
Heightened Crescentic Glomerulonephritis in Immune Challenged 129sv Mice Is TGF-β/Smad3 Dependent

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
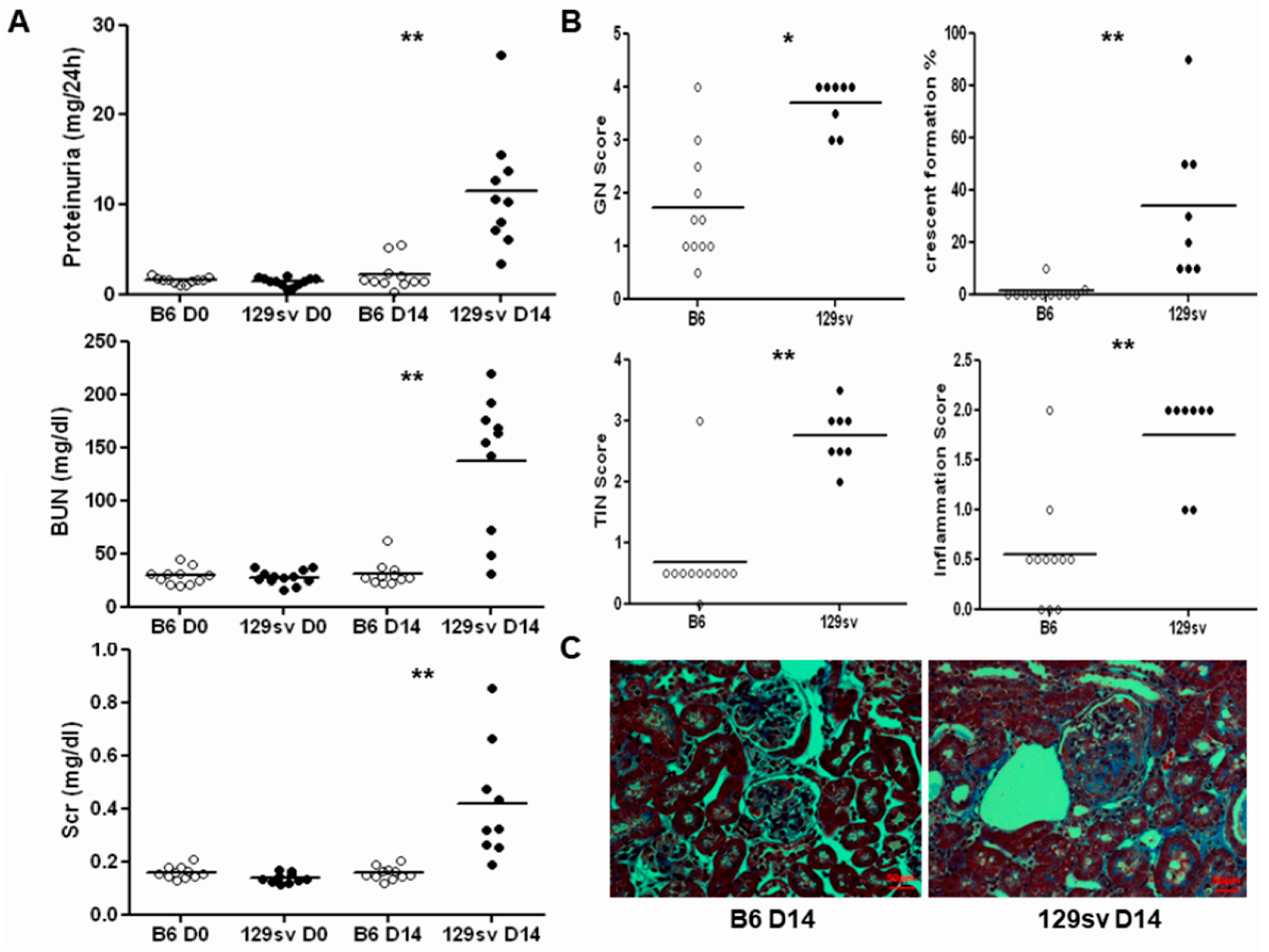
2.1. 129sv Mice Are More Susceptible to Anti-GBM Nephritis Than C57BL/6J Mice
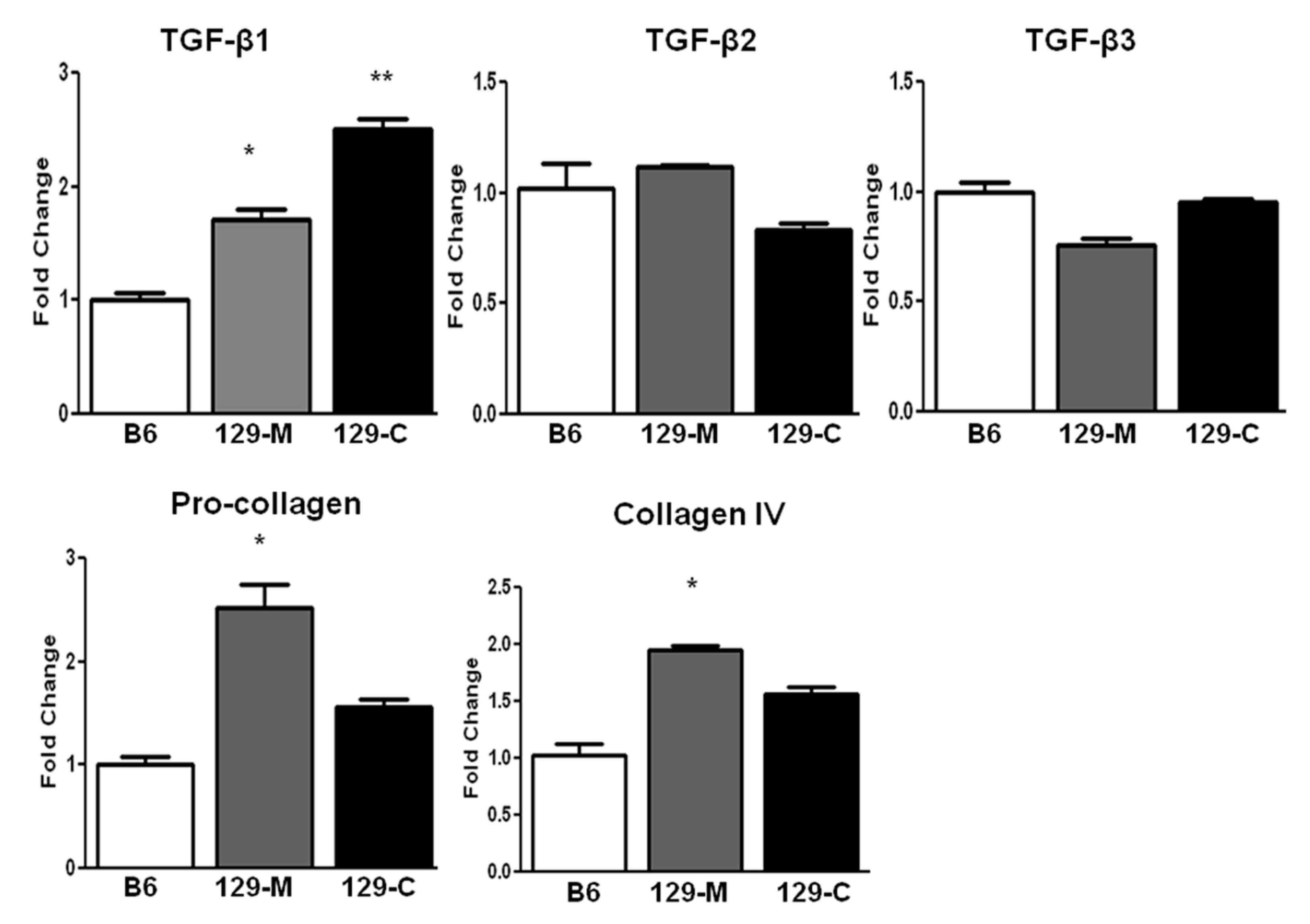
2.2. TGF-β1 and Col1a1/Col4a1 mRNA Expression Are Elevated in the Kidneys of 129sv Mice
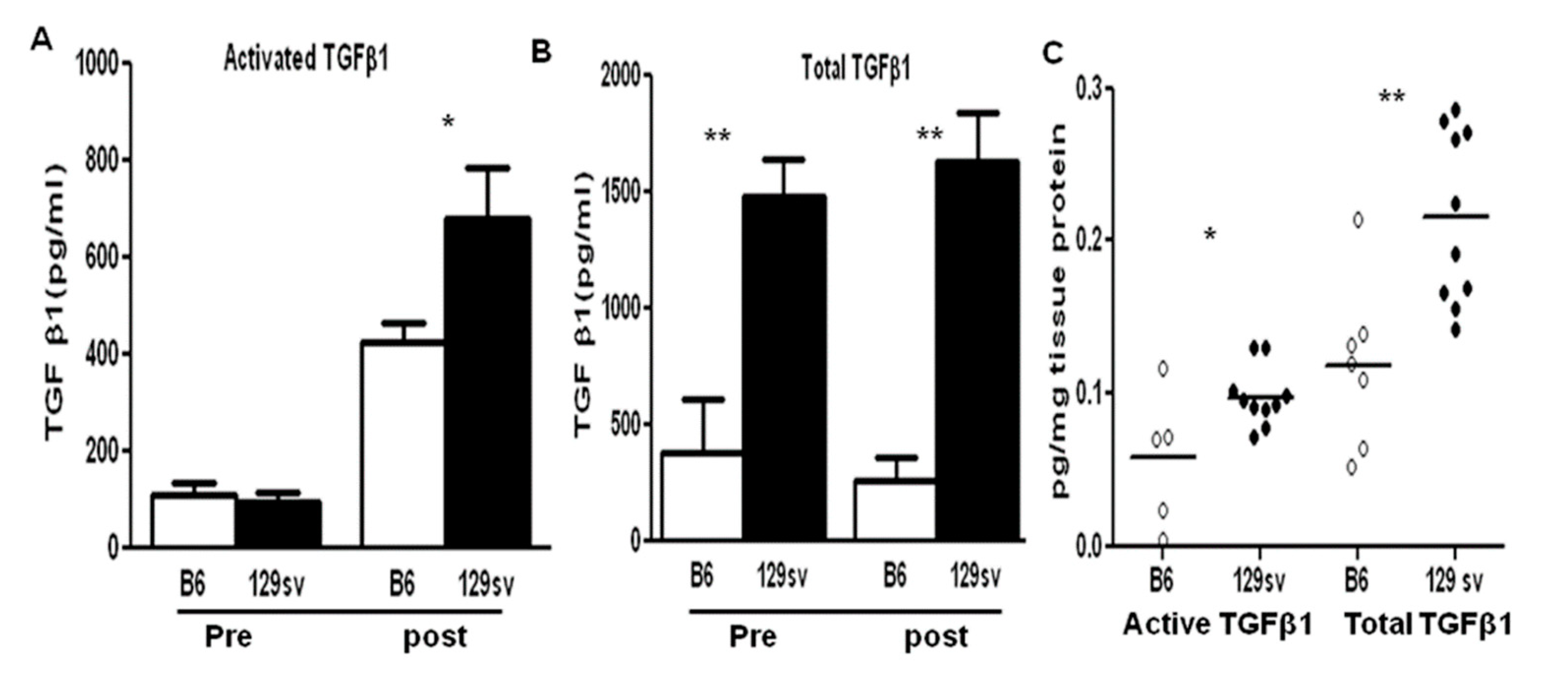
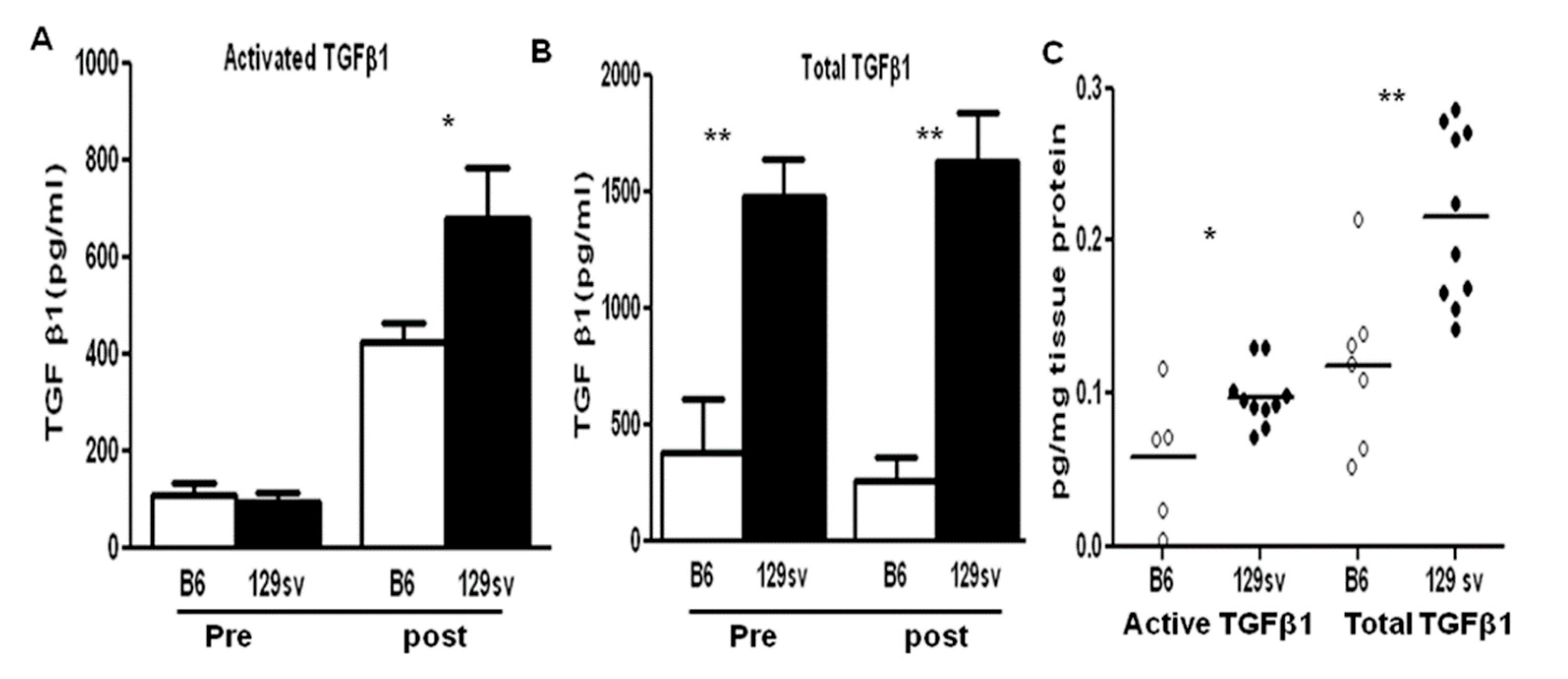
2.3. TGF-β1 Protein Levels Are Elevated in Both the Sera and Renal Tissue of 129sv Mice with Anti-GBM Nephritis
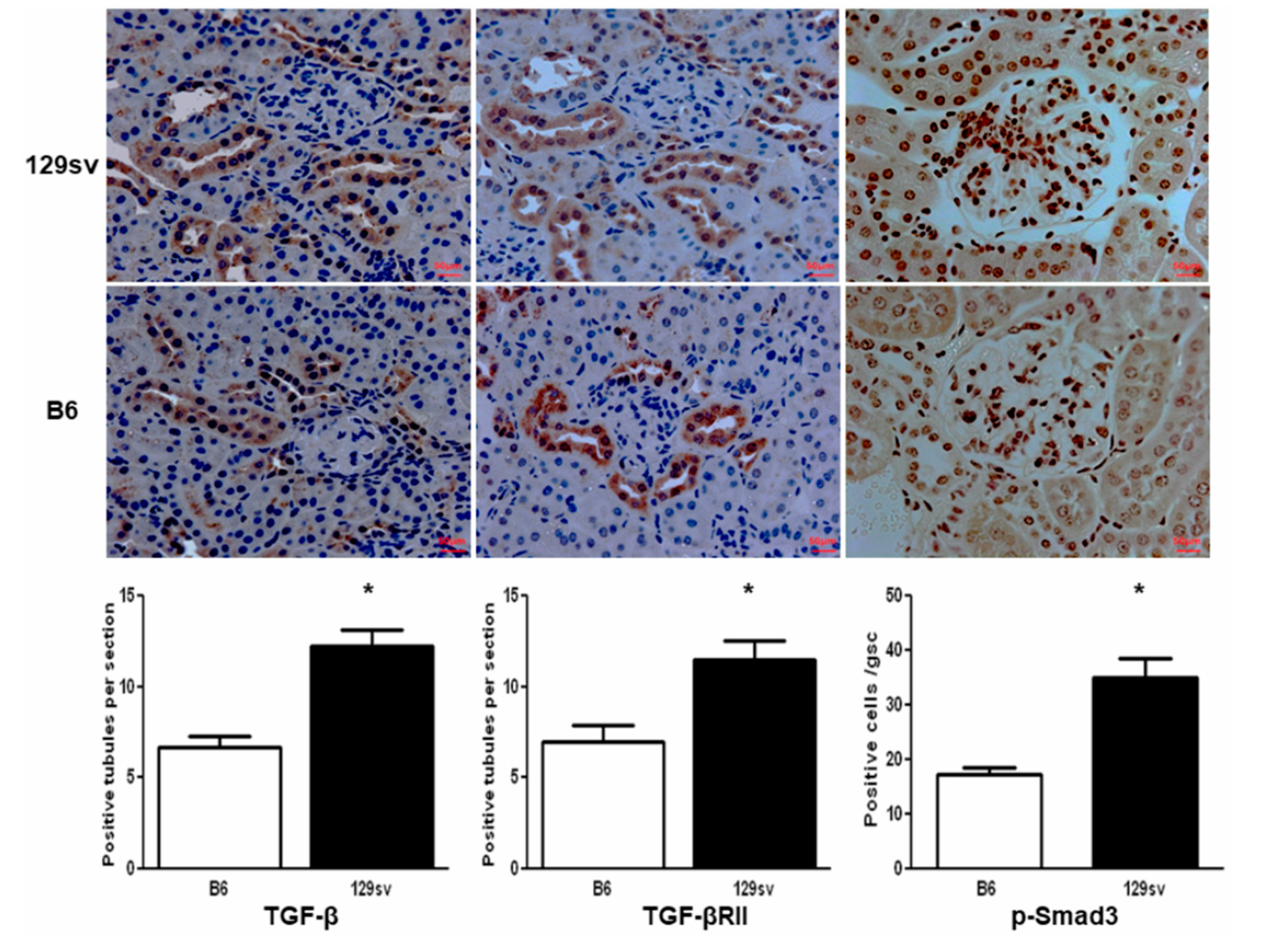
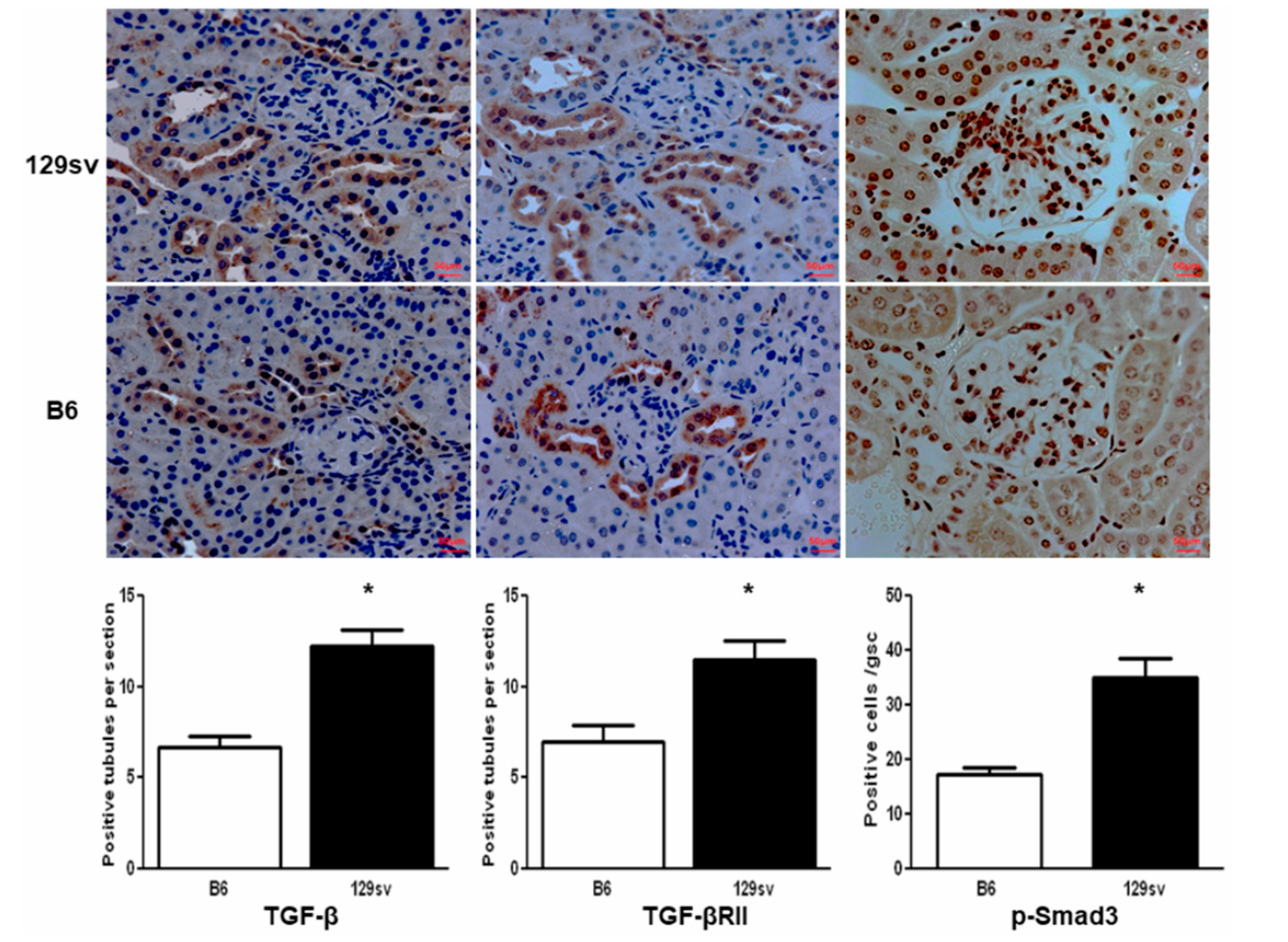
2.4. TGF-β1/SMAD Signaling Molecules Are Increased in the Kidneys of 129sv Mice
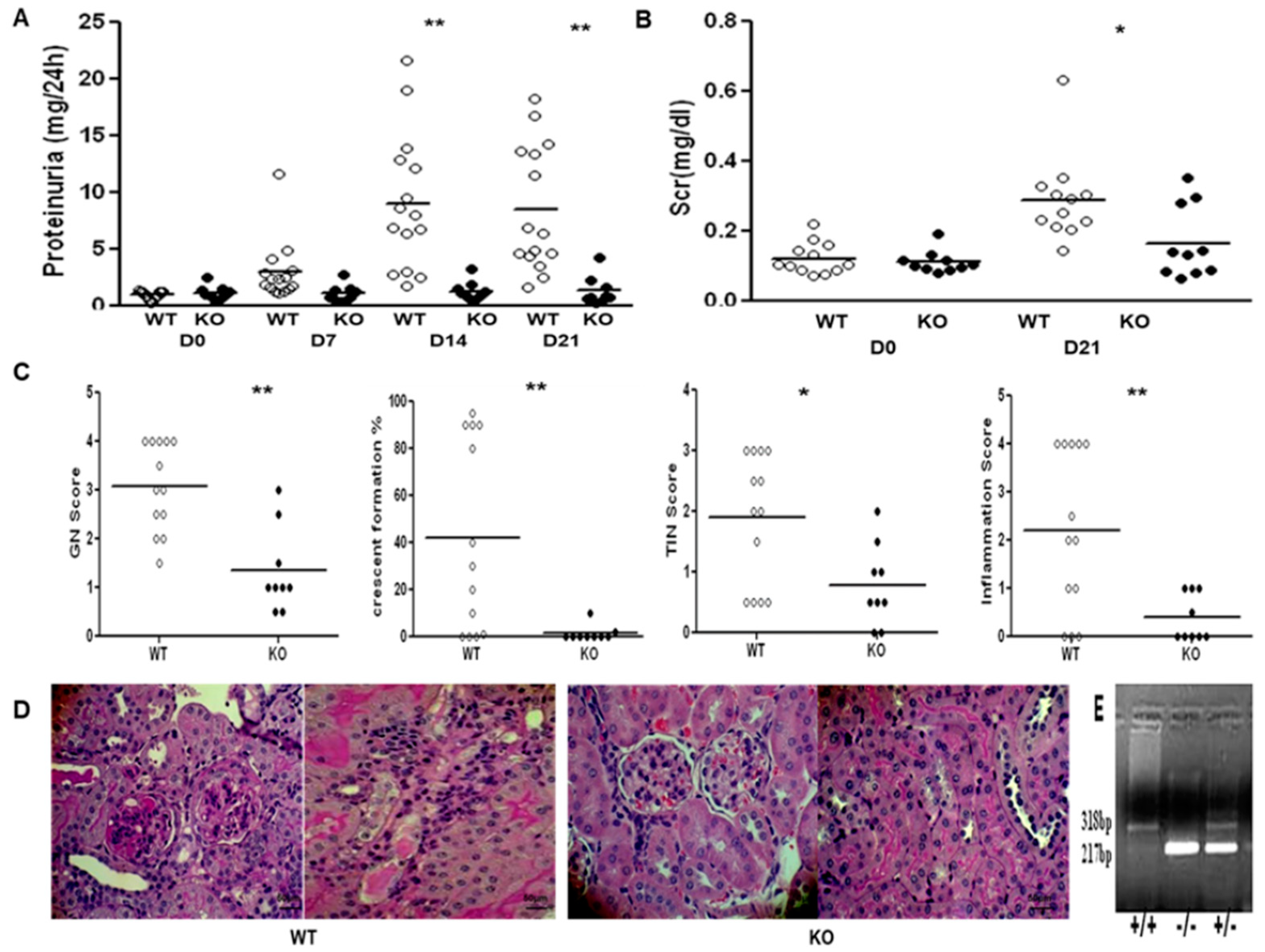
2.5. SMAD3 Deficiency Protects 129sv Mice from Anti-GBM Nephritis
3. Discussion
4. Materials and Methods
4.1. Animals and Anti-GBM Nephritis
4.2. Reagents
4.3. Genotyping of 129.smad3−/− Mice and Renal Tgf-β and Collagen mRNA Level Detection Using Real-Time PCR
4.4. Pathology and Immunohistochemistry
4.5. ELISA
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Koka, V.; Lan, H.Y. Transforming growth factor β and Smad signaling in kidney diseases. Nephrology 2005, 10, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, M.K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Nat. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.J.; Jha, S.; Ling, H.; Pozzi, A.; Ledbetter, S.; Fogo, A.B. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int. 2004, 65, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Mohan, C. Mechanisms of tissue injury the pathogenesis of lupus nephritis 2010. In Systemic Lupus Erythematosus, 5th ed.; Lahita, R.G., Koike, T., Buyon, J., Tsokos, G., Eds.; Elsevier: New York, NY, USA; pp. 453–473.
- Becker-Merok, A.; Eilertsen, G.Ø.; Nossent, J.C. Levels of transforming growth factor-beta are low in systemic lupus erythematosus patients with active disease. J. Rheumatol. 2010, 37, 2039–2045. [Google Scholar] [CrossRef]
- Hammad, A.M.; Youssef, H.M.; El-Arman, M.M. Transforming growth factor beta 1 in children with systemic lupus erythematosus: A possible relation with clinical presentation of lupus nephritis. Lupus 2006, 15, 608–612. [Google Scholar] [CrossRef]
- Atfy, M.; Amr, G.E.; Elnaggar, A.M.; Labib, H.A.; Esh, A.; Elokely, A.M. Impact of CD4+CD25high regulatory T-cells and FoxP3 expression in the peripheral blood of patients with systemic lupus erythematosus. Egypt J. Immunol. 2009, 16, 117–126. [Google Scholar]
- Chen, D.Y.; Chen, Y.M.; Chen, H.H.; Hsieh, C.W.; Lin, C.C.; Lan, J.L. The associations of circulating CD4 (+) CD25 (high) regulatory T cells and TGF-β with disease activity and clinical course in patients with adult-onset Still’s disease. Connect. Tissue Res. 2010, 51, 370–377. [Google Scholar] [CrossRef]
- Yang, C.W.; Hsueh, S.; Wu, M.S.; Lai, P.C.; Huang, J.Y.; Wu, C.H.; Hu, S.A.; Chen, J.F.; Huang, C.C. Glomerular transforming growth factor-beta1 mRNA as a marker of glomerulosclerosis-application in renal biopsies. Nephron 1997, 77, 290–297. [Google Scholar] [CrossRef]
- Saxena, V.; Lienesch, D.W.; Zhou, M.; Bommireddy, R.; Azhar, M.; Doetschman, T.; Singh, R.R. Dual roles of immunoregulatory cytokine TGF-beta in the pathogenesis of autoimmunity-mediated organ damage. J. Immunol. 2008, 180, 1903–1912. [Google Scholar] [CrossRef] [Green Version]
- Pusey, C.D. Anti-glomerular basement membrane disease. Kidney Int. 2003, 64, 1535–1550. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Du, Y.; Mohan, C. Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin. Immunol. 2007, 124, 109–118. [Google Scholar] [CrossRef]
- Zhou, A.; Ueno, H.; Shimomura, M.; Tanaka, R.; Shirakawa, T.; Nakamura, H.; Matsuo, M.; Iijima, K. Blockade of TGF-beta action ameliorates renal dysfunction and histologic progression in anti-GBM nephritis. Kidney Int. 2003, 64, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Song, C.Y.; Kim, B.C.; Hong, H.K.; Lee, H.S. TGF-b type II receptor deficiency prevents renal injury via decrease in ERK activity in crescentic glomerulonephritis. Kidney Int. 2007, 71, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Sharma, R.; Wang, H.; Zhou, X.J.; Mohan, C. Strain distribution pattern of susceptibility to immune-mediated nephritis. J. Immunol. 2004, 172, 5047–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Rahman, Z.S.; Xie, S.; Zhu, J.; Du, Y.; Qin, X.; Zhou, H.; Zhou, X.J.; Mohan, C. Strain distribution pattern of immune nephritis—A follow-up study. Int. Immunol. 2008, 20, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Massagu’e, J. TGF-Signal Transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar]
- Brown, K.A.; Pietenpol, J.A.; Moses, H.L. A Tale of Two Proteins: Differential Roles and Regulation of Smad2 and Smad3 in TGF-b Signaling. J. Cell. Biochem. 2007, 101, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Ka, S.M.; Huang, X.R.; Lan, H.Y.; Tsai, P.Y.; Yang, S.M.; Shui, H.A.; Chen, A. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 2007, 18, 1777–1788. [Google Scholar] [CrossRef]
- Yaswen, L.; Kulkarni, A.B.; Fredrickson, T.; Mittleman, B.; Schiffman, R.; Payne, S.; Longenecker, G.; Mozes, E.; Karlsson, S. Autoimmune manifestations in the transforming growth factor-beta 1 knockout mouse. Blood 1996, 87, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Raz, E.; Dudler, J.; Lotz, M.; Baird, S.M.; Berry, C.C.; Eisenberg, R.A.; Carson, D.A. Modulation of disease activity in murine systemic lupus erythematosus by cytokine gene delivery. Lupus 1995, 4, 286–292. [Google Scholar] [CrossRef]
- Jin, Y.X.; Xu, L.Y.; Guo, H.; Ishikawa, M.; Link, H.; Xiao, B.G. TGF-beta1 inhibits protracted-relapsing experimental autoimmune encephalomyelitis by activating dendritic cells. J. Autoimmun. 2000, 14, 213–220. [Google Scholar] [CrossRef]
- Piccirillo, C.A.; Chang, Y.; Prud’homme, G.J. TGF-beta1 somatic gene therapy prevents autoimmune disease in nonobese diabetic mice. J. Immunol. 1998, 161, 3950–3956. [Google Scholar] [PubMed]
- Chen, W.; Jin, W.; Cook, M.; Weiner, H.L.; Wahl, S.M. Oral delivery of group A streptococcal cell walls augments circulating TGF-beta and suppresses streptococcal cell wall arthritis. J. Immunol. 1998, 161, 6297–6304. [Google Scholar]
- Kanamaru, Y.; Nakao, A.; Mamura, M.; Suzuki, Y.; Shirato, I.; Okumura, K.; Tomino, Y.; Ra, C. Blockade of TGF-beta signaling in T cells prevents the development of experimental glomerulonephritis. J. Immunol. 2001, 166, 2818–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.R.; Chung, A.C.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-beta1 protects against crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Chung, A.C.; Wang, X.J.; Lai, K.N.; Lan, H.Y. Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Renal. Physiol. 2008, 295, F118–F127. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-β receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef]
- Sugimoto, H.; LeBleu, V.S.; Bosukonda, D.; Keck, P.; Taduri, G.; Bechtel, W.; Okada, H.; Carlson, W., Jr.; Bey, P.; Rusckowski, M.; et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 2012, 18, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Flyvbjerg, A.; Rasch, R.; Bak, M.; Logan, A. Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J. Endocrinol. 2001, 170, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. 2006 IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Merline, R.; Lazaroski, S.; Babelova, A.; Tsalastra-Greu, W.; Pfeilschifter, J.; Schlute, K.D.; Gunther, A.; Iozzo, R.V.; Schaefer, R.M.; Schaefer, L. Decorin deficiency in diabetic mice: Aggravation of nephropathy due to overexpression of profibrotic factors, enhanced apoptosis and mononuclear cell infiltration. J. Physiol. Pharmacol. 2009, 60 (Suppl. 4), 5–13. [Google Scholar]
- Zhang, Z.; Wu, F.; Zheng, F.; Li, H. Adenovirus-mediated decorin gene transfection has therapeutic effects in a streptozocin-induced diabetic rat model. Nephron. Exp. Nephrol. 2010, 116, e11–e21. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Mu, W.; Tomita, N.; Huang, X.R.; Li, J.H.; Zhu, H.J.; Morishita, R.; Johnson, R.J. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 2003, 14, 1535–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Zhen, J.; Du, Y.; Zhou, J.K.; Peng, A.; Vaziri, N.D.; Mohan, C.; Xu, Y.; Zhou, X.J. Green tea polyphenol (-)-epigallocatechin-3-gallate restores Nrf2 activity and ameliorates crescentic glomerulonephritis. PLoS ONE 2015, 10, e0119543. [Google Scholar]
- Qin, L.; Du, Y.; Ding, H.; Haque, A.; Hicks, J.; Pedroza, C.; Mohan, C. Bradykinin 1 receptor blockade subdues systemic autoimmunity, renal inflammation, and blood pressure in murine lupus nephritis. Arthritis Res. Ther. 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, A.; Ye, T.; Rakheja, D.; Tu, Y.; Wang, T.; Du, Y.; Zhou, J.K.; Vaziri, N.D.; Hu, Z.; Mohan, C.; et al. The green tea polyphenol (-)-epigallocatechin-3-gallate ameliorates experimental immune-mediated glomerulonephritis. Kidney Int. 2011, 80, 601–611. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Xie, C.; Ravikumar, S.; Orme, J.; Li, L.; Zhou, X.J.; Mohan, C. Heightened Crescentic Glomerulonephritis in Immune Challenged 129sv Mice Is TGF-β/Smad3 Dependent. Int. J. Mol. Sci. 2021, 22, 2059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042059
Du Y, Xie C, Ravikumar S, Orme J, Li L, Zhou XJ, Mohan C. Heightened Crescentic Glomerulonephritis in Immune Challenged 129sv Mice Is TGF-β/Smad3 Dependent. International Journal of Molecular Sciences. 2021; 22(4):2059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042059
Chicago/Turabian StyleDu, Yong, Chun Xie, Sneha Ravikumar, Jacob Orme, Li Li, Xin J Zhou, and Chandra Mohan. 2021. "Heightened Crescentic Glomerulonephritis in Immune Challenged 129sv Mice Is TGF-β/Smad3 Dependent" International Journal of Molecular Sciences 22, no. 4: 2059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042059