
Functional Characterization of Antithrombin Mutations by Monitoring of Thrombin Inhibition Kinetics

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Study Population
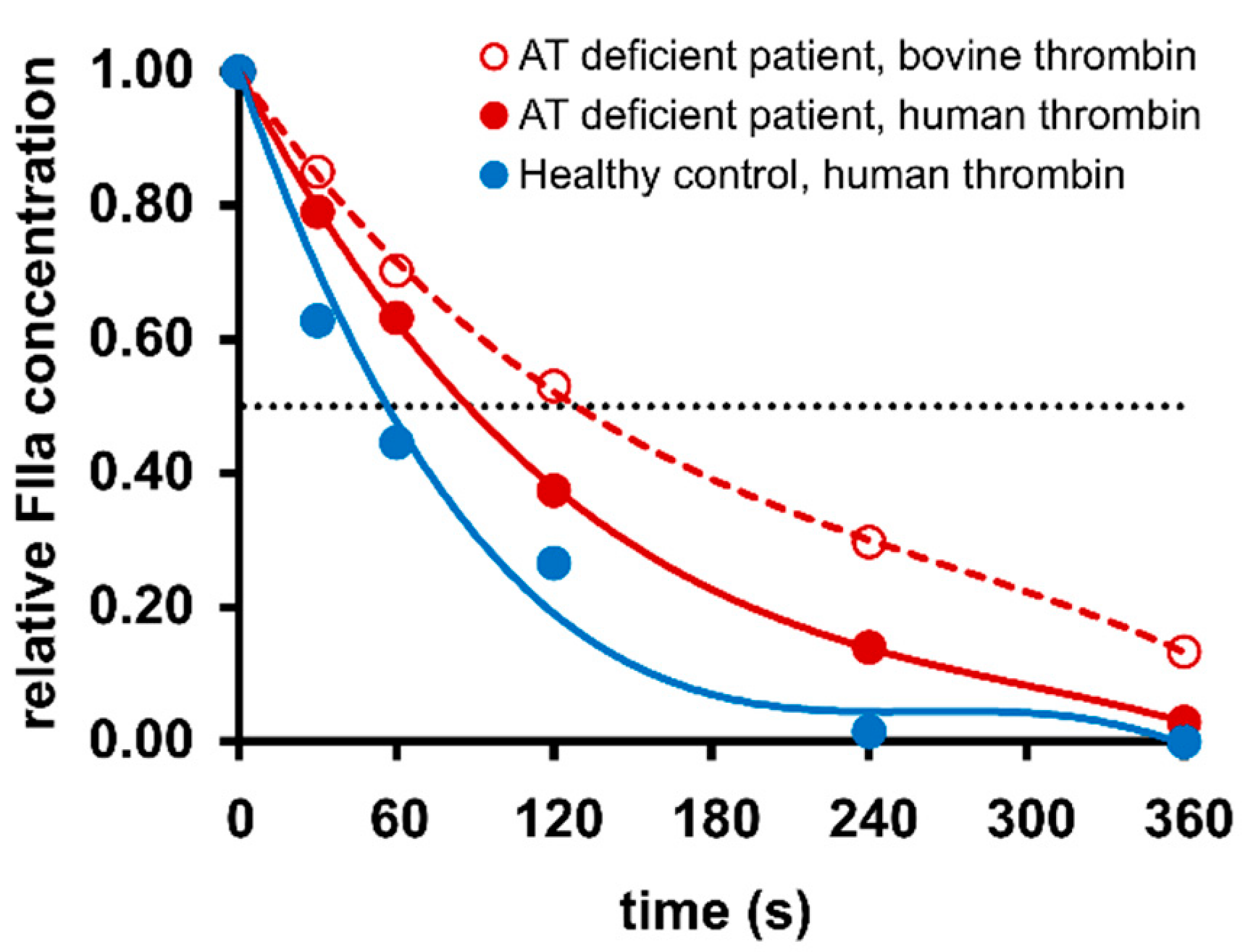
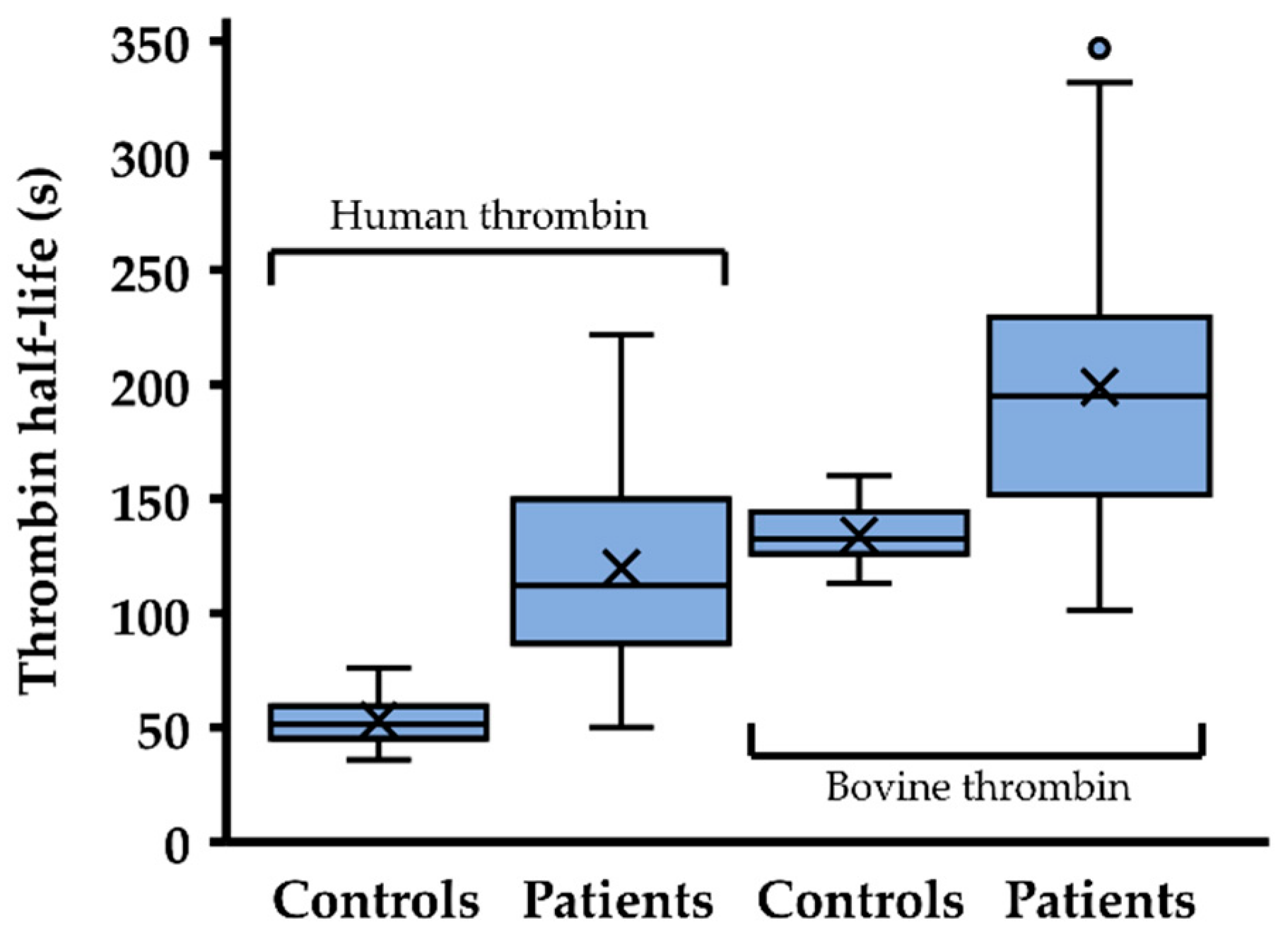
2.2. Thrombin Inhibition Capacity Is Reduced in Most Patients with Antithrombin Mutations
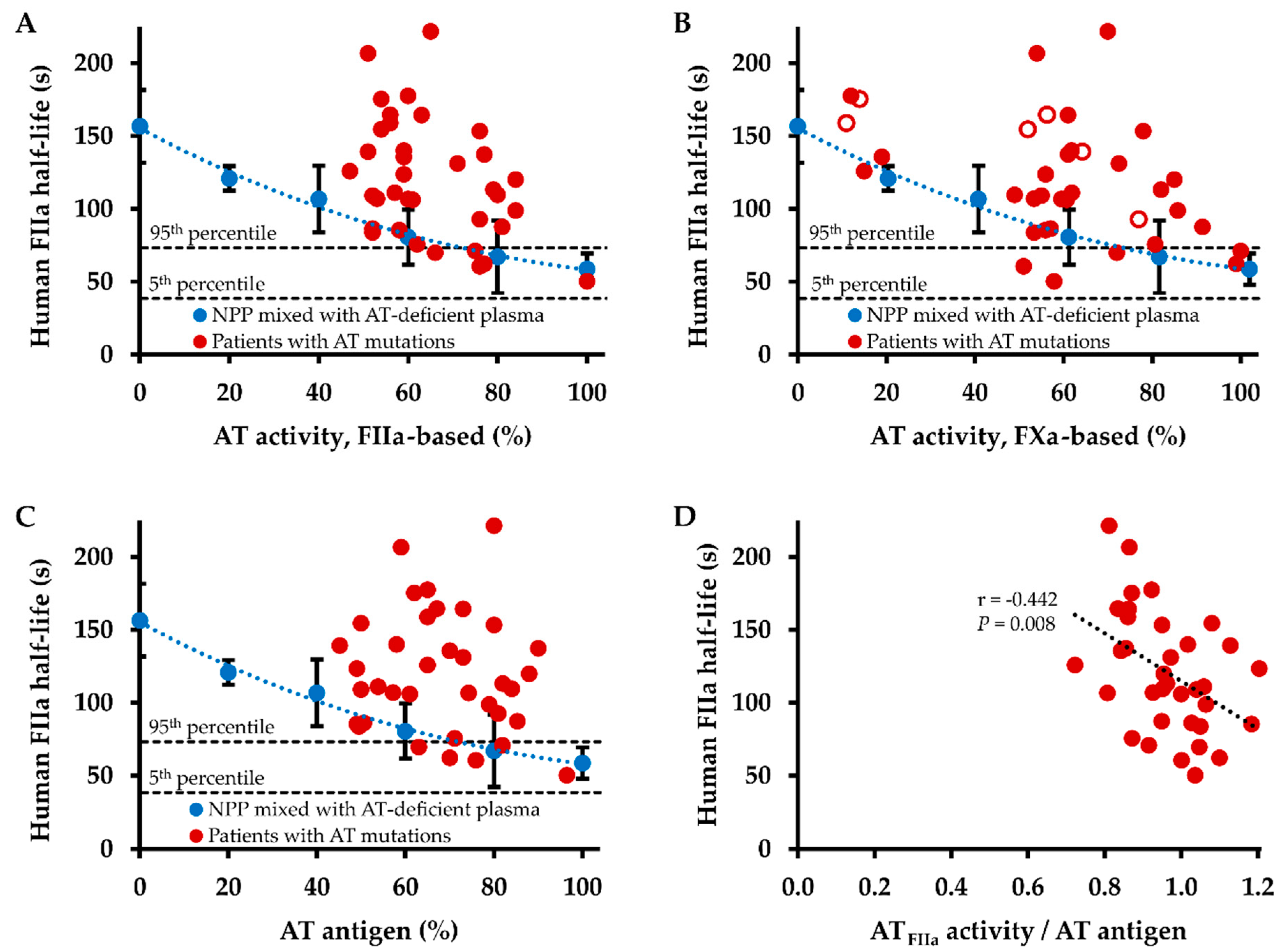
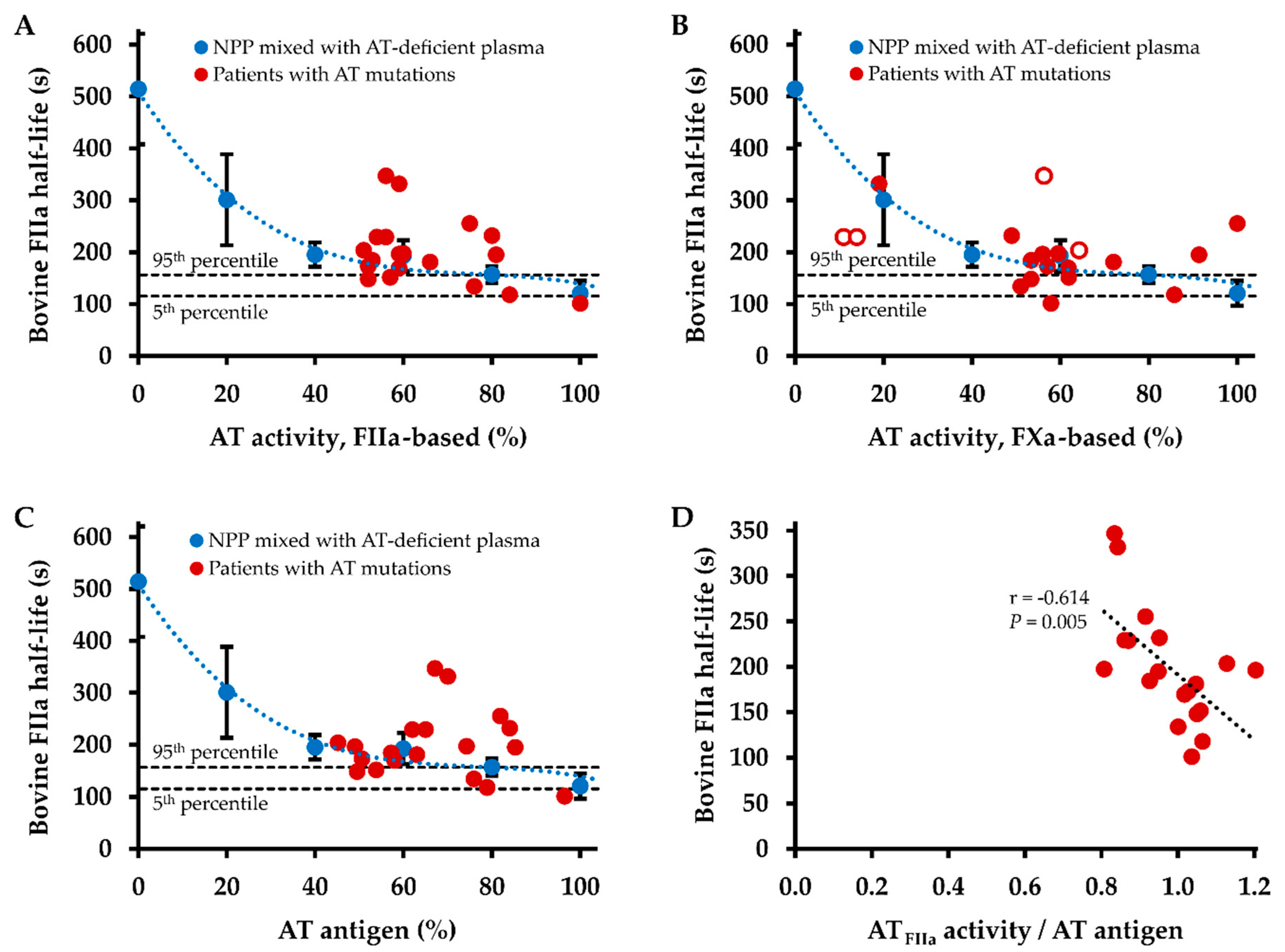
2.3. Influence of Antithrombin Level on Thrombin Inhibition Kinetics
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Patients and Samples
4.3. Laboratory Analysis of Blood Samples
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huntington, J.A. Natural inhibitors of thrombin. Thromb. Haemost. 2014, 111, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, J.C.; Beaulieu, L.M.; Huntington, J.A.; Church, F.C. Serpins in thrombosis, haemostasis and fibrinolysis. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 102–115. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Moll, S. Inherited antithrombin deficiency: A review. Haemophilia 2008, 14, 1229–1239. [Google Scholar] [CrossRef]
- Bauer, K.A.; Nguyen-Cao, T.M.; Spears, J.B. Issues in the Diagnosis and Management of Hereditary Antithrombin Deficiency. Ann. Pharmacother. 2016, 50, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Javela, K.; Engelbarth, S.; Hiltunen, L.; Mustonen, P.; Puurunen, M. Great discrepancy in antithrombin activity measured using five commercially available functional assays. Thromb. Res. 2013, 132, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Luxembourg, B.; Lindhoff-Last, E. Genomische Diagnostik thrombophiler Gerinnungsstörungen bei Frauen: Klinische Relevanz [Genomic diagnosis of thrombophilia in women: Clinical relevance]. Hamostaseologie 2007, 27, 22–31. [Google Scholar]
- Beeck, H.; Nagel, D.; Pindur, G.; Scharrer, I.; Preiss, A.; Seiler, D.; Hellstern, P. Measurement of antithrombin activity by thrombin-based and by factor Xa-based chromogenic substrate assays. Blood Coagul. Fibrinolysis 2000, 11, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.; Vicente, V. Puzzling questions on antithrombin: Diagnostic limitations and real incidence in venous and arterial thrombosis. Thromb. Res. 2015, 135, 1047–1048. [Google Scholar] [CrossRef]
- Merz, M.; Böhm-Weigert, M.; Braun, S.; Cooper, P.C.; Fischer, R.; Hickey, K.; Steffan, A.; Kemkes-Matthes, B.; Kitchen, S. Clinical multicenter evaluation of a new FXa-based Antithrombin assay. Int. J. Lab. Hematol. 2011, 33, 498–506. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Schulman, S.; Egberg, N.; Antovic, J.; Blombäck, N. Discrepancy between antithrombin activity methods revealed in Antithrombin Stockholm: Do factor Xa-based methods overestimate antithrombin activity in some patients? Blood 2002, 99, 2271–2272. [Google Scholar] [CrossRef]
- Harbrecht, U.; Fimmers, R.; Oldenburg, J.; Mayer, G.; Rühl, H.; Müller, J.; Pötzsch, B. Thrombin inhibition profiles in healthy individuals and thrombophilic patients. Thromb. Haemost. 2012, 107, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G. Thrombin generation in hemorrhage control and vascular occlusion. Circulation 2011, 124, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Becher, T.; Braunstein, J.; Berdel, P.; Gravius, S.; Rohrbach, F.; Oldenburg, J.; Mayer, G.; Pötzsch, B. Profiling of active thrombin in human blood by supramolecular complexes. Angew. Chem. Int. Ed. Engl. 2011, 50, 6075–6078. [Google Scholar] [CrossRef] [PubMed]
- Sas, G.; Petö, I.; Bánhegyi, D.; Blaskó, G.; Domján, G. Heterogeneity of the “classical” antithrombin III deficiency. Thromb. Haemost. 1980, 43, 133–136. [Google Scholar] [CrossRef]
- Daly, M.; O’Meara, A.; Hallinan, F.M. Identification and characterization of a new antithrombin III familial variant (AT Dublin) with possible increased frequency in children with cancer. Br. J. Haematol. 1987, 65, 457–462. [Google Scholar] [CrossRef]
- Caspers, M.; Pavlova, A.; Driesen, J.; Harbrecht, U.; Klamroth, R.; Kadar, J.; Fischer, R.; Matthes, B.K.; Oldenburg, J. Deficiencies of antithrombin, protein C and protein S-practical experience in genetic analysis of a large patient cohort. Thromb. Haemost. 2012, 108, 247–257. [Google Scholar] [CrossRef]
- Delev, D.; Geisen, C.; Spannagl, M.; Krause, M.; Miesbach, W.; Heller, C.; Bergmann, F.; Schmeink, U.; Grossmann, R.; Lindhoff-Last, E.; et al. Molecular basis of antithrombin deficiency. Thromb. Haemost. 2011, 105, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Duckert, F. Influence of heparin cofactor II (HCII) on the determination of antithrombin III (AT). Thromb. Res. 1985, 40, 571–576. [Google Scholar] [CrossRef]
- Gausman, J.N.; Marlar, R.A. Assessment of Hereditary Thrombophilia: Performance of Antithrombin (AT) Testing. Methods Mol. Biol. 2017, 1646, 161–167. [Google Scholar] [CrossRef]
- Owen, M.C.; Borg, J.Y.; Soria, C.; Soria, J.; Caen, J.; Carrell, R.W. Heparin binding defect in a new antithrombin III variant: Rouen, 47 Arg to His. Blood 1987, 69, 1275–1279. [Google Scholar] [CrossRef] [Green Version]
- Perry, D.; Daly, M.; Harper, P.; Tait, R.; Price, J.; Walker, I.; Carrell, R. Antithrombin Cambridge II, 384 Ala to Ser. Further evidence of the role of the reactive centre loop in the inhibitory function of the serpins. FEBS Lett. 1991, 285, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Corral, J.; Hernandez-Espinosa, D.; Soria, J.M.; Gonzalez-Conejero, R.; Ordonez, A.; Gonzalez-Porras, J.R.; Perez-Ceballos, E.; Lecumberri, R.; Sanchez, I.; Roldan, V.; et al. Antithrombin Cambridge II (A384S): An underestimated genetic risk factor for venous thrombosis. Blood 2007, 109, 4258–4263. [Google Scholar] [CrossRef]
- Olds, R.J.; Lane, D.A.; Boisclair, M.; Sas, G.; Bock, S.C.; Thein, S.L. Antithrombin Budapest 3. An antithrombin variant with reduced heparin affinity resulting from the substitution L99F. FEBS Lett. 1992, 300, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Tollefsen, D.M. Heparin cofactor II modulates the response to vascular injury. Arterioscler. Thrombin Vasc. Biol. 2007, 27, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kario KMatsuo, T.; Kobayashi, H. Heparin cofactor II deficiency in the elderly: Comparison with antithrombin III. Thromb. Res. 1992, 66, 489–498. [Google Scholar] [CrossRef]
- Toulon, P.; Bardin, J.M.; Blumenfeld, N. Increased heparin cofactor II levels in women taking oral contraceptives. Thromb. Haemost. 1990, 64, 365–368. [Google Scholar] [CrossRef]
- Puurunen, M.; Salo, P.; Engelbarth, S.; Javela, K.; Perola, M. Type II antithrombin deficiency caused by a founder mutation Pro73Leu in the Finnish population: Clinical picture. J. Thromb. Haemost. 2013, 11, 1844–1849. [Google Scholar] [CrossRef]
- Ozawa, T.; Takikawa, Y.; Niiya, K.; Fujiwara, T.; Suzuki, K.; Sato, S.; Sakuragawa, N. Antithrombin Morioka (Cys 95-Arg): A novel missense mutation causing type I antithrombin deficiency. Thromb. Haemost. 1997, 77, 403. [Google Scholar] [CrossRef] [PubMed]
- Rühl, H.; Schröder, L.; Müller, J.; Sukhitashvili, S.; Welz, J.; Kuhn, W.C.; Oldenburg, J.; Rudlowski, C.; Pötzsch, B. Impact of hormone-associated resistance to activated protein C on the thrombotic potential of oral contraceptives: A prospective observational study. PLoS ONE 2014, 9, e105007. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.J.; Schmolders, J.; Rommelspacher, Y.; Strauss, A.; Rühl, H.; Mayer, G.; Oldenburg, J.; Wirtz, D.C.; Müller, J.; Pötzsch, B. Activity Pattern Analysis Indicates Increased but Balanced Systemic Coagulation Activity in Response to Surgical Trauma. TH Open 2018, 2, e350–e356. [Google Scholar] [CrossRef] [Green Version]
- Rühl, H.; Berens, C.; Winterhagen, F.I.; Reda, S.; Müller, J.; Oldenburg, J.; Pötzsch, B. Increased Activated Protein C Response Rates Reduce the Thrombotic Risk of Factor V Leiden Carriers but Not of Prothrombin 20210G>A Carriers. Circ. Res. 2019, 125, 523–534. [Google Scholar] [CrossRef]
- Becher, T.; Müller, J.; Akin, I.; Baumann, S.; Stach, K.; Borggrefe, M.; Pötzsch, B.; Loßnitzer, D. Characterization of circulating thrombin in patients with septic shock: A prospective observational study. J. Thromb. Thrombolysis 2020, 50, 90–97. [Google Scholar] [CrossRef]
- Rühl, H.; Friemann, A.M.; Reda, S.; Schwarz, N.; Winterhagen, F.I.; Berens, C.; Müller, J.; Oldenburg, J.; Pötzsch, B. Activated Factor XI is Increased in Plasma in Response to Surgical Trauma but not to Recombinant Activated FVII-Induced Thrombin Formation. J. Atheroscler. Thromb. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rühl, H.; Reda, S.; Müller, J.; Oldenburg, J.; Pötzsch, B. Activated Factor X-Based versus Thrombin-Based Antithrombin Testing in Thrombophilia Workup in the DOAC Era. Thromb. Haemost. 2018, 118, 381–387. [Google Scholar] [CrossRef]
- XLSTAT, version 2019.2.1; Statistical and Data Analysis Solution; Addinsoft: Boston, MA, USA, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Human Thrombin Inhibition Assessed | Bovine Thrombin Inhibition Assessed | |
|---|---|---|---|
| Demographics | Mean age (range), years | 35 (4–82) | 34 (4–71) |
| Males/females | 12/24 | 5/14 | |
| Thrombosis history of study subjects 1 | Asymptomatic subjects | 11 | 4 |
| Deep vein thrombosis | 19 | 13 | |
| Pulmonary embolism | 11 | 5 | |
| Other venous thrombosis | 5 | 4 | |
| Arterial thrombosis | 2 | 1 | |
| SERPINC1 mutations 2 | c.89T>A | 3 | - |
| c.391C>T (thereof homozygous) | 11 (5) | 6 (3) | |
| c.462_464del | 3 | 3 | |
| c.1058C>T | 2 | 2 | |
| c.1153G>A | 3 | 2 | |
| c.74G>A, c.236G>A, c.470A>G, c.481C>T, c.1347del G, c.1355T>C | 1 each | 1 each | |
| c.248T>A, c.331T>A, c.379T>C, c.569A>C, c.721G>T, c.805G>A, c.1246G>T, exon 1–7 large deletion | 1 each | - |
| Nucleotide Exchange 1 | Amino Acid Exchange 1 | Antithrombin Activity, Thrombin-Based 2 | Half-Life of Human Thrombin 2 | Half-Life of Bovine Thrombin 2 |
|---|---|---|---|---|
| c.236G>A | p.Arg79His | 100% | 50.2 s | 101.2 s |
| c.1246G>T | p.Ala416Ser | 77% | 62.3 s | - |
| c.74G>A | p.Gly25Asp | 66% | 69.8 s | 181.1 s |
| c.805G>A | p.Glu269Lys | 62% | 75.7 s | - |
| c.1347del G | p.Leu449fsX9 | 81% | 87.5 s | 195.0 s |
| c.462_464del | p.Phe155fs | 52 (52–53)% | 92.3 (83.8–107.0) s | 168.5 (148.1–184.5) s |
| c.1355T>C | p.Ile452Thr | 84% | 98.8 s | 117.9 s |
| c.391C>T | p.Leu131Phe | 77 (71–84)% | 104.9 (60.6–137.4) s | 207.3 (134.3–255.5) s |
| c.248T>A | p.Leu83Gln | 61% | 106.1 s | - |
| c.1153G>A | p.Gly385Ser | 58 (57–59)% | 106.7 (85.5–123.6) s | 174.1 (151.6–196.6) s |
| c.721G>T | p.Glu241Ter | 52% | 109.1 s | - |
| c.89T>A | p.Val30Glu | 77 (76–79)% | 119.7 (92.7–153.3) s | - |
| c.1058C>T | p.Pro353Leu | 58 (56–60)% | 135.6 (106.7–164.5) s | 272.2 (197.4–347.0) s |
| c.481C>T | p.Arg161Ter | 51% | 139.2 s | 203.8 s |
| c.470A>G | p.Lys157Arg | 59% | 140.0 s | 169.7 s |
| Exon 1–7 large deletion | - | 54% | 154.5 s | - |
| c.391C>T (homozygous) | p.Leu131Phe | 55 (47–60)% | 154.7 (125.9–177.6) s | 263.4 (229.0–331.7) s |
| c.569A>C | p.Tyr190Ser | 63% | 164.3 s | - |
| c.379T>A | p.Cys127Arg | 51% | 206.6 s | - |
| c.331T>A | p.Ser111Thr | 65% | 221.7 s | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reda, S.; Müller, J.; Pavlova, A.; Pezeshkpoor, B.; Oldenburg, J.; Pötzsch, B.; Rühl, H. Functional Characterization of Antithrombin Mutations by Monitoring of Thrombin Inhibition Kinetics. Int. J. Mol. Sci. 2021, 22, 2119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042119
Reda S, Müller J, Pavlova A, Pezeshkpoor B, Oldenburg J, Pötzsch B, Rühl H. Functional Characterization of Antithrombin Mutations by Monitoring of Thrombin Inhibition Kinetics. International Journal of Molecular Sciences. 2021; 22(4):2119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042119
Chicago/Turabian StyleReda, Sara, Jens Müller, Anna Pavlova, Behnaz Pezeshkpoor, Johannes Oldenburg, Bernd Pötzsch, and Heiko Rühl. 2021. "Functional Characterization of Antithrombin Mutations by Monitoring of Thrombin Inhibition Kinetics" International Journal of Molecular Sciences 22, no. 4: 2119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042119