase and matrix metalloproteinase activity. Arthritis Res. Ther. 2017, 19, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Fitzgerald, J.B.; DiMicco, M.A.; Cheng, D.M.; Flannery, C.R.; Sandy, J.D.; Plaas, A.H.; Grodzinsky, A.J. Co-culture of mechanically injured cartilage with joint capsule tissue alters chondrocyte expression patterns and increases adamts5 production. Arch. Biochem. Biophys. 2009, 489, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Patwari, P.; Lin, S.N.; Kurz, B.; Cole, A.A.; Kumar, S.; Grodzinsky, A.J. Potent inhibition of cartilage biosynthesis by coincubation with joint capsule through an il-1-independent pathway. Scand. J. Med. Sci. Sports 2009, 19, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Beekhuizen, M.; Bastiaansen-Jenniskens, Y.M.; Koevoet, W.; Saris, D.B.; Dhert, W.J.; Creemers, L.B.; van Osch, G.J. Osteoarthritic synovial tissue inhibition of proteoglycan production in human osteoarthritic knee cartilage: Establishment and characterization of a long-term cartilage-synovium coculture. Arthritis Rheumatol. 2011, 63, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Hoff, P.; Buttgereit, F.; Burmester, G.R.; Jakstadt, M.; Gaber, T.; Andreas, K.; Matziolis, G.; Perka, C.; Rohner, E. Osteoarthritis synovial fluid activates pro-inflammatory cytokines in primary human chondrocytes. Int. Orthop. 2013, 37, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavedi, S.; Diaz-Rodriguez, P.; Erndt-Marino, J.D.; Hahn, M.S. A three-dimensional chondrocyte-macrophage coculture system to probe inflammation in experimental osteoarthritis. Tissue Eng. Part A 2017, 23, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Utomo, L.; Bastiaansen-Jenniskens, Y.M.; Verhaar, J.A.; van Osch, G.J. Cartilage inflammation and degeneration is enhanced by pro-inflammatory (m1) macrophages in vitro, but not inhibited directly by anti-inflammatory (m2) macrophages. Osteoarthr. Cartil. 2016, 24, 2162–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.I.; Argyle, D.J.D. In vitro models for the study of osteoarthritis. Vet. J. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imgenberg, J.; Rolauffs, B.; Grodzinsky, A.J.; Schunke, M.; Kurz, B. Estrogen reduces mechanical injury-related cell death and proteoglycan degradation in mature articular cartilage independent of the presence of the superficial zone tissue. Osteoarthr. Cartil. 2013, 21, 1738–1745. [Google Scholar] [CrossRef] [Green Version]
- Quinn, T.M.; Maung, A.A.; Grodzinsky, A.J.; Hunziker, E.B.; Sandy, J.D. Physical and biological regulation of proteoglycan turnover around chondrocytes in cartilage explants. Implications for tissue degradation and repair. Ann. N. Y. Acad. Sci. 1999, 878, 420–441. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Sun, H.J.; Wang, Y.H.; Zhang, Z. Relationships of common polymorphisms in IL-6, IL-1a, and IL-1b genes with susceptibility to osteoarthritis: A meta-analysis. Clin. Rheumatol. 2015, 34, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Zhai, G.; Hart, D.J.; Kato, B.S.; Wang, H.; Williams, F.M.; Spector, T.D. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The chingford study. Arthritis Rheumatol. 2009, 60, 2037–2045. [Google Scholar] [CrossRef] [Green Version]
- de Hooge, A.S.; van de Loo, F.A.; Bennink, M.B.; Arntz, O.J.; de Hooge, P.; van den Berg, W.B. Male il-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr. Cartil. 2005, 13, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Daheshia, M.; Yao, J.Q. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J. Rheumatol. 2008, 35, 2306–2312. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, M.; Baron, B. The role of tnf-alpha in rheumatoid arthritis: A focus on regulatory t cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting interleukin-6 signaling in clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2004, 43 (Suppl. 3), iii2–iii9. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.; Wu, Z.; Li, M.; Shao, M.; Hu, T. Interleukin-1beta is a potential therapeutic target for periodontitis: A narrative review. Int. J. Oral. Sci. 2020, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Altobelli, E.; Angeletti, P.M.; Piccolo, D.; De Angelis, R. Synovial fluid and serum concentrations of inflammatory markers in rheumatoid arthritis, psoriatic arthritis and osteoarthitis: A systematic review. Curr. Rheumatol. Rev. 2017, 13, 170–179. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000 Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.M.; Samuel, R.O.; Gomes-Filho, J.E.; Dezan-Junior, E.; Cintra, L.T. The role of il-6 on apical periodontitis: A systematic review. Int. Endod. J. 2014, 47, 615–621. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tanaka, T. Interleukin 6 and rheumatoid arthritis. BioMed Res. Int. 2014, 2014, 698313. [Google Scholar] [CrossRef] [Green Version]
- Taams, L.S. Interleukin-17 in rheumatoid arthritis: Trials and tribulations. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Yamshchikov, V.F.; Mishina, M.; Cominelli, F. A possible role of IL-1ra 3’-untranslated region in modulation of protein production. Cytokine 2002, 17, 98–107. [Google Scholar] [CrossRef]
- Wu, X.; Kondragunta, V.; Kornman, K.S.; Wang, H.Y.; Duff, G.W.; Renner, J.B.; Jordan, J.M. Il-1 receptor antagonist gene as a predictive biomarker of progression of knee osteoarthritis in a population cohort. Osteoarthr. Cartil. 2013, 21, 930–938. [Google Scholar] [CrossRef] [Green Version]
- van Meegeren, M.E.; Roosendaal, G.; Coeleveld, K.; Nieuwenhuizen, L.; Mastbergen, S.C.; Lafeber, F.P. A single intra-articular injection with il-4 plus il-10 ameliorates blood-induced cartilage degeneration in haemophilic mice. Br. J. Haematol. 2013, 160, 515–520. [Google Scholar] [CrossRef]
- Nabbe, K.C.; van Lent, P.L.; Holthuysen, A.E.; Sloetjes, A.W.; Koch, A.E.; Radstake, T.R.; van den Berg, W.B. Local il-13 gene transfer prior to immune-complex arthritis inhibits chondrocyte death and matrix-metalloproteinase-mediated cartilage matrix degradation despite enhanced joint inflammation. Arthritis Res. Ther. 2005, 7, R392–R401. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.S.; Leiferman, E.M.; Frisch, K.E.; Brickson, S.L.; Murphy, W.L.; Baer, G.S.; Vanderby, R. Interleukin expression after injury and the effects of interleukin-1 receptor antagonist. PLoS ONE 2013, 8, e71631. [Google Scholar] [CrossRef]
- Elsaid, K.A.; Ubhe, A.; Shaman, Z.; D’Souza, G. Intra-articular interleukin-1 receptor antagonist (il1-ra) microspheres for posttraumatic osteoarthritis: In vitro biological activity and in vivo disease modifying effect. J. Exp. Orthop. 2016, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.S.; Leiferman, E.M.; Frisch, K.E.; Duenwald-Kuehl, S.E.; Brickson, S.L.; Murphy, W.L.; Baer, G.S.; Vanderby, R. Interleukin-1 receptor antagonist modulates inflammation and scarring after ligament injury. Connect. Tissue Res. 2014, 55, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, F.E.; Corp, N.; Kingsbury, S.R.; Frobell, R.; Englund, M.; Felson, D.T.; Levesque, M.; Majumdar, S.; Wilson, C.; Beard, D.J.; et al. Towards prevention of post-traumatic osteoarthritis: Report from an international expert working group on considerations for the design and conduct of interventional studies following acute knee injury. Osteoarthr. Cartil. 2019, 27, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arden, N.; Richette, P.; Cooper, C.; Bruyere, O.; Abadie, E.; Branco, J.; Brandi, M.L.; Berenbaum, F.; Clerc, C.; Dennison, E.; et al. Can we identify patients with high risk of osteoarthritis progression who will respond to treatment? A focus on biomarkers and frailty. Drugs Aging 2015, 32, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Spil, W.E.; Szilagyi, I.A. Osteoarthritis year in review 2019: Biomarkers (biochemical markers). Osteoarthr. Cartil. 2020, 28, 296–315. [Google Scholar] [CrossRef] [Green Version]
- Anitua, E.; Sanchez, M.; de la Fuente, M.; Azofra, J.; Zalduendo, M.; Aguirre, J.J.; Andia, I. Relationship between investigative biomarkers and radiographic grading in patients with knee osteoarthritis. Int. J. Rheumatol. 2009, 2009, 747432. [Google Scholar] [CrossRef] [PubMed]
- Deveza, L.A.; Kraus, V.B.; Collins, J.E.; Guermazi, A.; Roemer, F.W.; Nevitt, M.C.; Hunter, D.J. Is synovitis detected on non-contrast-enhanced magnetic resonance imaging associated with serum biomarkers and clinical signs of effusion? Data from the osteoarthritis initiative. Scand. J. Rheumatol. 2018, 47, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bobacz, K.; Maier, R.; Fialka, C.; Ekhart, H.; Woloszczuk, W.; Geyer, G.; Erlacher, L.; Smolen, J.; Graninger, W.B. Is pro-matrix metalloproteinase-3 a marker for posttraumatic cartilage degradation? Osteoarthr. Cartil. 2003, 11, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Conaghan, P.G.; D’Agostino, M.A.; Le Bars, M.; Baron, G.; Schmidely, N.; Wakefield, R.; Ravaud, P.; Grassi, W.; Martin-Mola, E.; So, A.; et al. Clinical and ultrasonographic predictors of joint replacement for knee osteoarthritis: Results from a large, 3-year, prospective EULAR study. Ann. Rheum. Dis. 2010, 69, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Group, M.K.; Jones, M.H.; Oak, S.R.; Andrish, J.T.; Brophy, R.H.; Cox, C.L.; Dunn, W.R.; Flanigan, D.C.; Fleming, B.C.; Huston, L.J.; et al. Predictors of radiographic osteoarthritis 2 to 3 years after anterior cruciate ligament reconstruction: Data from the moon on-site nested cohort. Orthop. J. Sports Med. 2019, 7, 2325967119867085. [Google Scholar]
- Bodkin, S.G.; Werner, B.C.; Slater, L.V.; Hart, J.M. Post-traumatic osteoarthritis diagnosed within 5 years following ACI reconstruction. Knee Surg. Sports Traumatol. Arthrosc. 2020, 28, 790–796. [Google Scholar] [CrossRef]
- Khan, T.; Alvand, A.; Prieto-Alhambra, D.; Culliford, D.J.; Judge, A.; Jackson, W.F.; Scammell, B.E.; Arden, N.K.; Price, A.J. Acl and meniscal injuries increase the risk of primary total knee replacement for osteoarthritis: A matched case-control study using the clinical practice research datalink (CPRD). Br. J. Sports Med. 2019, 53, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Bittersohl, B.; Hosalkar, H.S.; Miese, F.R.; Schibensky, J.; Konig, D.P.; Herten, M.; Antoch, G.; Krauspe, R.; Zilkens, C. Zonal t2* and t1gd assessment of knee joint cartilage in various histological grades of cartilage degeneration: An observational in vitro study. BMJ Open 2015, 5, e006895. [Google Scholar] [CrossRef] [Green Version]
- Tschaikowsky, M.; Selig, M.; Brander, S.; Balzer, B.N.; Hugel, T.; Rolauffs, B. Proof-of-concept for the detection of early osteoarthritis pathology by clinically applicable endomicroscopy and quantitative ai-supported optical biopsy. Osteoarthr. Cartil. 2020. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, M.; Luck, S.; Martin, P.; Felka, T.; Aicher, W.; Rolauffs, B.; Schmidt, V. Modeling chondrocyte patterns by elliptical cluster processes. J. Struct. Biol. 2012, 177, 447–458. [Google Scholar] [CrossRef]
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 120), 48–56. [Google Scholar] [PubMed]
- Minguzzi, M.; Cetrullo, S.; D’Adamo, S.; Silvestri, Y.; Flamigni, F.; Borzi, R.M. Emerging players at the intersection of chondrocyte loss of maturational arrest, oxidative stress, senescence and low-grade inflammation in osteoarthritis. Oxid. Med. Cell Longev. 2018, 2018, 3075293. [Google Scholar] [CrossRef] [Green Version]
- Ershler, W.B. Interleukin-6: A cytokine for gerontologists. J. Am. Geriatr. Soc. 1993, 41, 176–181. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Rezus, E.; Cardoneanu, A.; Burlui, A.; Luca, A.; Codreanu, C.; Tamba, B.I.; Stanciu, G.D.; Dima, N.; Badescu, C.; Rezus, C. The link between inflammaging and degenerative joint diseases. Int. J. Mol. Sci. 2019, 20, 614. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, J.L.; Woodhouse, L.J.; Nettel-Aguirre, A.; Emery, C.A. Outcomes associated with early post-traumatic osteoarthritis and other negative health consequences 3-10 years following knee joint injury in youth sport. Osteoarthr. Cartil. 2015, 23, 1122–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, E.M. Joint injury causes knee osteoarthritis in young adults. Curr. Opin. Rheumatol. 2005, 17, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Lees, S.; Golub, S.B.; Last, K.; Zeng, W.; Jackson, D.C.; Sutton, P.; Fosang, A.J. Bioactivity in an aggrecan 32-mer fragment is mediated via toll-like receptor 2. Arthritis Rheumatol. 2015, 67, 1240–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.M. An aggrecan fragment drives osteoarthritis pain through toll-like receptor 2. JCI Insight 2018, 3, e95704. [Google Scholar] [CrossRef]
- Han, B.; Li, Q.; Wang, C.; Patel, P.; Adams, S.M.; Doyran, B.; Nia, H.T.; Oftadeh, R.; Zhou, S.; Li, C.Y.; et al. Decorin regulates the aggrecan network integrity and biomechanical functions of cartilage extracellular matrix. ACS Nano 2019, 13, 11320–11333. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Buckwalter, J.A.; Martin, J.A. Damps synergize with cytokines or fibronectin fragment on inducing chondrolysis but lose effect when acting alone. Mediat. Inflamm. 2017, 2017, 2642549. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Guo, D.; Homandberg, G.A.; Buckwalter, J.A.; Martin, J.A. A single blunt impact on cartilage promotes fibronectin fragmentation and upregulates cartilage degrading stromelysin-1/matrix metalloproteinase-3 in a bovine ex vivo model. J. Orthop. Res. 2014, 32, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, Y.; Abe, T.; Asawa, Y.; Nishizawa, S.; Saijo, H.; Hikita, A.; Hoshi, K. Influence of damage-associated molecular patterns from chondrocytes in tissue-engineered cartilage. Tissue Eng. Part A 2020. [Google Scholar] [CrossRef]
- Aulin, C.; Lassacher, T.; Palmblad, K.; Erlandsson Harris, H. Early stage blockade of the alarmin hmgb1 reduces cartilage destruction in experimental OA. Osteoarthr. Cartil. 2020, 28, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Wenzhao, L.; Jiangdong, N.; Deye, S.; Muliang, D.; Junjie, W.; Xianzhe, H.; Mingming, Y.; Jun, H. Dual regulatory roles of hmgb1 in inflammatory reaction of chondrocyte cells and mice. Cell Cycle 2019, 18, 2268–2280. [Google Scholar] [CrossRef]
- Rosenberg, J.H.; Rai, V.; Dilisio, M.F.; Sekundiak, T.D.; Agrawal, D.K. Increased expression of damage-associated molecular patterns (DAMPS) in osteoarthritis of human knee joint compared to hip joint. Mol. Cell. Biochem. 2017, 436, 59–69. [Google Scholar] [CrossRef]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Peilin, W.; Songsong, T.; Chengyu, Z.; Zhi, C.; Chunhui, M.; Yinxian, Y.; Lei, Z.; Min, M.; Zongyi, W.; Mengkai, Y.; et al. Directed elimination of senescent cells attenuates development of osteoarthritis by inhibition of c-iap and xiap. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2618–2632. [Google Scholar] [CrossRef]
- Vinatier, C.; Dominguez, E.; Guicheux, J.; Carames, B. Role of the inflammation-autophagy-senescence integrative network in osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Martin, J.A.; Brown, T.; Heiner, A.; Buckwalter, J.A. Post-traumatic osteoarthritis: The role of accelerated chondrocyte senescence. Biorheology 2004, 41, 479–491. [Google Scholar] [PubMed]
- Philipot, D.; Guerit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. P16ink4a and its regulator mir-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of pdgf-aa. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, O.H.; Wilson, D.R.; Clement, C.C.; Rathod, S.; Cherry, C.; Powell, B.; Lee, Z.; Khalil, A.M.; Green, J.J.; Campisi, J.; et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef]
- Eskelinen, A.S.A.; Tanska, P.; Florea, C.; Orozco, G.A.; Julkunen, P.; Grodzinsky, A.J.; Korhonen, R.K. Mechanobiological model for simulation of injured cartilage degradation via pro-inflammatory cytokines and mechanical stimulus. PLoS Comput. Biol. 2020, 16, e1007998. [Google Scholar] [CrossRef]
- Heijink, A.; Gomoll, A.H.; Madry, H.; Drobnic, M.; Filardo, G.; Espregueira-Mendes, J.; Van Dijk, C.N. Biomechanical considerations in the pathogenesis of osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Griffin, T.M.; Guilak, F. The role of mechanical loading in the onset and progression of osteoarthritis. Exerc. Sport. Sci. Rev. 2005, 33, 195–200. [Google Scholar] [CrossRef]
- Perruccio, A.V.; Chandran, V.; Power, J.D.; Kapoor, M.; Mahomed, N.N.; Gandhi, R. Systemic inflammation and painful joint burden in osteoarthritis: A matter of sex? Osteoarthr. Cartil. 2017, 25, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, S.; Mrosewski, I.; Silawal, S.; Schulze-Tanzil, G. The interrelation of osteoarthritis and diabetes mellitus: Considering the potential role of interleukin-10 and in vitro models for further analysis. Inflamm. Res. 2018, 67, 285–300. [Google Scholar] [CrossRef]
- Courties, A.; Sellam, J. Osteoarthritis and type 2 diabetes mellitus: What are the links? Diabetes Res. Clin. Pract. 2016, 122, 198–206. [Google Scholar] [CrossRef]
- Louati, K.; Vidal, C.; Berenbaum, F.; Sellam, J. Association between diabetes mellitus and osteoarthritis: Systematic literature review and meta-analysis. RMD Open 2015, 1, e000077. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Panoutsopoulou, K.; Southam, L.; Rayner, N.W.; Day-Williams, A.G.; Lopes, M.C.; Boraska, V.; Esko, T.; Evangelou, E.; Hoffman, A.; et al. Identification of new susceptibility loci for osteoarthritis (arcogen): A genome-wide association study. Lancet 2012, 380, 815–823. [Google Scholar] [PubMed] [Green Version]
- Reynard, L.N.; Barter, M.J. Osteoarthritis year in review 2019: Genetics, genomics and epigenetics. Osteoarthr. Cartil. 2020, 28, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, J.C.; Selig, M.; Hart, M.L.; Kurz, B.; Rolauffs, B. Articular chondrocyte phenotype regulation through the cytoskeleton and the signaling processes that originate from or converge on the cytoskeleton: Towards a novel understanding of the intersection between actin dynamics and chondrogenic function. Int. J. Mol. Sci. 2021. submitted. [Google Scholar]




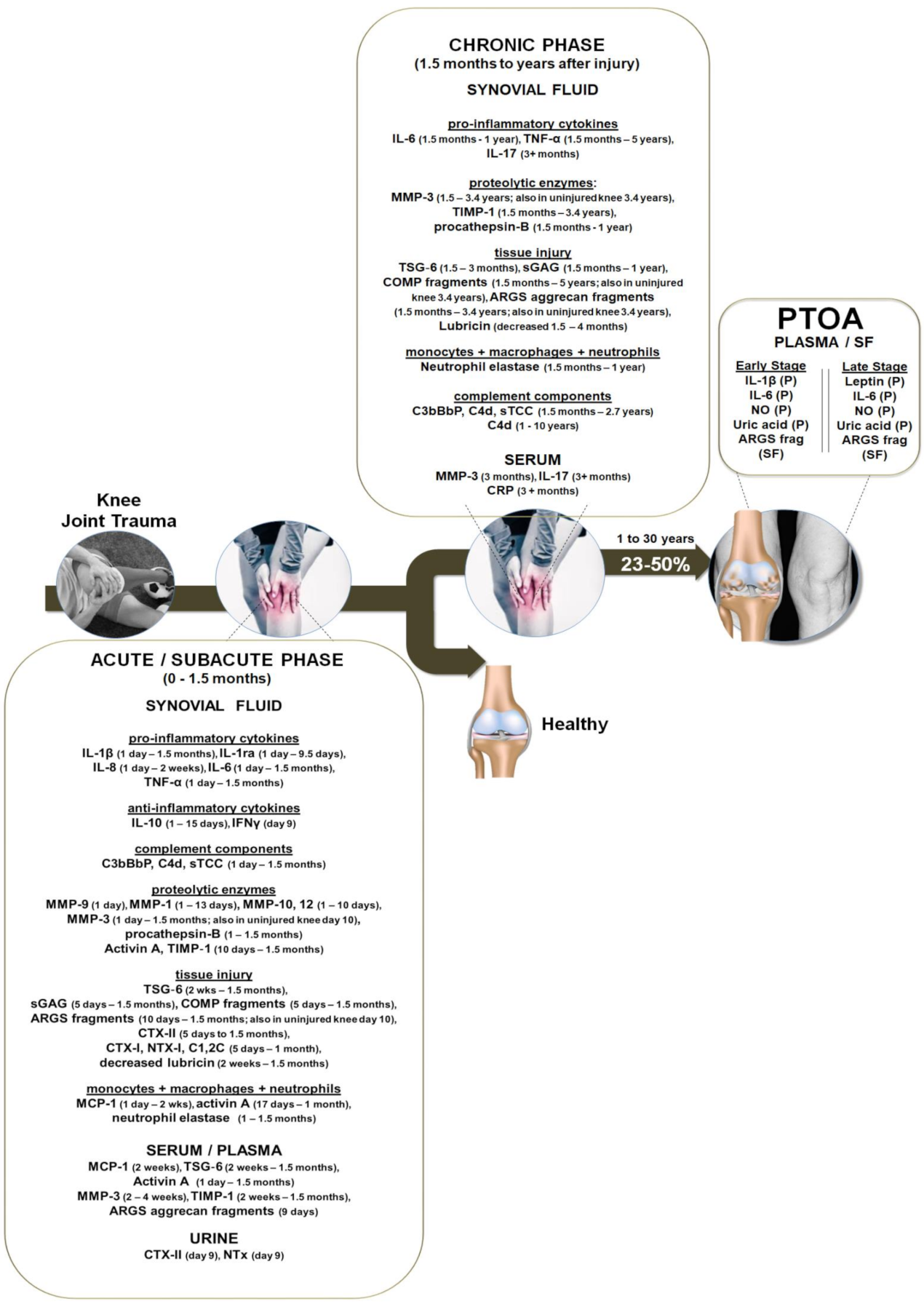{kind=link}
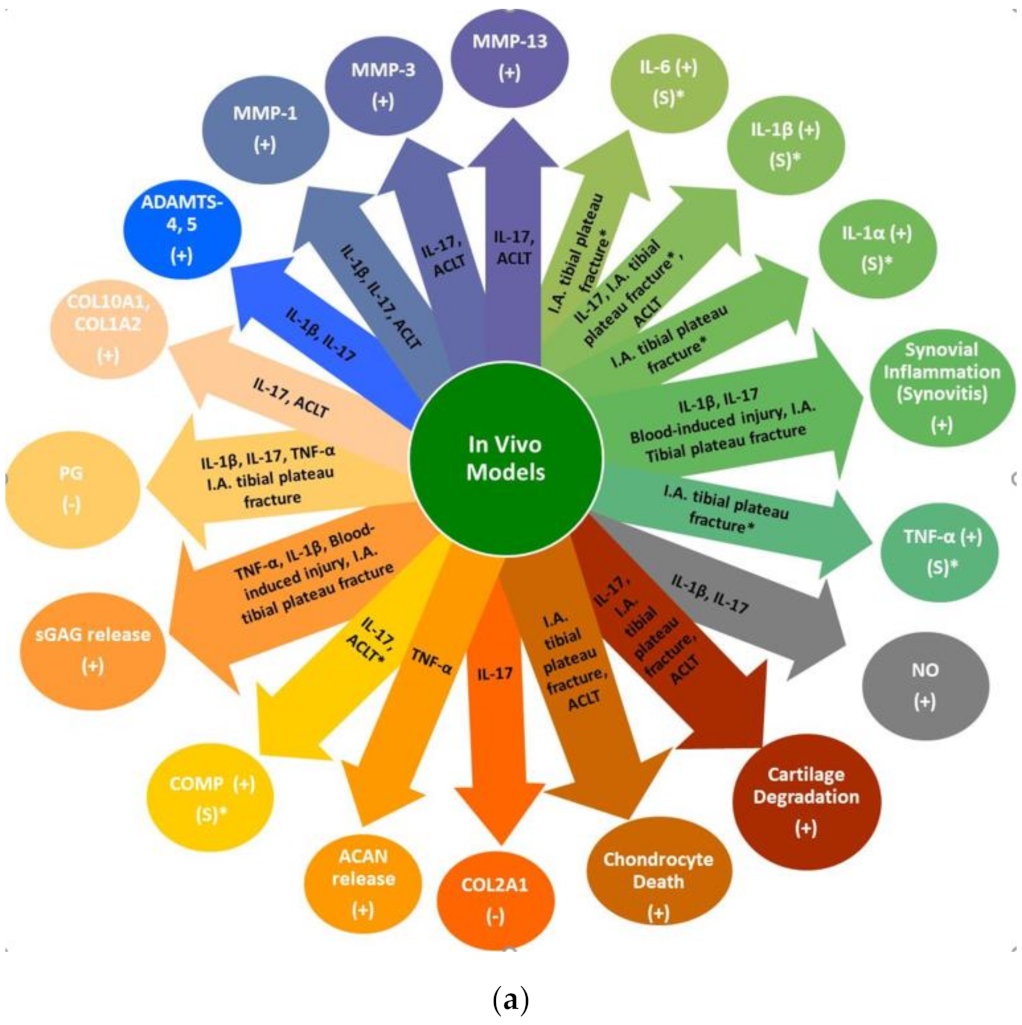{kind=link}
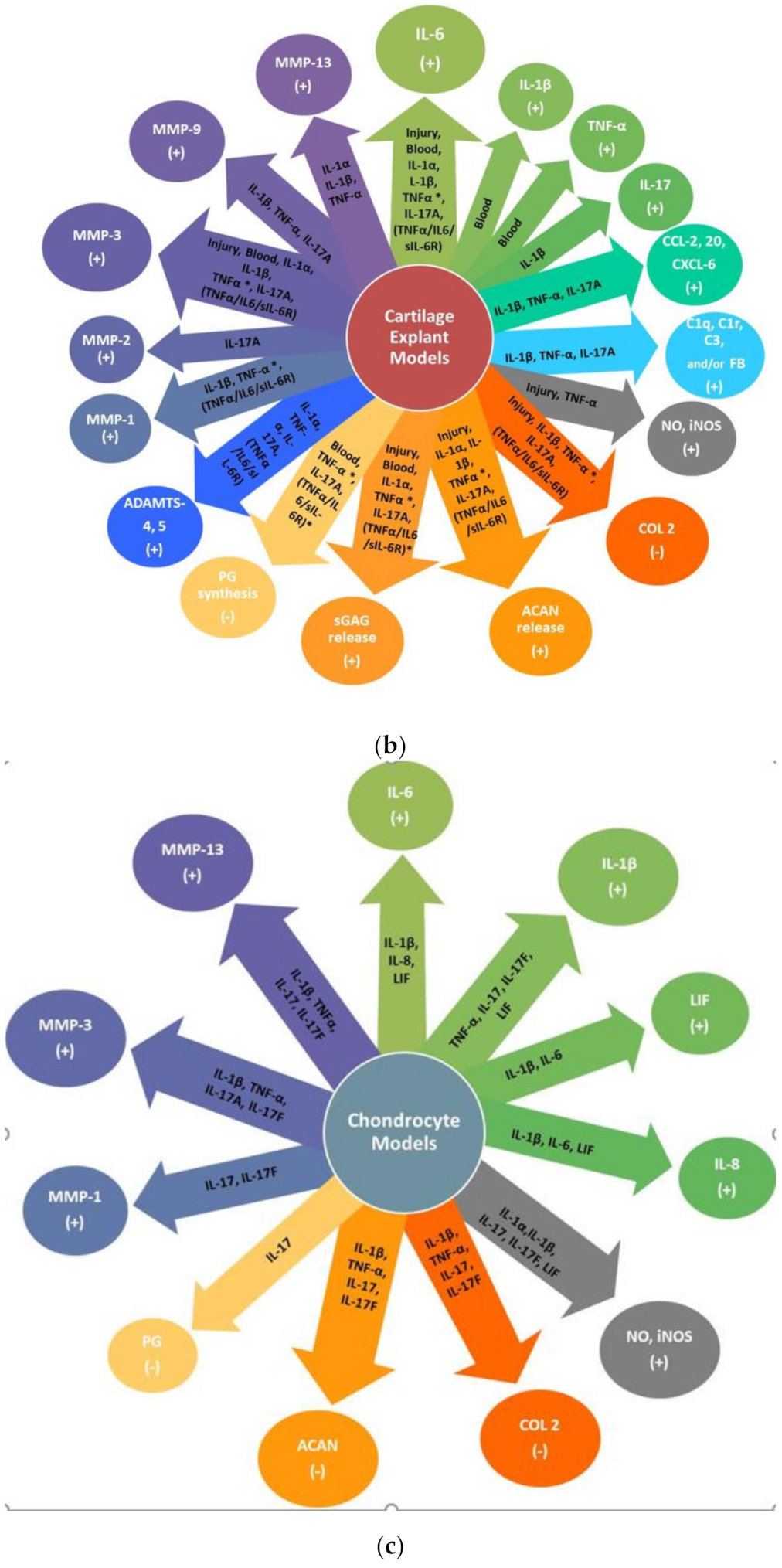{kind=link}
| Study | Q1 | Q 2 | Q 3 | Q 4 | Q 5 | Q 6 | Q 7 | Q 8 | Q 9 | Q 10 | Q 11 | Q 12 | Q 13 | Q 14 | Score | Quality |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haller [21] | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 1 | 1 | 1 | 0 | 10 | good |
| Haller [22] | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 1 | 1 | 1 | 0 | 10 | good |
| Swärd [23] | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 1 | 10 | good |
| Bigoni [24] | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 0 | 10 | good |
| Irie [25] | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 0 | NR | 0 | 0 | 8 | fair |
| Elsaid [26] | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | NA | 0 | NR | 0 | 1 | 10 | good |
| Catterall [27] | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | NA | 0 | NR | 1 | 0 | 7 | fair |
| Watt [28] | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 1 | 12 | good |
| Lattermann [29] | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | NA | 1 | NR | U | 0 | 7 | fair |
| Dahlberg [30] | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 0 | 10 | good |
| Higuchi [31] | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 0 | 9 | fair |
| Struglics [32] | 1 | 1 | 1 | 0 | U | 1 | 1 | 1 | 1 | NA | 0 | U | 1 | 1 | 9 | fair |
| Sarafan [33] | 1 | 0 | 1 | U | 0 | 1 | 1 | 0 | 1 | NA | 1 | NR | 1 | 0 | 7 | fair |
| Lohmander [34] | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | NA | 1 | 8 | fair |
| Lohmander [35] | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | NA | 1 | 8 | fair |
| Struglics [36] | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | NA | 1 | NR | 1 | 1 | 10 | good |
| Struglics [37] | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | NA | 1 | NR | 0 | 1 | 10 | good |
| Larsson [38] | 1 | 1 | NA | 1 | 0 | 1 | 1 | 1 | 1 | NA | 1 | 1 | 1 | 0 | 10 | good |
| Panina [39] | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | NA | 0 | NR | 1 | 1 | 10 | fair |
| Study | Type of Injuries | Comparison Group | Phase after Injury | Markers Measured | Age Mean Median (Range) | Cohort Size | Sex | Confounding Factor |
|---|---|---|---|---|---|---|---|---|
| Haller [21] | 24 patients had low and 21 high energy acute tibial plateau fracture | Contralateral uninjured knee | A | IFN-γ, IL-1β, IL-1RA, IL-2, IL-4, IL-6, IL-7, IL-8, IL-10, IL-12(p70), IL-13, IL-17A, TNF-α, MCP-1, MIP-1β | 42 (20–60) | 45 | 31 males 14 females | |
| Haller [22] | 24 patients had low and 21 high energy acute tibial plateau fracture | Contralateral uninjured knee | A | MMP-1, -2, -3, -7, -9, -10, -12, -13 | 42 (20–60) | 45 | 31 males 14 females | |
| Swärd [23] | 100% had hemarthrosis; 57 ACL tear, 3 medial meniscus injuries, 1 posterior cruciate ligament tear, 5 patellar dislocations; 1 suspected tibial fracture; 3 MCL tear, 2 lateral collateral ligament tears, 35 rotational knee traumas; 3 knee contusions; 1 knee hyperextension. 15 of these had associated MCLT | Age- and gender-matched healthy uninjured controls | A, S | sGAG, ARGS aggrecan fragments, OCL, SPARC, OPN, IL-1β, IL-6, IL-8, TNF-α | 25 (13–64) | 111 | 83 males 28 females | 16 patients had a previous injury (mean 5 years prior to injury) and 7 had previous knee surgery |
| Bigoni [24] | 18 patients had ACL tear, 30 ACL tear associated + meniscal injury | Healthy uninjured controls | A, S, C | IL-1, IL-1ra, IL-6, IL-8, IL-10, TNF-α | 30 (14–55) | 48 | 48 males | Unequal distribution of the number of patients at the different time points measured; Did not include any female subjects |
| Irie [25] | ACL tear | OA & post- meniscectomy including hydrarthrosis | A, S | TNF-α, IL-1β, IL-6, IL-8, IL-1ra, IL-10 | 27 (13–55) | 34 | 20 males 14 females | Unequal distribution of the number of patients at the different time points measured |
| Elsaid [26] | ACL tear | Contralateral uninjured knee | S, C | Lubricin, IL-1β, TNF-α, IL-6, Procathepsin B, Neutrophil elastase, sGAG | 24 (15–47) | 30 | 19 males 11 females | Unequal distribution of patients at different time points; No sub-stratification of the injured groups based on time after injury |
| Catterall [27] | ACL tear + other knee joint tissue damage including bone contusions, medial collateral ligament tears, meniscal tears and chondral defects | None (samples were measured over time) | A, S | IL-1β, CRP, sGAG, ARGS-aggrecan, FA846, CS846, C2C, CTxII, C1,2C, CTxI, NTX, CPII, Osteocalcin, β-Aspartate, D-Asx, D-Serine, sCD44, COMP, Tenascin C, Lubricin, MMP-3 | 23 (19–26) | 11 | 6 males 5 females | Half of the patients at 2nd time point were given i.a. IL-1ra but authors reported no statistical differences between IL-1ra vs. placebo arms for all measured biomarkers |
| Watt [28] | 74% had hemarthrosis; 61 combined ACL + meniscal tear, 34 severe trauma (1 or more injuries including meniscal tear, cruciate ligament rupture, collateral ligament tear, posterolateral corner injury, traumatic chondral defects, articular or periarticular fracture, or patellofemoral or tibiofemoral dislocation with severe knee trauma defined as combined (>1) ligament rupture, fracture or dislocation); 28 a single ACL or MCL rupture, 27 meniscal tears | Macroscopically intact cartilage from lower limb tumor amputation or transplant donations | A, S, C | Activin A, CRP, IL-1β, IL-6, MCP-1, MMP-3, TIMP-1, TSG-6 | 25 (16–50) | 150 | 121 males 29 females | Wide time range for baseline visit (0–8 weeks) |
| Lattermann [29] | Isolated ACL injury (no more than a grade 1 MCL injury defined clinically) | None (samples were measured over time) | A, S | IL-1α, IL-1β, IL-1ra, COMP fragments, CTX-II, sGAG, MMP-1, MMP-3, MMP-9, NTX-I, TSG-6 | NR (18–32) | 41 | NR | Controls were not included |
| Dahlberg [30] | 31 patients had a combination of cruciate ligament, collateral ligament and meniscus injuries, 15 isolated meniscus injuries, 8 cartilage lesions without ligament injuries; remaining had fibrillations + occasional clefts in the joint cartilage surface in one knee compartment | Contralateral uninjured knee or healthy uninjured controls | A, S, C | Aggrecan fragments, COMP, MMP-3, TIMP-1 | 28 (18–40) | 54 | NR | 4 patients had undergone ligament reconstruction more than 3 years before |
| Higuchi [31] | Complete ACL rupture | Age-matched healthy uninjured controls | A, S, C | IL-1β, TNF-α, IL-6, MMP-3, TIMP-1 | 26 (17–42) | 32 | 20 males 12 females | Wide time range for injury group (2–134 weeks) and with no sub-stratification of the injured groups for most of the markers measured; Performed mostly correlations |
| Struglics [32] | Acute ruptured ACL | Healthy uninjured controls | A, S, C | TNF-α, IL-6, IL-8, IL-10, IFN-γ, ARGS, CTX-II, NTX-I | NR (21–31) | 121 | 152 males 31 females | Wide time range for baseline visit (0–6 weeks) |
| Sarafan [33] | Intra-articular fractures | Healthy uninjured controls | C | IL-17, CRP | NR (28–42) | 20 | 9 males 11 females | Confusing inclusion/exclusion criteria; While the authors report that fracture patients endured pain for at least 3 months and samples were taken at the time of knee joint surgery, it was not clear when the samples were collected |
| Lohmander [34] | ACL rupture isolated or combined with another ligament or meniscus tear or an isolated meniscus tear | Healthy uninjured controls | A, S, C | CTX-II, Aggrecan fragments, MMP-1, MMP-3, TIMP-1 | 37 (14–70) | 247 males 82 females | ||
| Lohmander [35] | 159 patients had ACL rupture +/− injury to meniscus; 129 medial or lateral meniscus tear | Healthy uninjured controls | A, S, C | Aggrecan and COMP fragments, CPII, MMP-1, MMP-3, TIMP-1, BSP | 33 (14–70) | 221 males 67 females | ||
| Struglics [36] | Patients were recently injured (0–12 weeks after injury; 98% had hemarthrosis) or had an old injury (1–37 years after injury): 39 had ACL, 4 PCL, 69 ACL injury + meniscal tear, 37 ACL injury + meniscal tear + other ligament injuries, 47 ACL injury + other ligament injuries, 58 isolated meniscal tear, 7 meniscal tear + non-ACL ligament injuries, 13 patellar dislocation +/− soft tissue injuries, 7 other injuries + medial or lateral collateral ligament tears; 2 + give-way), 10 no signs of soft tissue injury | Healthy uninjured controls, OA, RA or PPA | A, S, C, PTOA | C4d, C3bBbP, sTCC, sGAG, IL-1β, IL-6, IL-8, TNF-α, ARGS neoepitope of aggrecan, Osteocalcin, SPARC, COMP, C2C, Osteopontin | Recently injury 26 (13–64) Old injury 32 (18–65) | 219 75 | 164 males 55 females 50 males 25 females | Only 8% of the old injury group were diagnosed with PTOA; Sub-stratification of the group was only divided into two groups: 1–3 years (median of 2.0 years) and 3.01–36.9 years (median of 5.1 years) with the latter group having a wide range of time after injury |
| Struglics [37] | ACL injury | Healthy uninjured controls | A, S, C | COMP | 26 (21–31) | 121 | 90 males 31 females | |
| Larsson [38] | ACL rupture +/− meniscus tear | Healthy uninjured controls | A, S, C, PTOA | Aggrecan, ARGS aggrecan fragments, sGAG | Acute knee injury 27 (16–59) Chronic knee injury 40 (16–70) | 192 | 56 males 13 females 95 males 28 females | Chronic knee injury group had a wide range of time from 3 months–36 years (mean of 1 year) after injury and 97% had PTOA |
| Panina [39] | All patients suffered a knee injury ≤ 1 year following a meniscus injury and were diagnosed with early- (Kellgren–Lawrence grade 1–2) or late-stage (grade 3–4) PTOA | Healthy uninjured controls | PTOA | IL-1β, IL-6, IL-18, TNF-α, Leptin, NO | NR (26–65) | 134 | 60 males 74 females | 77% of patients were in the early-stage PTOA group; Control data was only included for plasma samples; not for SF |
| Cytokine | Concentration in Synovial Fluid | Phase Present after Knee Trauma | Concentration Used in Laboratory Models | Fold Change |
|---|---|---|---|---|
| IL-1β | 0–25 pg/mL | Acute, Subacute, PTOA | 25–1 × 105 pg/mL | 50–400× |
| TNF-α | 7–20 pg/mL | Acute, Subacute, Chronic | 100–1 × 106 pg/mL | 77–5000× |
| IL-6 | 1–66,099 pg/mL | Acute, Subacute, Chronic, PTOA | 100–2 × 106 pg/mL | 3–5× |
| IL-17 | 2–8 pg/mL | Chronic, PTOA | 100–1 × 106 pg/mL | 12–52× |
| IL-1β | TNF-α | IL-6 | IL-17 | ||
|---|---|---|---|---|---|
| STRENGTH OF ASSOCIATION | Association between risk factor and outcome | Convincing [21,23,24,25,26,27,29,31,39,208] | Convincing [23,24,25,26,28,32,36,37] | Convincing [21,23,24,25,26,28,31,32,36,37,39,208,209] | Convincing [21,33,55,56,57,58,59,60] |
| CONSISTENCY OF FINDINGS | The same findings are observed among different locations, populations or in different study designs including different types of injuries | Convincing While some studies showed that IL-1β was significantly increased vs. [21,23,24,25,26,27,29,31,39], some studies showed that it was below the detection for some patients [24,26,28,29,31] | Convincing [23,24,25,26,28,32,36,37] | Convincing [21,23,24,25,26,28,31,32,36,37,39] | Convincing [21,33] |
| SPECIFICITY OF ASSOCIATION | The factor influences the outcome | Credible Clinically present from 24 h to 1.5 months in A, S phases [21,23,24,25,26,27,29,31] and in early PTOA [39] indicating that it influences disease progression Conflicting data: Some studies showed that IL-1β was not present [28] or only present in some patients [24,26,29,31] Application of IL-1β in vivo, ex vivo & in vitro mirrors clinical symptoms (Figure 2A–C) | Convincing Clinically present from 24 h to 5 years in A, S, C phases [23,24,25,26,28,32,36,37] Application of TNF-α in vivo, ex vivo and in vitro mirrors clinical symptoms (Figure 2A–C) | Convincing Clinically present from 24 h to 1 year in A, S, C phases and in early & late PTOA [21,23,24,25,26,28,31,32,36,37,39] IL-6 KO mice develop more advanced knee OA [210] and application of IL-6 ex vivo and in vitro mirrors clinical symptoms (Figure 2A–C) | Credible There are only 2 clinical studies showing that IL-17 is present from 24h to 9.5 days in the A phase [21] and C phase (3 or more months) after injury [33] Application of IL-17 in vivo, ex vivo and in vitro mirrors clinical symptoms (Figure 2A–C) |
| TEMPORAL SEQUENCE OF ASSOCIATION | The factor precedes the outcome | Convincing A, S, Early PTOA [21,23,24,25,26,27,29,31,39] | Convincing A, S, C Phases [23,24,25,26,28,32,36] | Convincing A, S, C, Early & Late PTOA [21,23,24,25,26,28,31,32,36,39] | Convincing A, C Phases [21,33] |
| BIOLOGICAL GRADIENT | Dose-response relationship, where longer or higher exposure leads to an increased risk of disease | Convincing Early PTOA was associated with a highIL-1β plasma concentration [39] | Convincing Correlationbetween TNF-α concentrations and ARGS fragments in the SF up to 5 years after knee trauma [32] suggesting that long exposure to TNF-α increases the risk of disease | Convincing Early and late PTOA were associated with high IL-6 concentrations PTOA [39] | Convincing Levels of IL-17 in the SF correlate with severity of knee OA [58] Presence of IL-17 in synovial fluid identifies a subset of patients with end-stage knee OA [60] |
| BIOLOGICAL PLAUSIBILITY | Presence of a potential biological mechanism | Convincing [12,211] | Convincing [12,212] | Convincing [191,213] | Convincing [187,189] |
| COHERENCE | The current studies agree and do not conflict with previously reported evidence and the biology of disease | Credible Clarification is needed on why some patients have high concentrations of IL-1β present, while others do not | Convincing | Convincing | Credible More clinical studies need to measure IL-17 in the SF and serum at the various phases after knee trauma and in clinical PTOA |
| EXPERIMENTAL EVIDENCE | Evidence drawn from experimental models agree with clinical data | Convincing | Convincing | Convincing | Convincing |
| ANALOGY | Analogous examples that lead to the same outcome | Convincing RA, periodontitis [214,215] | Convincing RA, psoriatic arthritis ankylosing spondylitis, Crohn’s disease [216,217] | Convincing RA, psoriatic arthritis, periodontitis [216,218,219] | Convincing RA, psoriatic arthritis, ankylosing spondylitis, periodontitis [184,216,220,221] |
| CAUSAL EFFECT | 7/9 EVIDENCE MET | 9/9 EVIDENCE MET | 9/9 EVIDENCE MET | 7/9 EVIDENCE MET |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khella, C.M.; Asgarian, R.; Horvath, J.M.; Rolauffs, B.; Hart, M.L. An Evidence-Based Systematic Review of Human Knee Post-Traumatic Osteoarthritis (PTOA): Timeline of Clinical Presentation and Disease Markers, Comparison of Knee Joint PTOA Models and Early Disease Implications. Int. J. Mol. Sci. 2021, 22, 1996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041996
Khella CM, Asgarian R, Horvath JM, Rolauffs B, Hart ML. An Evidence-Based Systematic Review of Human Knee Post-Traumatic Osteoarthritis (PTOA): Timeline of Clinical Presentation and Disease Markers, Comparison of Knee Joint PTOA Models and Early Disease Implications. International Journal of Molecular Sciences. 2021; 22(4):1996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041996
Chicago/Turabian StyleKhella, Christine M., Rojiar Asgarian, Judith M. Horvath, Bernd Rolauffs, and Melanie L. Hart. 2021. "An Evidence-Based Systematic Review of Human Knee Post-Traumatic Osteoarthritis (PTOA): Timeline of Clinical Presentation and Disease Markers, Comparison of Knee Joint PTOA Models and Early Disease Implications" International Journal of Molecular Sciences 22, no. 4: 1996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041996