
Effect of Oxaliplatin, Olaparib and LY294002 in Combination on Triple-Negative Breast Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
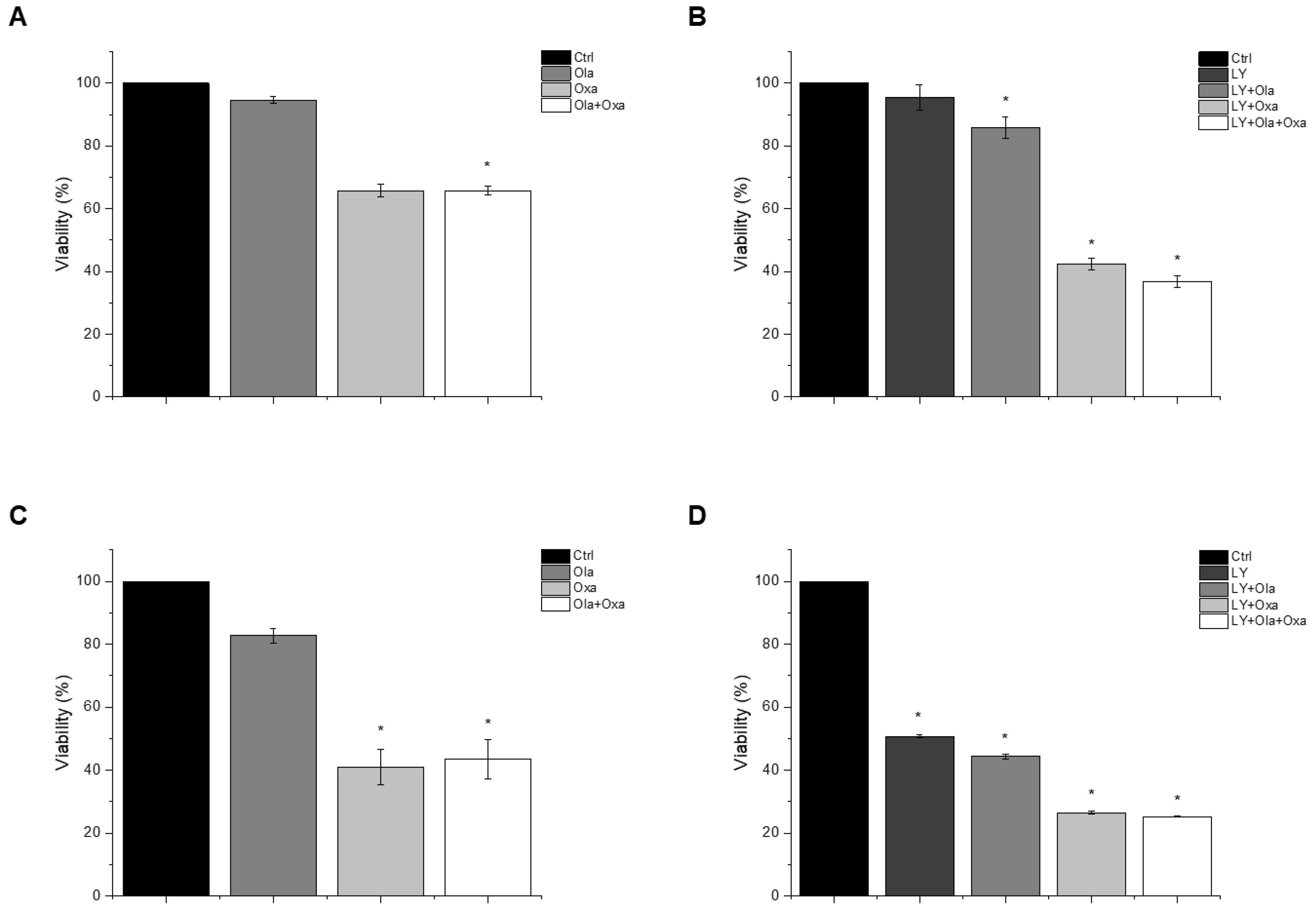
2.1. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on Cell Viability
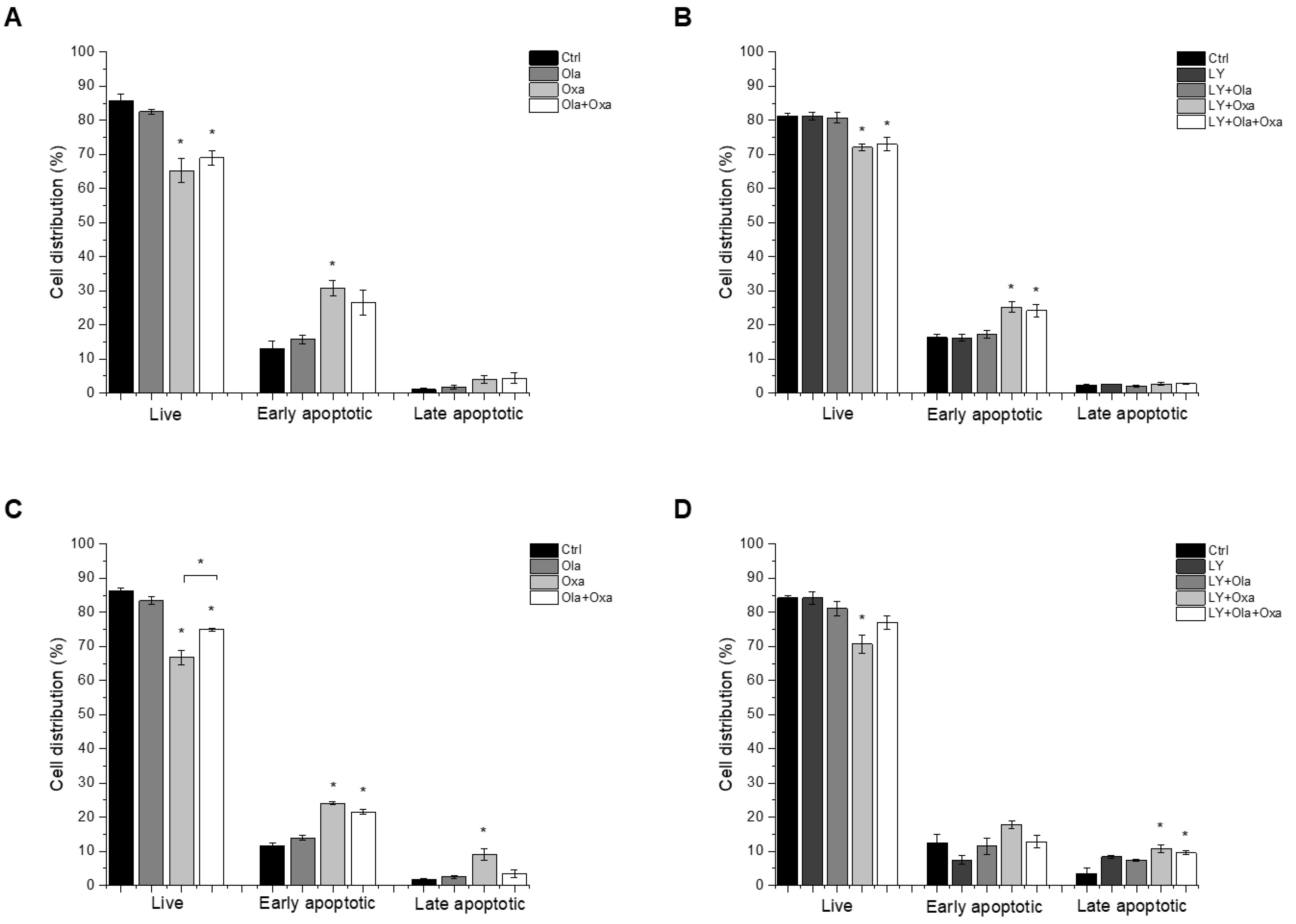
2.2. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on Cell Death Processes
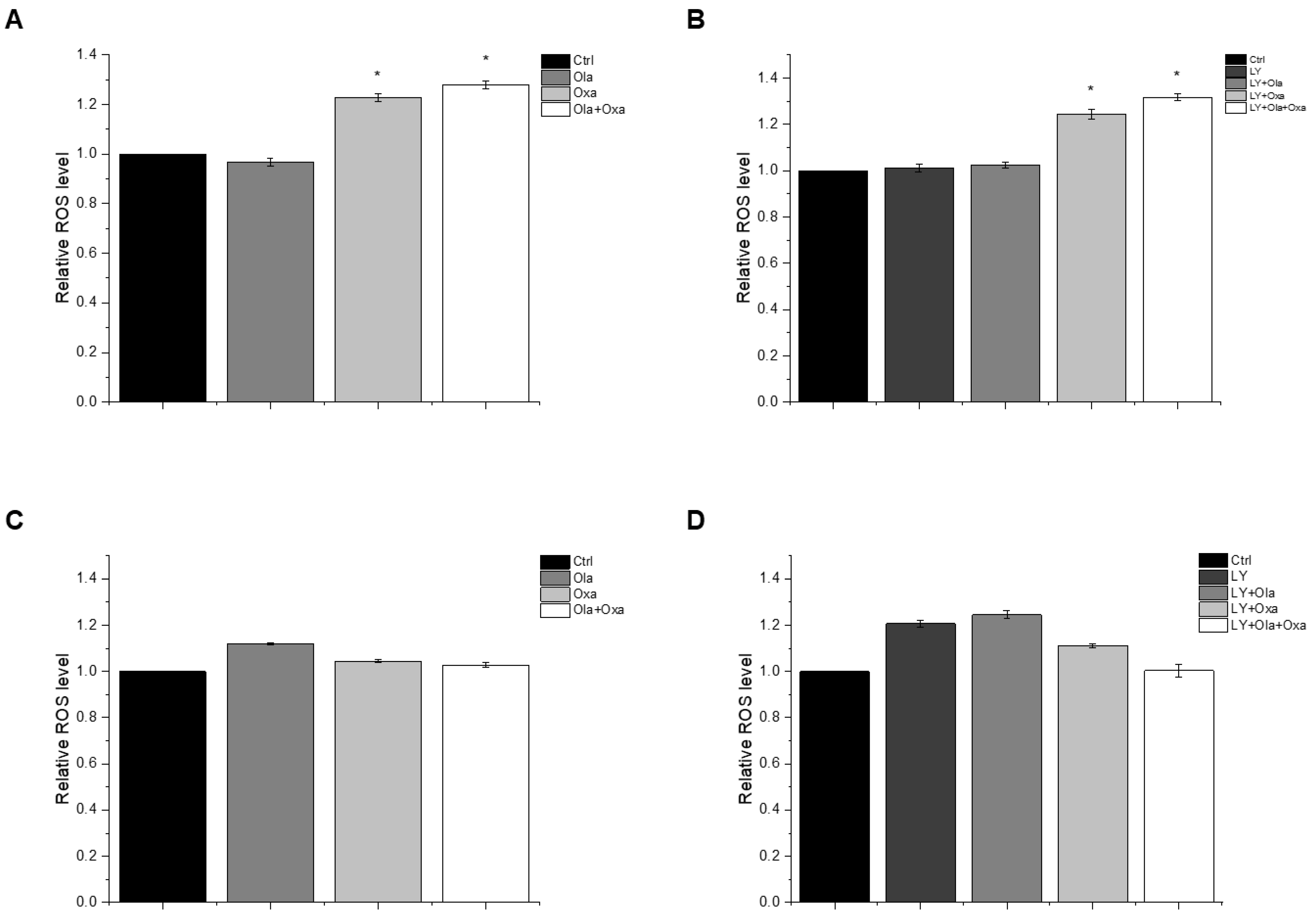
2.3. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on ROS Production
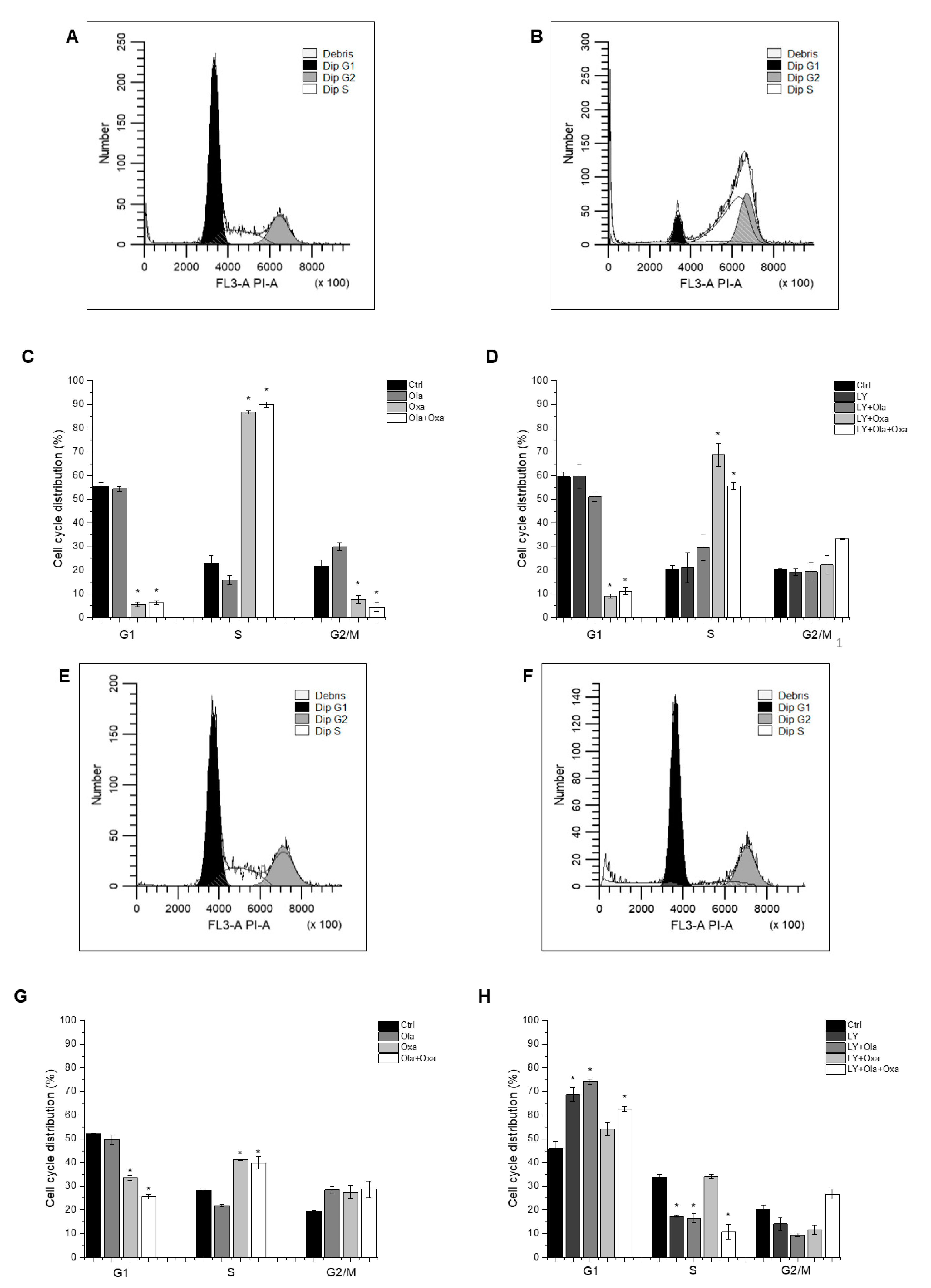
2.4. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on the Cell Cycle
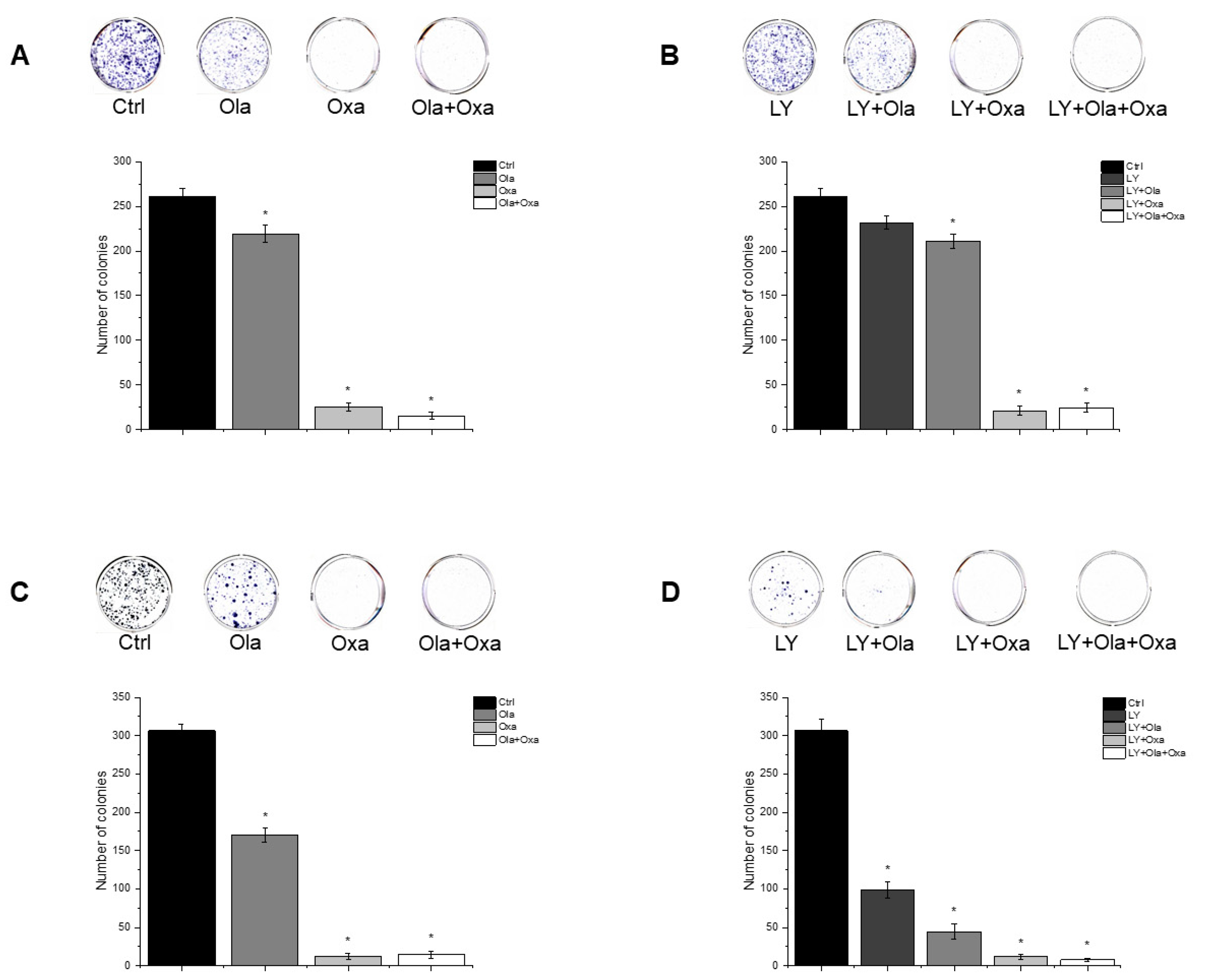
2.5. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on Colony Formation
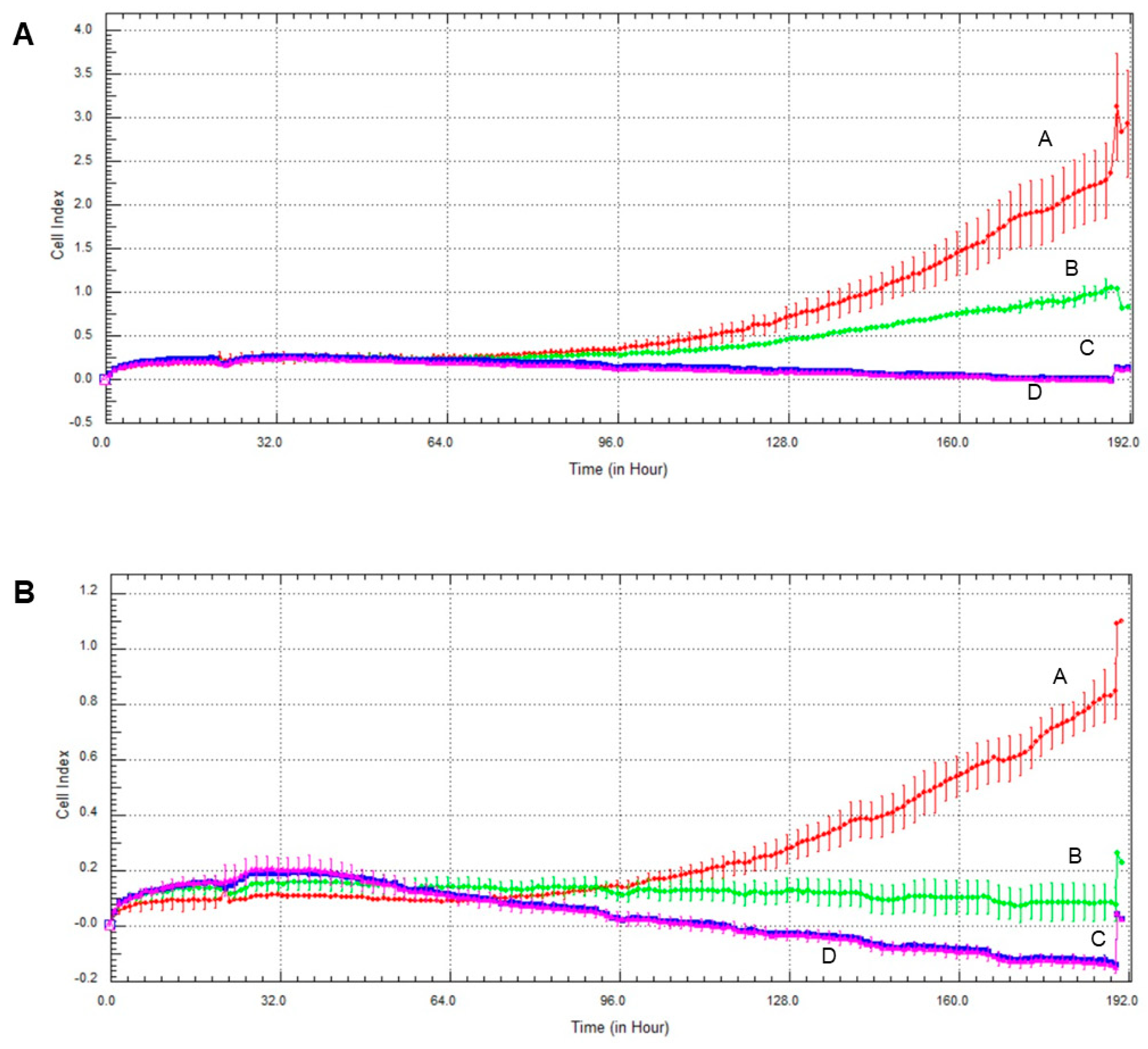
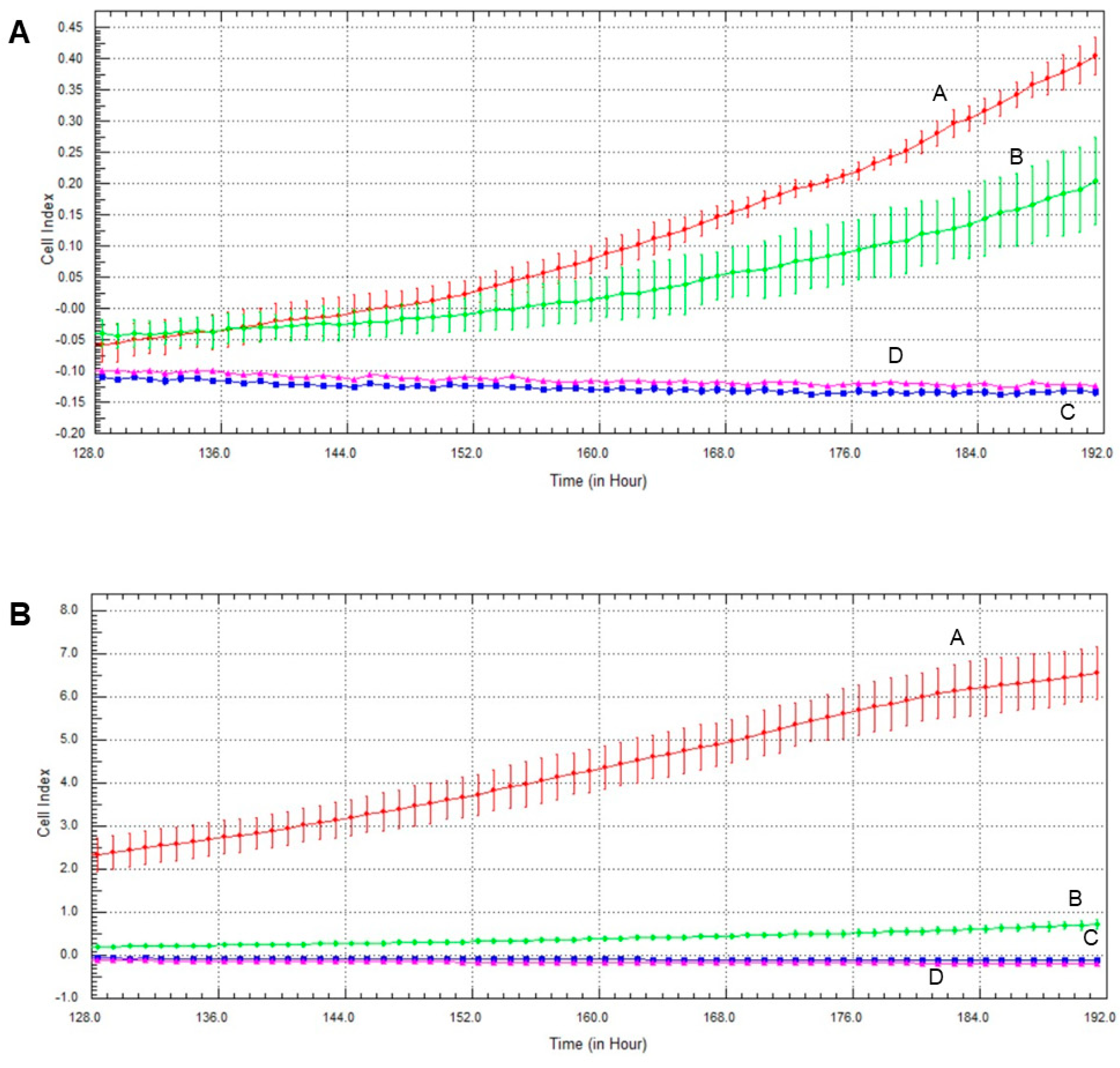
2.6. Effect of Olaparib and Oxaliplatin on Invasive Growth
2.7. Effect of Olaparib, Oxaliplatin and Akt Pathway Inhibitor on Invasive Growth
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Cultures
4.3. Survival Assay
4.4. Apoptosis Assay
4.5. Assay for Reactive Oxygen Species
4.6. Cell Cycle Analysis
4.7. Clonogenic Assay
4.8. Growth Measurement
4.9. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, A. Breast Cancer Metastasis and Drug Resistance, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2019; Volume 1152, p. 1. [Google Scholar]
- Donepudi, M.S.; Kondapalli, K.; Amos, S.J.; Venkanteshan, P. Breast cancer statistics and markers. J. Cancer Res. Ther. 2014, 10, 506. [Google Scholar]
- Dai, X.; Xiang, L.; Bai, Z. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J. Cancer 2016, 7, 1281–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareyeldin, R.M.; Gupta, I.; Al-Hashimi, I.; Al-Thawadi, H.A.; Farsi, H.F.A.; Vranic, S.; Moustafa, A.E.A. Gene expression and miRNAs profiling: Function and regulation in human epidermal growth factor receptor 2 (HER2)-positive breast cancer. Cancers 2019, 11, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeferlin, L.A.; Chalfant, C.E.; Park, M.A. Challenges in the treatment of triple negative and HER2-overexpressing breast cancer. J. Surg. Sci. 2013, 1, 3–7. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 74, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, R.K.; Parker, J.; Ahmed, S.; Qasem, E.; Mohammed, A.A.; Zeeshan, M.; Jehangir, E. Review of cisplatin and oxaliplatin in current immunogenic and monoclonalantibody treatments. World J. Oncol. 2014, 5, 97–108. [Google Scholar] [PubMed] [Green Version]
- Kang, X.; Xiao, H.H.; Song, H.Q.; Jing, X.B.; Yan, L.S.; Qi, R. Advances in drug delivery system for platinum agents based combination therapy. Cancer Biol. Med. 2015, 12, 362–374. [Google Scholar]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [Green Version]
- Seetharam, R.N.; Sood, A.; Goel, S. Oxaliplatin: Pre-clinical perspectives on the mechanisms of action, response and resistance. Ecancermedicalscience 2009, 3, 153. [Google Scholar]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Turk, A.; Wisinski, K.B. PARP Inhibition in BRCA-mutant breast cancer. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Ringley, J.T.; Moore, D.C.; Patel, J.; Rose, M.S. Poly (ADP-ribose) polymerase inhibitors in the management of ovarian cancer: A Drug Class Review. Pharm. Ther. 2018, 43, 549–556. [Google Scholar]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Lux, M.P.; Fasching, P.A.; Schrauder, M.G.; Hein, A.; Jud, S.M.; Rauh, C.; Beckmann, M.W. The PI3K pathway: Background and Ttreatment approaches. Breast Care 2016, 11, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Arafa, E.S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinine up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019, 30, x12–x20. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A History of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Cseh, A.M.; Fabian, Z.; Quintana-Cabrera, R.; Szabo, A.; Eros, K.; Soriano, M.E.; Gallyas, F.; Scorrano, L.; Sumegi, B. PARP inhibitor PJ34 protects mitochondria and induces DNA-damage mediated apoptosis in combination with cisplatin or temozolomide in B16F10 melanoma cells. Front. Physiol. 2019, 10, 538. [Google Scholar] [CrossRef] [Green Version]
- Garutti, M.; Pelizzari, G.; Bartoletti, M.; Malfatti, M.C.; Gerratana, L.; Tell, G.; Puglisi, F. Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors. Int. J. Mol. Sci. 2019, 20, 3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O'Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallyas, F., Jr.; Sumegi, B. Mitochondrial Protection by PARP Inhibition. Int. J. Mol. Sci. 2020, 21, 2767. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Hays, J.L.; Chiou, V.L.; Annunziata, C.M.; Swisher, E.M.; Harrell, M.I.; Yu, M.; Gordon, N.; Sissung, T.M.; Ji, J.; et al. Phase I/Ib study of olaparib and carboplatin in women with triple negative breast cancer. Oncotarget 2017, 8, 79175–79187. [Google Scholar] [CrossRef] [Green Version]
- Michels, J.; Vitale, I.; Senovilla, L.; Enot, D.P.; Garcia, P.; Lissa, D.; Olaussen, K.A.; Brenner, C.; Soria, J.C.; Castedo, M.; et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2015, 12, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-H.; Xiong, M.; Chen, X.-P.; Xiao, Z.-Y.; Zhao, Y.-F.; Huang, Z.-Y. PJ34, an inhibitor of PARP-1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol. Rep. 2008, 20, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Movahhed-Komeili, T.; Fouladdel, S.; Barzegar, E.; Atashpour, S.; Ghahremani, M.H.; Ostad, S.N.; Madjd, Z.; Azizi, E. PI3K/Akt inhibition and down-regulation of BCRP resensitize MCF7 breast cancer cell line to mitotraxane chemotherapy. IRAN J. Basic Med. Sci. 2015, 18, 472–477. [Google Scholar]
- Imai, Y.; Yoshimori, M.; Fukuda, K.; Yamagishi, H.; Ueda, Y. The PI3K/Akt inhibitor LY294002 reverses BCRP-mediated drug resistance without affecting BCRP translocation. Oncol. Rep. 2012, 27, 1703–1709. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, Q.; Hu, Y.; Zhang, J. Antitumor effects and mechanisms of olaparib in combination with carboplatin and BKM120 on human triple-negative breast cancer cells. Oncol. Rep. 2018, 40, 3223–3234. [Google Scholar] [CrossRef]
- Stankovic, J.S.K.; Selakovic, D.; Mihailovic, V.; Rosic, G. Antioxidant Supplementation in the Treatment of Neurotoxicity Induced by Platinum-Based Chemotherapeutics-A Review. Int. J. Mol. Sci. 2020, 21, 7753. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Zheng, Y.; Yan, Y.; Bargonetti, J.; Foster, D.A. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 2006, 25, 7305–7310. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.W.; Shewach, D.S. Radiosensitization by gemcitabine in p53 wild-type and mutant MCF7 breast carcinoma cell lines. Clin. Cancer Res. 2001, 7, 2581–2589. [Google Scholar]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.; Rida, P.C.G.; Aneja, R. Heading off with the herd: How cancer cells might maneuver supernumerary centrosomes for directional migration. Cancer Metastasis Rev. 2013, 32, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Golubev, A. Transition probability in cell proliferation, stochasticity in cell differentiation, and the restriction point of the cell cycle in one package. Prog. Biophys. Mol. Biol. 2012, 110, 87–96. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreidesz, K.; Koszegi, B.; Kovacs, D.; Bagone Vantus, V.; Gallyas, F.; Kovacs, K. Effect of Oxaliplatin, Olaparib and LY294002 in Combination on Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 2056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042056
Andreidesz K, Koszegi B, Kovacs D, Bagone Vantus V, Gallyas F, Kovacs K. Effect of Oxaliplatin, Olaparib and LY294002 in Combination on Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(4):2056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042056
Chicago/Turabian StyleAndreidesz, Kitti, Balazs Koszegi, Dominika Kovacs, Viola Bagone Vantus, Ferenc Gallyas, and Krisztina Kovacs. 2021. "Effect of Oxaliplatin, Olaparib and LY294002 in Combination on Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 4: 2056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042056