
A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis—And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder


Abstract
:1. Introduction
1.1. Oxidant Stress and Intracellular Calcium Signals—Roles in Alzheimer’s Pathogenesis
1.2. Phycocyanobilin—A Phyconutrient NADPH Oxidase Inhibitor
1.3. Phase Two Induction and Support for Glutathione Synthesis
2. A Comprehensive Antioxidant Strategy May Aid Prevention of Alzheimer’s Disease
2.1. Astaxanthin—Antioxidant Protection for Calcium-Overloaded Mitochondria
2.2. Controlling Amyloid β Production via Modulation of BACE1 and ADAM10 Expression
2.3. Antioxidants May Support Astrocyte Glutamate Uptake
2.4. Antioxidants May Sustain Activity of Amyloid β-Degrading Proteases
2.5. Could Antioxidants Aid Expulsion of Amyloid β from the Brain?
2.6. Antioxidants May Support Cerebrovascular Endothelial Nitric Oxide Synthase Activity
3. Potential Enhancers of Amyloid β Neurotoxicity
3.1. Microglial Production of Interleukin-1β Potentiates Amyloid β Neurotoxicity
3.2. Magnesium Deficiency May Up-Regulate Amyloid β Neurotoxicity
4. Toward an Integrated Nutraceutical/Lifestyle Strategy for Amyloid β Neurotoxicity in Alzheimer’s Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| NAC | N-acetylcysteine |
| RYR2 | ryanodine receptors |
| ER | endoplasmic reticulum |
| IRS-1 | insulin receptor substrate-1 |
| GLT-1 | glutamate |
| PCB | Phycocyanobilin |
| MLT | melatonin |
| HO-1 | heme oxygenase-1 |
| H2S | hydrogen sulfide |
| CBS | cystathionine beta-synthase |
| Se | selenium |
| AST | astaxanthin |
| ETC | electron transport chain |
| APP | amyloid precursor protein |
| sGC | soluble guanylate cyclase |
| NO | nitric oxide |
| PPARα | peroxisome proliferator-activated receptor α |
| PKC | protein kinase C |
| IDE | insulin-degrading enzyme |
| P-GP | P-glycoprotein |
| eNOS | endothelial nitric oxide synthase |
| DDAH | dimethylarginine dimethylaminohydrolase |
| ADMA | asymmetric dimethylarginine |
| IL-1β | interleukin-1β |
| Mg | Magnesium |
| DHA | docosahexaenoic acid |
| NMDA | N-methyl-D-aspartate |
| JNK | c-Jun N-terminal kinase |
| ASK1 | apoptosis signal-regulating kinase 1 |
| NADPH | Reduced form of Nicotinamide adenine dinucleotide phosphate |
| LRP1 | lipoprotein receptor-related protein 1 |
| GFAP | glial fibrillary acidic protein |
| EGCG | epigallocatechin-3-gallate |
| SREBP-2 | sterol regulatory element-binding protein 2 |
| RAGE | receptor for advanced glycation end products |
| LTP | Long-term potentiation |
| TLR | Toll-like receptor |
| HMGB1 | high-mobility group box protein 1 |
| FGF21 | fibroblast growth factor 21 |
| LA | Lipoic acid |
References
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schröder, U.H.; Fändrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SanMartin, C.D.; Veloso, P.; Adasme, T.; Lobos, P.; Bruna, B.; Galaz, J.; García, A.; Hartel, S.; Hidalgo, C.; Paula-Lima, A.C. RyR2-Mediated Ca(2+) Release and Mitochondrial ROS Generation Partake in the Synaptic Dysfunction Caused by Amyloid β Peptide Oligomers. Front. Mol. Neurosci. 2017, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Guo, H.; Guo, C.; Zhao, S.; Gong, D.; Zhao, Y. Involvement of IRE α signaling in the hippocampus in patients with mesial temporal lobe epilepsy. Brain Res. Bull. 2011, 84, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Aljanabi, N.M.; Mamtani, S.; Al-Ghuraibawi, M.M.H.; Yadav, S.; Nasr, L. Alzheimer’s and Hyperglycemia: Role of the Insulin Signaling Pathway and GSK-3 Inhibition in Paving a Path to Dementia. Cureus 2020, 12, e6885. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, D.; Wang, Y.; Jiang, T.; Hu, S.; Zhang, M.; Yu, X.; Gong, C.-X. Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J. Alzheimers Dis. 2013, 33, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3 β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; O’Neal, M.A.; Hu, J.; Kavalali, E.T.; Bezprozvanny, I.; Malter, J.S. Pin1 mediates Aβ 42-induced dendritic spine loss. Sci. Signal. 2018, 11, eaap8734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Q.; Santini, F.; Breese, R.; Ross, D.; Zhang, X.D.; Stone, D.J.; Ferrer, M.; Townsend, M.; Wolfe, A.L.; Seager, M.A.; et al. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J. Biol. Chem. 2010, 285, 7619–7632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [Green Version]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimers Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.P.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1712. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sun, B.; Institoris, A.; Zhan, X.; Guo, W.; Song, Z.; Liu, Y.; Hiess, F.; Boyce, A.K.J.; Ni, M.; et al. Limiting RyR2 Open Time Prevents Alzheimer’s Disease-Related Neuronal Hyperactivity and Memory Loss but Not β-Amyloid Accumulation. Cell Rep. 2020, 32, 108169. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, M.A.; Stallings, N.R.; Malter, J.S. Alzheimer’s Disease, Dendritic Spines, and Calcineurin Inhibitors: A New Approach? ACS Chem. Neurosci. 2018, 9, 1233–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimers Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006, 291, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Roberts, S.J.; Datla, S.; Dusting, G.J. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension 2006, 48, 950–957. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F. Clinical potential of Spirulina as a source of phycocyanobilin. J. Med. Food 2007, 10, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Inoguchi, T.; Sasaki, S.; Maeda, Y.; McCarty, M.F.; Fujii, M.; Ikeda, N.; Kobayashi, K.; Sonoda, N.; Takayanagi, R. Phycocyanin and phycocyanobilin from Spirulina platensis protect against diabetic nephropathy by inhibiting oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R110–R120. [Google Scholar] [CrossRef] [Green Version]
- Terry, M.J.; Maines, M.D.; Lagarias, J.C. Inactivation of phytochrome- and phycobiliprotein-chromophore precursors by rat liver biliverdin reductase. J. Biol. Chem. 1993, 268, 26099–26106. [Google Scholar] [CrossRef]
- Romay, C.; Armesto, J.; Remirez, D.; González, R.; Ledon, N.; García, I. Antioxidant and anti-inflammatory properties of C-phycocyanin from blue-green algae. Inflamm. Res. 1998, 47, 36–41. [Google Scholar] [CrossRef]
- Bannu, S.M.; Lomada, D.; Gulla, S.; Chandrasekhar, T.; Reddanna, P.; Reddy, M.C. Potential Therapeutic Applications of C-Phycocyanin. Curr. Drug Metab. 2019, 20, 967–976. [Google Scholar] [CrossRef]
- Mysliwa-Kurdziel, B.; Solymosi, K. Phycobilins and Phycobiliproteins Used in Food Industry and Medicine. Mini Rev. Med. Chem. 2017, 17, 1173–1193. [Google Scholar] [CrossRef] [Green Version]
- Barbalace, M.C.; Malaguti, M.; Giusti, L.; Lucacchini, A.; Hrelia, S.; Angeloni, C. Anti-Inflammatory Activities of Marine Algae in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Huang, Y.; Zhang, R.; Cai, T.; Cai, Y. Medical Application of Spirulina platensis Derived C-Phycocyanin. Evid. Based Complement. Alternat. Med. 2016, 2016, 7803846. [Google Scholar] [PubMed] [Green Version]
- Lima, F.A.V.; Joventino, I.P.; Joventino, F.P.; Cordeiro de Almeida, A.; Neves, K.R.T.; do Carmo, M.R.; Leal, L.K.A.M.; de Andrade, G.M. Neuroprotective Activities of Spirulina platensis in the 6-OHDA Model of Parkinson’s Disease Are Related to Its Anti-Inflammatory Effects. Neurochem. Res. 2017, 42, 3390–3400. [Google Scholar] [CrossRef]
- Tobón-Velasco, J.C.; Palafox-Sánchez, V.; Mendieta, L.; García, E.; Santamaría, A.; Chamorro-Cevallos, G.; Daniel Limón, I. Antioxidant effect of Spirulina (Arthrospira) maxima in a neurotoxic model caused by 6-OHDA in the rat striatum. J. Neural Transm. 2013, 120, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Pabon, M.M.; Jernberg, J.N.; Morganti, J.; Contreras, J.; Hudson, C.E.; Klein, R.L.; Bickford, P.C. A spirulina-enhanced diet provides neuroprotection in an α-synuclein model of Parkinson’s disease. PLoS ONE 2012, 7, e45256. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, G.; Pérez-Albiter, M.; Serrano-García, N.; Mares-Sámano, J.J.; Rojas, P. Spirulina maxima pretreatment partially protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Nutr. Neurosci. 2006, 9, 207–212. [Google Scholar] [CrossRef]
- Pavón-Fuentes, N.; Marín-Prida, J.; Llópiz-Arzuaga, A.; Falcón-Cama, V.; Campos-Mojena, R.; Cervantes-Llanos, M.; Beatriz Piniella-Matamoros, B.; Pentón-Arias, E.; Pentón-Rol, G. Phycocyanobilin reduces brain injury after endothelin-1- induced focal cerebral ischaemia. Clin. Exp. Pharmacol. Physiol. 2020, 47, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Thaakur, S.; Sravanthi, R. Neuroprotective effect of Spirulina in cerebral ischemia-reperfusion injury in rats. J. Neural Transm. 2010, 117, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Marín-Prida, J.; Pavón-Fuentes, N.; Llópiz-Arzuaga, A.; Fernández-Massó, J.R.; Delgado-Roche, L.; Mendoza-Marí, Y.; Santana, S.P.; Cruz-Ramírez, A.; Valenzuela-Silva, C.; Nazábal-Gálvez, M.; et al. Phycocyanobilin promotes PC12 cell survival and modulates immune and inflammatory genes and oxidative stress markers in acute cerebral hypoperfusion in rats. Toxicol. Appl. Pharmacol. 2013, 272, 49–60. [Google Scholar] [CrossRef]
- Pentón-Rol, G.; Marín-Prida, J.; Pardo-Andreu, G.; Martínez-Sánchez, G.; Acosta-Medina, E.F.; Valdivia-Acosta, A.; Lagumersindez-Denis, N.; Rodríguez-Jiménez, E.; Llópiz-Arzuaga, A.; López-Saura, P.A.; et al. C-Phycocyanin is neuroprotective against global cerebral ischemia/reperfusion injury in gerbils. Brain Res. Bull. 2011, 86, 42–52. [Google Scholar]
- Lo, C.M.; Carroll, K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling: From chemistry to functional significance. Antioxid. Redox Signal. 2013, 18, 1557–1593. [Google Scholar] [CrossRef]
- Dickinson, D.A.; Forman, H.J. Glutathione in defense and signaling: Lessons from a small thiol. Ann. NY Acad. Sci. 2002, 973, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005, 7, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Parsons, Z.D.; Gates, K.S. Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 2013, 52, 6412–6423. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dinkova-Kostova, A.T.; Talalay, P. Coordinate regulation of enzyme markers for inflammation and for protection against oxidants and electrophiles. Proc. Natl. Acad. Sci. USA 2008, 105, 15926–15931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Agyeman, A.S.; Visvanathan, K.; Groopman, J.D.; Chen, J.G.; Chen, T.Y.; Fahey, J.W.; Talalay, P. Keap1-nrf2 signaling: A target for cancer prevention by sulforaphane. Top. Curr. Chem. 2013, 329, 163–177. [Google Scholar] [PubMed] [Green Version]
- Abed, D.A.; Goldstein, M.; Albanyan, H.; Jin, H.; Hu, L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Othman, M.S.; Fareid, M.A.; Abdel Hameed, R.S.; Abdel Moneim, A.E. The Protective Effects of Melatonin on Aluminum-Induced Hepatotoxicity and Nephrotoxicity in Rats. Oxid. Med. Cell Longev. 2020, 2020, 7375136. [Google Scholar] [CrossRef]
- GarcÃa, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF- κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J. 2015, 29, 3863–3875. [Google Scholar] [CrossRef] [PubMed]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells 2020, 9, 1861. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3β pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.H.; Hua, N.; Zang, X.; Huang, T.; He, L. Melatonin ameliorates Aβ (1–42) -induced Alzheimer’s cognitive deficits in mouse model. J. Pharm. Pharmacol. 2018, 70, 70–80. [Google Scholar] [CrossRef]
- Song, C.; Li, M.; Xu, L.; Shen, Y.; Yang, H.; Ding, M.; Liu, X.; Xie, Z. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 2018, 29, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Jürgenson, M.; Zharkovskaja, T.; Noortoots, A.; Morozova, M.; Beniashvili, A.; Zapolski, M.; Zharkovsky, A. Effects of the drug combination memantine and melatonin on impaired memory and brain neuronal deficits in an amyloid-predominant mouse model of Alzheimer’s disease. J. Pharm. Pharmacol. 2019, 71, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Qiu, X.; Wang, Y.; Liu, J.; Li, Q.; Jiang, H.; Li, S.; Song, C. Long-term oral melatonin alleviates memory deficits, reduces amyloid-β deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice. Neurosci. Lett. 2020, 735, 135192. [Google Scholar] [CrossRef] [PubMed]
- Strasky, Z.; Zemankova, L.; Nemeckova, I.; Rathouska, J.; Wong, R.J.; Muchova, L.; Subhanova, I.; Vanikova, J.; Vanova, K.; Vitek, L.; et al. Spirulina platensis and phycocyanobilin activate atheroprotective heme oxygenase-1: A possible implication for atherogenesis. Food Funct. 2013, 4, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, W.; Qin, S. Therapeutic effect of phycocyanin on acute liver oxidative damage caused by X-ray. Biomed. Pharmacother. 2020, 130, 110553. [Google Scholar] [CrossRef] [PubMed]
- Phelan, D.; Winter, G.M.; Rogers, W.J.; Lam, J.C.; Denison, M.S. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 1998, 357, 155–163. [Google Scholar] [CrossRef]
- Vítek, L. Bilirubin as a signaling molecule. Med. Res. Rev. 2020, 40, 1335–1351. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Oxidative stress and ageing: Is ageing a cysteine deficiency syndrome? Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2355–2372. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, S.; Dean, O.; Copolov, D.L.; Malhi, G.S.; Berk, M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert Opin. Biol. Ther. 2008, 8, 1955–1962. [Google Scholar] [CrossRef]
- More, J.; Galusso, N.; Veloso, P.; Montecinos, L.; Finkelstein, J.P.; Sanchez, G.; Paula-Lima, A. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer’s Disease Rat Model. Front. Aging Neurosci. 2018, 10, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNicolantonio, J.J.; OKeefe, J.H.; McCarty, M.F. Boosting endogenous production of vasoprotective hydrogen sulfide via supplementation with taurine and N-acetylcysteine: A novel way to promote cardiovascular health. Open Heart 2017, 4, e000600. [Google Scholar] [CrossRef] [PubMed]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60. [Google Scholar] [CrossRef]
- Cao, L.; Cao, X.; Zhou, Y.; Nagpure, B.V.; Wu, Z.-Y.; Hu, L.F.; Yang, Y.; Sethi, G.; Moore, P.K.; Bian, J.-S. Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ 1–42 synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav. Immun. 2018, 73, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef]
- Yang, Y.J.; Zhao, Y.; Yu, B.; Xu, G.-G.; Wang, W.; Zhan, J.-Q.; Tang, Z.-Y.; Wang, T.; Wei, B. GluN2B-containing NMDA receptors contribute to the beneficial effects of hydrogen sulfide on cognitive and synaptic plasticity deficits in APP/PS1 transgenic mice. Neuroscience 2016, 335, 170–183. [Google Scholar] [CrossRef] [PubMed]
- He, X.L.; Yan, N.; Chen, X.S.; Qi, Y.W.; Yan, Y.; Cai, Z. Hydrogen sulfide down-regulates BACE1 and PS1 via activating PI3K/Akt pathway in the brain of APP/PS1 transgenic mouse. Pharmacol. Rep. 2016, 68, 975–982. [Google Scholar] [CrossRef]
- He, X.L.; Yan, N.; Zhang, H.; Qi, Y.-W.; Zhu, L.-J.; Liu, M.-J.; Yan, Y. Hydrogen sulfide improves spatial memory impairment and decreases production of Aβ in APP/PS1 transgenic mice. Neurochem. Int. 2014, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. A diet rich in taurine, cysteine, folate, B(12) and betaine may lessen risk for Alzheimer’s disease by boosting brain synthesis of hydrogen sulfide. Med. Hypotheses 2019, 132, 109356. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, B.; Li, Y.; Sun, F.; Li, P.; Xia, W.; Zhou, X.; Li, Q.; Wang, X.; Chen, J.; et al. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension 2016, 67, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Qu, J.; Li, Q.; Cui, M.; Wang, J.; Zhang, K.; Liu, X.; Feng, H.; Chen, Y. Taurine supplementation reduces neuroinflammation and protects against white matter injury after intracerebral hemorrhage in rats. Amino Acids 2018, 50, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, H.V.; Yoon, J.H.; Kang, B.R.; Cho, S.M.; Lee, S.; Kim, J.Y.; Kim, J.W.; Cho, Y.; Woo, J.; et al. Taurine in drinking water recovers learning and memory in the adult APP/PS1 mouse model of Alzheimer’s disease. Sci. Rep. 2014, 4, 7467. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Lee, S.; Choi, S.L.; Kim, H.Y.; Baek, S.; Kim, Y. Taurine Directly Binds to Oligomeric Amyloid-β and Recovers Cognitive Deficits in Alzheimer Model Mice. Adv. Exp. Med. Biol. 2017, 975, 233–241. [Google Scholar] [PubMed]
- Stadtman, T.C. Selenium biochemistry. Mammalian selenoenzymes. Ann. NY Acad. Sci. 2000, 899, 399–402. [Google Scholar] [CrossRef]
- Ye, Y.; Qu, J.; Pu, Y.; Rao, S.; Xu, F.; Wu, C. Selenium Biofortification of Crop Food by Beneficial Microorganisms. J. Fungi 2020, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Duffield-Lillico, A.J.; Dalkin, B.L.; Reid, M.E.; Turnbull, B.W.; Slate, E.H.; Jacobs, E.T.; Marshall, J.R.; Clark, L.C. Nutritional Prevention of Cancer Study Group. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: An analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003, 91, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Alexander, J.; Aaseth, J. Supplementation with Selenium and Coenzyme Q10 Reduces Cardiovascular Mortality in Elderly with Low Selenium Status. A Secondary Analysis of a Randomised Clinical Trial. PLoS ONE 2016, 11, e0157541. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Myung, S.K.; Jeon, Y.J.; Kim, Y.; Chang, Y.J.; Ju, W.; Seo, H.G.; Huh, B.Y. Effects of selenium supplements on cancer prevention: Meta-analysis of randomized controlled trials. Nutr. Cancer 2011, 63, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Berr, C.; Arnaud, J.; Akbaraly, T.N. Selenium and cognitive impairment: A brief-review based on results from the EVA study. Biofactors 2012, 38, 139–144. [Google Scholar] [CrossRef]
- Yang, Y.W.; Liou, S.H.; Hsueh, Y.M.; Lyu, W.-S.; Liu, C.-S.; Liu, H.-J.; Chung, M.-C.; Hung, P.-H.; Chung, C.-J. Risk of Alzheimer’s disease with metal concentrations in whole blood and urine: A case-control study using propensity score matching. Toxicol. Appl. Pharmacol. 2018, 356, 8–14. [Google Scholar] [CrossRef]
- Sun, H. Associations of Spatial Disparities of Alzheimer’s Disease Mortality Rates with Soil Selenium and Sulfur Concentrations and Four Common Risk Factors in the United States. J. Alzheimers Dis. 2017, 58, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, K.; Sun, X.; Qin, S.; Wu, M.; Qin, L.; Wang, Y.; Li, Z.; Zhong, X.; Wei, X. A cross-sectional study of blood selenium concentration and cognitive function in elderly Americans: National Health and Nutrition Examination Survey 2011–2014. Ann. Hum. Biol. 2020, 47, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Judd, P.A.; Long, A.; Butcher, M.; Caygill, C.P.; Diplock, A.T. Vegetarians and vegans may be most at risk from low selenium intakes. BMJ 1997, 314, 1834. [Google Scholar] [CrossRef] [Green Version]
- Sobiecki, J.G. Vegetarianism and colorectal cancer risk in a low-selenium environment: Effect modification by selenium status? A possible factor contributing to the null results in British vegetarians. Eur. J. Nutr. 2017, 56, 1819–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Liu, X.; Osawa, T. Astaxanthin protects neuronal cells against oxidative damage and is a potent candidate for brain food. Forum Nutr. 2009, 61, 129–135. [Google Scholar]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Calcium Signal. Dis. 2007, 45, 481–506. [Google Scholar]
- Huang, C.; Wen, C.; Yang, M.; Li, A.; Fan, C.; Gan, D.; Li, Q.; Zhao, J.; Zhu, L.; Lu, D. Astaxanthin Improved the Cognitive Deficits in APP/PS1 Transgenic Mice Via Selective Activation of mTOR. J. Neuroimmune Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Li, Q.; Zhang, T.; Wang, D.; Yang, L.; Xu, J.; Yanagita, T.; Xue, C.; Chang, Y.; Wang, Y. Effects of Astaxanthin and Docosahexaenoic-Acid-Acylated Astaxanthin on Alzheimer’s Disease in APP/PS1 Double-Transgenic Mice. J. Agric. Food Chem. 2018, 66, 4948–4957. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Zagórska, A.; Dulak, J. HIF-1: The knowns and unknowns of hypoxia sensing. Acta Biochim. Pol. 2004, 51, 563–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.N.; Xi, M.M.; Guo, Y.; Hai, C.X.; Yang, W.L.; Qin, X.J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014, 224, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Dachs, G.U.; Currie, M.J.; Vissers, M.C. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med. 2014, 69, 308–317. [Google Scholar] [CrossRef]
- Fedele, A.O.; Whitelaw, M.L.; Peet, D.J. Regulation of gene expression by the hypoxia-inducible factors. Mol. Interv. 2002, 2, 229–243. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [Green Version]
- Guglielmotto, M.; Monteleone, D.; Giliberto, L.; Fornaro, M.; Borghi, R.; Tamagno, E.; Tabaton, M. Amyloid-beta42 activates the expression of BACE1 through the JNK pathway. J. Alzheimers Dis. 2011, 27, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Saadipour, K.; Tiberi, A.; Lombardo, S.; Grajales, E.; Montroull, L.; Mañucat-Tan, N.B.; LaFrancois, J.; Cammer, M.; Mathews, P.M.; Scharfman, H.E.; et al. Regulation of BACE1 expression after injury is linked to the p75 neurotrophin receptor. Mol. Cell. Neurosci. 2019, 99, 103395. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.D.; Wang, R.; Li, J.J.; Zhang, Y.W.; Xu, H.; Liao, F.F. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol. Neurodegener. 2011, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, S.A.; d’Uscio, L.V.; Katusic, Z.S. Supplementation of nitric oxide attenuates AbPP and BACE1 protein in cerebral microcirculation of eNOS-deficient mice. J. Alzheimers Dis. 2013, 33, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006, 20, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsouri, L.; Lim, Y.M.; Blondrath, K.; Eleftheriadou, I.; Lombardero, L.; Birch, A.M.; Mirzaei, N.; Irvine, E.E.; Mazarakis, N.D.; Sastre, M. PPARgamma-coactivator-1alpha gene transfer reduces neuronal loss and amyloid-beta generation by reducing beta-secretase in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2016, 113, 12292–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsouri, L.; Parr, C.; Bogdanovic, N.; Willem, M.; Sastre, M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ -dependent mechanism. J. Alzheimers Dis. 2011, 25, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Stasch, J.P.; Schmidt, P.M.; Nedvetsky, P.I.; Nedvetskaya, T.Y.; Kumar, A.H.S.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Invest. 2006, 116, 2552–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef]
- Vesely, D.L. Biotin enhances guanylate cyclase activity. Science 1982, 216, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Kamiyama, M.; Kamiyama, S.; Horiuchi, K.; Ohinata, K.; Shirakawa, H.; Furukawa, Y.; Komai, M. Antihypertensive effect of biotin in stroke-prone spontaneously hypertensive rats. Br. J. Nutr. 2008, 99, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarty, M.F. In type 1 diabetics, high-dose biotin may compensate for low hepatic insulin exposure, promoting a more normal expression of glycolytic and gluconeogenic enyzymes and thereby aiding glycemic control. Med. Hypotheses 2016, 95, 45–48. [Google Scholar] [CrossRef]
- McCarty, M.F. cGMP may have trophic effects on beta cell function comparable to those of cAMP, implying a role for high-dose biotin in prevention/treatment of diabetes. Med. Hypotheses 2006, 66, 323–328. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. Neuroprotective potential of high-dose biotin. Med. Hypotheses 2017, 109, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ferguson, A.; Cervinski, M.A.; Lynch, K.L.; Kyle, P.B. AACC Guidance Document on Biotin Interference in Laboratory Tests. J. Appl. Lab. Med. 2020, 5, 575–587. [Google Scholar] [CrossRef]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Kim, J.Y.; Jun, H.J.; Kim, S.-J.; Lee, J.-H.; Hoang, M.H.; Hwang, K.-Y.; Um, S.-J.; Chang, H.I.; Lee, S.-J. The natural carotenoid astaxanthin, a PPAR-α agonist and PPAR-γ antagonist, reduces hepatic lipid accumulation by rewiring the transcriptome in lipid-loaded hepatocytes. Mol. Nutr. Food Res. 2012, 56, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Fanaee-Danesh, E.; Gali, C.C.; Tadic, J.; Zandl-Lang, M.; Kober, A.C.; Agujetas, V.R.; de Dios, C.; Tam-Amersdorfer, C.; Stracke, A.; Albrecher, N.M.; et al. Astaxanthin exerts protective effects similar to bexarotene in Alzheimer’s disease by modulating amyloid-beta and cholesterol homeostasis in blood-brain barrier endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2224–2245. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-beta(1–42) across the blood-brain barrier. J. Clin. Invest. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, H.; Zhang, X.; Meng, X.; Xu, P.; Zou, X.; Qu, S. Amyloid-beta peptide decreases expression and function of glutamate transporters in nervous system cells. Int. J. Biochem. Cell Biol. 2017, 85, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wyssenbach, A.; Quintela, T.; Llavero, F.; Zugaza, J.L.; Matute, C.; Alberdi, E. Amyloid β-induced astrogliosis is mediated by β1-integrin via NADPH oxidase 2 in Alzheimer’s disease. Aging Cell 2016, 15, 1140–1152. [Google Scholar] [CrossRef]
- Kalandadze, A.; Wu, Y.; Robinson, M.B. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J. Biol. Chem. 2002, 277, 45741–45750. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, A.L.; Gonzãlez, M.I.; Krizman-Genda, E.N.; Susarla, B.T.; Robinson, M.B. Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem. Int. 2008, 53, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Villarreal, J.; García-Tardón, N.; Ibáñez, I.; Giménez, C.; Zafra, F. Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 2012, 60, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- García-Tardón, N.; González-González, I.M.; Martínez-Villarreal, J.; Fernández-Sánchez, E.; Giménez, C.; Zafra, F. Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4–2-dependent ubiquitination but not phosphorylation. J. Biol. Chem. 2012, 287, 19177–19187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, M.; Takase, H.; Nunome, M.; Enomoto, H.; Ito, I.-I.; Gong, J.-S.; Michikawa, M. Amyloid-β Reduces Exosome Release from Astrocytes by Enhancing JNK Phosphorylation. J. Alzheimers Dis. 2016, 53, 1433–1441. [Google Scholar] [CrossRef]
- Gao, K.; Wang, C.R.; Jiang, F.; Wong, A.Y.K.; Su, N.; Jiang, J.H.; Chai, R.C.; Vatcher, G.; Teng, J.; Chen, J.; et al. Traumatic scratch injury in astrocytes triggers calcium influx to activate the JNK/c-Jun/AP-1 pathway and switch on GFAP expression. Glia 2013, 61, 2063–2077. [Google Scholar] [CrossRef]
- Gwoździńska, P.; Buchbinder, B.A.; Mayer, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of β-ENaC in a Human Alveolar Epithelial Cell Line. Front. Immunol. 2017, 8, 591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, K.R.; Bhalla, V.; Oyster, N.M.; Wijngaarden, M.A.; Lee, J.K.; Li, H.; Chandran, S.; Xia, X.; Huang, Z.; Chalkley, R.J.; et al. Phosphopeptide screen uncovers novel phosphorylation sites of Nedd4–2 that potentiate its inhibition of the epithelial Na+ channel. J. Biol. Chem. 2010, 285, 21671–21678. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Yang, L.; Du, H.; Sun, Q.; Wang, X.; Cong, L.; Liu, X.; Yin, L.; Li, S.; Du, Y. Insulin Attenuates Beta-Amyloid-Associated Insulin/Akt/EAAT Signaling Perturbations in Human Astrocytes. Cell Mol. Neurobiol. 2016, 36, 851–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, T.; Alexopoulos, P.; Tsolakidou, A.; Guo, L.H.; Henriksen, G.; Yousefi, B.H.; Förstl, H.; Sorg, C.; Kurz, A.; Drzezga, A.; et al. Cerebrospinal fluid BACE1 activity and brain amyloid load in Alzheimer’s disease. Sci. World J. 2012, 2012, 712048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewers, M.; Cheng, X.; Zhong, Z.; Nural, H.F.; Walsh, C.; Meindl, T.; Teipel, S.J.; Buerger, K.; He, P.; Shen, Y.; et al. Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease. J. Alzheimers Dis. 2011, 25, 373–381. [Google Scholar] [CrossRef]
- Tyler, S.J.; Dawbarn, D.; Wilcock, G.K.; Allen, S.J. alpha- and beta-secretase: Profound changes in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 299, 373–376. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid β -protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain--implications for Alzheimer diseaser. Nat. Rev. Neurol. 2016, 12, 248. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Zlokovic, B.V. Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Hartz, A.M.S.; Bernard, J.; Wolf, A.; Kachlmeier, A.; Mahringer, A.; Weggen, S.; Pahnke, J.; Pietrzik, C.U. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav. Immun. 2018, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, H.; Tomiyama, T.; Mori, H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 2003, 350, 113–116. [Google Scholar] [CrossRef]
- Yasojima, K.; McGeer, E.G.; McGeer, P.L. Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001, 919, 115–121. [Google Scholar] [CrossRef]
- Yasojima, K.; Akiyama, H.; McGeer, E.G.; McGeer, P.L. Reduced neprilysin in high plaque areas of Alzheimer brain: A possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 2001, 297, 97–100. [Google Scholar] [CrossRef]
- Wang, D.S.; Lipton, R.B.; Katz, M.J.; Davies, P.; Buschke, H.; Kuslansky, G.; Verghese, J.; Younkin, S.G.; Eckman, C.; Dickson, D.W. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J. Neuropathol. Exp. Neurol. 2005, 64, 378–385. [Google Scholar] [CrossRef] [Green Version]
- El-Amouri, S.S.; Zhu, H.; Yu, J.; Marr, R.; Verma, I.M.; Kindy, M.S. Neprilysin: An enzyme candidate to slow the progression of Alzheimer’s disease. Am. J. Pathol. 2008, 172, 1342–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Marr, R.A.; Rockenstein, E.; Crews, L.; Adame, A.; Potkar, R.; Patrick, C.; Gage, F.H.; Verma, I.M.; Masliah, E. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice. BMC Neurosci. 2008, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüttenrauch, M.; Baches, S.; Gerth, J.; Bayer, T.A.; Weggen, S.; Wirths, O. Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1291–1302. [Google Scholar]
- Wang, R.; Wang, S.; Malter, J.S.; Wang, D.S. Effects of HNE-modification induced by Abeta on neprilysin expression and activity in SH-SY5Y cells. J. Neurochem. 2009, 108, 1072–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Wei, C.; Huang, W.; Bennett, D.A.; Dickson, D.W.; Wang, R.; Wang, D. Distinct subcellular patterns of neprilysin protein and activity in the brains of Alzheimer’s disease patients, transgenic mice and cultured human neuronal cells. Am. J. Transl. Res. 2013, 5, 608–621. [Google Scholar] [PubMed]
- Wang, D.S.; Iwata, N.; Hama, E.; Saido, T.C.; Dickson, D.W. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 2003, 310, 236–241. [Google Scholar] [CrossRef]
- Wang, R.; Malter, J.S.; Wang, D.S. N-acetylcysteine prevents 4-hydroxynonenal- and amyloid-beta-induced modification and inactivation of neprilysin in SH-SY5Y cells. J. Alzheimers Dis 2010, 19, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Qian, J.; Liu, J.; Zhao, R.; Li, B.; Wang, R. Identification of the sites of 4-hydroxy-2-nonenal and neprilysin adduction using a linear trap quadrapole Velos Pro-Orbitrap Elite mass spectrometer. Eur. J. Mass Spectrom. 2016, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Han, J.S. The antioxidant xanthorrhizol prevents amyloid-β-induced oxidative modification and inactivation of neprilysin. Biosci. Rep. 2018, 38, BSR20171611. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Naito, Y.; Sakuma, K.; Kuchide, M.; Tokuda, H.; Maoka, T.; Toyokuni, S.; Oka, S.; Yasuhara, M.; Yoshikawa, T. Astaxanthin limits exercise-induced skeletal and cardiac muscle damage in mice. Antioxid. Redox Signal. 2003, 5, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Inokuchi, Y.; Shimazawa, M.; Otsubo, K.; Ishibashi, T.; Hara, H. Astaxanthin, a dietary carotenoid, protects retinal cells against oxidative stress in-vitro and in mice in-vivo. J. Pharm. Pharmacol. 2008, 60, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.M.; Girens, R.E.; Larson, S.K.; Jones, M.R.; Restivo, J.L.; Holtzman, D.M.; Cirrito, J.R.; Yuede, C.M.; Zimmerman, S.D.; Timson, B.F. A spectrum of exercise training reduces soluble Aβ in a dose-dependent manner in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 85, 218–224. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, D.; Zhang, X.; Li, T.; Li, J.; Tang, Y.; Le, W. Hypoxia-induced down-regulation of neprilysin by histone modification in mouse primary cortical and hippocampal neurons. PLoS ONE 2011, 6, e19229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Hou, T.; Wang, W.; Luo, Y.; Yan, F.; Jia, J. The Effect of Chronic Cerebral Hypoperfusion on Amyloid-β Metabolism in a Transgenic Mouse Model of Alzheimer’s Disease (PS1V97L). J. Alzheimers Dis. 2018, 62, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Rong, C.; Chen, Y.; Yang, C.; Hu, Q.; Mo, Y.; Zhang, C.; Gu, X.; Zhang, L.; He, W.; et al. (-)-Epigallocatechin-3-gallate attenuates cognitive deterioration in Alzheimer’s disease model mice by upregulating neprilysin expression. Exp. Cell Res. 2015, 334, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, J.; Jiang, C.; Wang, S.; Que, R.; An, L. Up-regulation of neprilysin mediates the protection of fructo-oligosaccharides against Alzheimer’s disease. Food Funct. 2020, 11, 6565–6572. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Lehmann, J.; Mett, J.; Zimmer, V.C.; Grösgen, S.; Stahlmann, C.P.; Hundsdörfer, B.; Haupenthal, V.J.; Rothhaar, T.L.; Herr, C.; et al. Impact of Vitamin D on amyloid precursor protein processing and amyloid- beta peptide degradation in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Pietrzik, C.U. Endothelial LRP1—A Potential Target for the Treatment of Alzheimer’s Disease: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2637–2651. [Google Scholar] [CrossRef]
- Gali, C.C.; Fanaee-Danesh, E.; Zandl-Lang, M.; Albrecher, N.M.; Tam-Amersdorfer, C.; Stracke, A.; Sachdev, V.; Reichmann, F.; Sun, Y.; Avdili, A.; et al. Amyloid-beta impairs insulin signaling by accelerating autophagy-lysosomal degradation of LRP-1 and IR- β in blood-brain barrier endothelial cells in vitro and in 3XTg-AD mice. Mol. Cell. Neurosci. 2019, 99, 103390. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Aβ 1–42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H., III; Miller, D.S.; Bauer, B. Aβ 40 Reduces P-Glycoprotein at the Blood-Brain Barrier through the Ubiquitin-Proteasome Pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Choi, S.M.; Whitcomb, D.J.; Kim, B.C. Adiponectin controls the apoptosis and the expression of tight junction proteins in brain endothelial cells through AdipoR1 under beta amyloid toxicity. Cell Death Dis. 2017, 8, e3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Brenn, A.; Grube, M.; Peters, M.; Fischer, A.; Jedlitschky, G.; Kroemer, H.K.; Warzok, R.W.; Vogelgesang, S. Beta-Amyloid Downregulates MDR1-P-Glycoprotein (Abcb1) Expression at the Blood-Brain Barrier in Mice. Int. J. Alzheimers Dis. 2011, 2011, 690121. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Wang, G.; Zhou, P.; Zhou, J.; Pitstick, R.; Previti, M.L.; Younkin, L.; Younkin, S.G.; Van Nostrand, W.E.; Cho, S.; et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc. Natl. Acad. Sci. USA 2011, 108, 5063–5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wen, L.; Martin, M.; Hsu, C.Y.; Fang, L.; Lin, F.M.; Lin, T.Y.; Geary, M.J.; Geary, G.G.; Zhao, Y.; et al. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation 2015, 131, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Shin, S.M. Induction of Lipin1 by ROS-Dependent SREBP-2 Activation. Toxicol. Res. 2017, 33, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorente-Cortés, V.; Costales, P.; Bernués, J.; Camino-Lopez, S.; Badimon, L. Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J. Mol. Biol. 2006, 359, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Cortés, V.; Royo, T.; Otero-Viñas, M.; Berrozpe, M.; Badimon, L. Sterol regulatory element binding proteins downregulate LDL receptor-related protein (LRP1) expression and LRP1-mediated aggregated LDL uptake by human macrophages. Cardiovasc. Res. 2007, 74, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Akkaya, B.G.; Zolnerciks, J.K.; Ritchie, T.K.; Bauer, B.; Hartz, A.M.; Sullivan, J.A.; Linton, K.J. The multidrug resistance pump ABCB1 is a substrate for the ubiquitin ligase NEDD4–1. Mol. Membr. Biol. 2015, 32, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Pieper, R.O.; Li, D.; Wei, P.; Liu, M.; Woo, S.Y.; Aldape, K.D.; Sawaya, R.; Xie, K.; Huang, S. FoxM1B regulates NEDD4–1 expression, leading to cellular transformation and full malignant phenotype in immortalized human astrocytes. Cancer Res. 2010, 70, 2951–2961. [Google Scholar] [CrossRef] [Green Version]
- Kwak, Y.D.; Wang, B.; Li, J.J.; Wang, R.; Deng, Q.; Diao, S.; Chen, Y.; Xu, R.; Masliah, E.; Xu, H.; et al. Upregulation of the E3 ligase NEDD4–1 by oxidative stress degrades IGF-1 receptor protein in neurodegeneration. J. Neurosci. 2012, 32, 10971–10981. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Carr, J.R.; Wang, Z.; Nogueira, V.; Hay, N.; Tyner, A.L.; Lau, L.F.; Costa, R.H.; Raychaudhuri, P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009, 28, 2908–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, C.; Liu, X.; Qiu, C.; Zheng, J. FoxM1 is regulated by both HIF-1α and HIF- 2α and contributes to gastrointestinal stromal tumor progression. Gastric Cancer 2019, 22, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Tang, L.; Zhu, Q.; Yi, F.; Zhang, F.; Li, P.L.; Li, N. Hypoxia-inducible factor-1α contributes to the profibrotic action of angiotensin II in renal medullary interstitial cells. Kidney Int. 2011, 79, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Lee, E.K.; Kim, D.H.; Yu, B.P.; Chung, H.Y. Kaempferol modulates pro-inflammatory NF-kappaB activation by suppressing advanced glycation endproducts-induced NADPH oxidase. Age 2010, 32, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Xie, Y.; Satyanarayanan, S.K.; Zeng, H.; Liu, Q.; Huang, M.; Ma, Y.; Wan, J.B.; Yao, X.; Su, K.P.; et al. Omega-3 polyunsaturated fatty acids promote brain-to-blood clearance of β-Amyloid in a mouse model with Alzheimer’s disease. Brain Behav. Immun. 2020, 85, 35–45. [Google Scholar] [CrossRef]
- Chan, J.P.; Wong, B.H.; Chin, C.F.; Galam, D.L.A.; Foo, J.C.; Wong, L.C.; Ghosh, S.; Wenk, M.R.; Cazenave-Gassiot, A.; Silver, D.L. The lysolipid transporter Mfsd2a regulates lipogenesis in the developing brain. PLoS Biol. 2018, 16, e2006443. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, G.; Ma, M.; Qiu, N.; Zhu, L.; Li, J. Fatty acids modulate the expression levels of key proteins for cholesterol absorption in Caco-2 monolayer. Lipids Health Dis. 2018, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dacks, P.A.; Shineman, D.W.; Fillit, H.M. Current evidence for the clinical use of long-chain polyunsaturated n-3 fatty acids to prevent age-related cognitive decline and Alzheimer’s disease. J. Nutr. Health Aging 2013, 17, 240–251. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Qiu, J.; Li, Y.; Wang, J.; Jiao, J. Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: A dose-response meta-analysis of 21 cohort studies. Am. J. Clin. Nutr. 2016, 103, 330–340. [Google Scholar] [CrossRef]
- Nock, T.G.; Chouinard-Watkins, R.; Plourde, M. Carriers of an apolipoprotein E epsilon 4 allele are more vulnerable to a dietary deficiency in omega-3 fatty acids and cognitive decline. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1068–1078. [Google Scholar] [CrossRef]
- Santhanam, A.V.; d’Uscio, L.V.; He, T.; Das, P.; Younkin, S.G.; Katusic, Z.S. Uncoupling of endothelial nitric oxide synthase in cerebral vasculature of Tg2576 mice. J. Neurochem. 2015, 134, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.P.; Druhan, L.J.; Guzman, J.E.; Parinandi, N.; Zhang, L.; Green-Church, K.B.; Cardounel, A.J. Mechanism of 4-HNE mediated inhibition of hDDAH-1: Implications in no regulation. Biochemistry 2008, 47, 1819–1826. [Google Scholar] [CrossRef]
- Dragovich, M.A.; Chester, D.; Fu, B.M.; Wu, C.; Xu, Y.; Goligorsky, M.S.; Zhang, X.F. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am. J. Physiol. Cell Physiol. 2016, 311, C846–C853. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, C.F.; Santos, R.M.; Barbosa, R.M.; Cadenas, E.; Radi, R.; Laranjinha, J. Neurovascular coupling in hippocampus is mediated via diffusion by neuronal-derived nitric oxide. Free Radic. Biol Med. 2014, 73, 421–429. [Google Scholar] [CrossRef]
- Michetti, M.; Salamino, F.; Melloni, E.; Pontremoli, S. Reversible inactivation of calpain isoforms by nitric oxide. Biochem. Biophys. Res. Commun. 1995, 207, 1009–1014. [Google Scholar] [CrossRef]
- Samengo, G.; Avik, A.; Fedor, B.; Whittaker, D.; Myung, K.H.; Wehling-Henricks, M.; Tidball, J.G. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell 2012, 11, 1036–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Li, Y.; Wang, M.; Zhou, G.; Zhang, W. Effect of protein S-nitrosylation on autolysis and catalytic ability of μ-calpain. Food Chem. 2016, 213, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Katusic, Z.S. Loss of Endothelial Nitric Oxide Synthase Promotes p25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer’s Disease. Circ. Res. 2016, 119, 1128–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.S.; Annamalai, B.; Choi, S.; Singh, I.; Singh, A.K. S-nitrosoglutathione reduces tau hyper-phosphorylation and provides neuroprotection in rat model of chronic cerebral hypoperfusion. Brain Res. 2015, 1624, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukacs, Z.; Jambrecina, A.; Spickler, W.; Schulze, F.; Böger, R.H. Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Waugh, W.H.; Daeschner, C.W., III; Files, B.A.; McConnell, M.E.; Strandjord, S.E. Oral citrulline as arginine precursor may be beneficial in sickle cell disease: Early phase two results. J. Natl. Med. Assoc. 2001, 93, 363–371. [Google Scholar] [PubMed]
- Allerton, T.D.; Proctor, D.N.; Stephens, J.M.; Dugas, T.R.; Spielmann, G.; Irving, B.A. l-Citrulline Supplementation: Impact on Cardiometabolic Health. Nutrients 2018, 10, 921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selley, M.L. Increased concentrations of homocysteine and asymmetric dimethylarginine and decreased concentrations of nitric oxide in the plasma of patients with Alzheimer’s disease. Neurobiol. Aging 2003, 24, 903–907. [Google Scholar] [CrossRef]
- Arlt, S.; Schulze, F.; Eichenlaub, M.; Maas, R.; Lehmbeck, J.T.; Schwedhelm, E.; Jahn, H.; Böger, R.H. Asymmetrical dimethylarginine is increased in plasma and decreased in cerebrospinal fluid of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 26, 58–64. [Google Scholar] [CrossRef]
- Ginguay, A.; Regazzetti, A.; Laprevote, O.; Moinard, C.; De Bandt, J.P.; Cynober, L.; Billard, J.M.; Allinquant, B.; Dutar, P. Citrulline prevents age-related LTP decline in old rats. Sci. Rep. 2019, 9, 20138. [Google Scholar] [CrossRef] [PubMed]
- Haul, S.; Gödecke, A.; Schrader, J.; Haas, H.L.; Luhmann, H.J. Impairment of neocortical long-term potentiation in mice deficient of endothelial nitric oxide synthase. J. Neurophysiol. 1999, 81, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, K.G.; Hardingham, N.R.; Fox, K. Postsynaptic action potentials are required for nitric-oxide-dependent long-term potentiation in CA1 neurons of adult GluR1 knock-out and wild-type mice. J. Neurosci. 2008, 28, 14031–14041. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Gambuzza, M.E.; Sofo, V.; Salmeri, F.M.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disord. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–10107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017, 60, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, G.; Wang, B.; Yin, H.; Fang, X.; He, F.; Zhao, D.; Liu, Q.; Shi, L. IL-1R(-/-) alleviates cognitive deficits through microglial M2 polarization in AD mice. Brain Res. Bull. 2020, 157, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Simãµes, A.P.; Duarte, J.A.; Agasse, F.; Canas, P.M.; Tomé, A.R.; Agostinho, P.; Cunha, R.A. Blockade of adenosine A2A receptors prevents interleukin-1β-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. J. Neuroinflammation 2012, 9, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Sun, L.; Hayashi, Y.; Liu, X.; Koyama, S.; Wu, Z.; Nakanishi, H. Acute p38-mediated inhibition of NMDA-induced outward currents in hippocampal CA1 neurons by interleukin-1beta. Neurobiol. Dis. 2010, 38, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ruan, X.; Zhang, S.; Sun, X. Effect of interleukin-1 beta on the elevation of cytoplasmic free calcium of the cultured hippocampal neurons induced by L-glutamate. J. Tongji Med. Univ. 1999, 19, 120–123. [Google Scholar] [PubMed]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [CrossRef]
- Zhang, R.; Yamada, J.; Hayashi, Y.; Wu, Z.; Koyama, S.; Nakanishi, H. Inhibition of NMDA-induced outward currents by interleukin-1beta in hippocampal neurons. Biochem. Biophys. Res. Commun. 2008, 372, 816–820. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, C.Y.; Fujita, T.; Luo, S.W.; Kumamoto, E. Enhancement by interleukin-1β of AMPA and NMDA receptor-mediated currents in adult rat spinal superficial dorsal horn neurons. Mol. Pain 2013, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136 (Suppl. 1), 10–17. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. HMGB1-Mediated Neuroinflammatory Responses in Brain Injuries: Potential Mechanisms and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 4609. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; Lerner, A. The Second Phase of Brain Trauma Can Be Controlled by Nutraceuticals that Suppress DAMP-Mediated Microglial Activation. Expert Rev. Neurother. 2021. in review process. [Google Scholar]
- Grimm, M.O.W.P.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin D and Its Analogues Decrease Amyloid- β (Aβ) Formation and Increase Aβ-Degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Liu, W.; Zhou, H.Y.; Gui, Y.R.; Yang, Y.H.; Wu, M.J.; Xiao, Y.F.; Shang, J.T.; Long, G.F.; Shu, X.J. Epigallocatechin-3-gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr. Med. Sci. 2020, 40, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Sharman, M.J.; Gyengesi, E.; Liang, H.; Chatterjee, P.; Karl, T.; Li, Q.X.; Wenk, M.R.; Halliwell, B.; Martins, R.N.; Münch, G. Assessment of diets containing curcumin, epigallocatechin-3-gallate, docosahexaenoic acid and α-lipoic acid on amyloid load and inflammation in a male transgenic mouse model of Alzheimer’s disease: Are combinations more effective? Neurobiol. Dis. 2019, 124, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Klakotskaia, D.; Ajit, D.; Weisman, G.A.; Wood, W.G.; Sun, G.Y.; Serfozo, P.; Simonyi, A.; Schachtman, T.R. Beneficial effects of dietary EGCG and voluntary exercise on behavior in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2015, 44, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Han, K.; Kong, J.J.; Zhang, X.M.; Sha, S.; Ren, G.R.; Cao, Y.P. (-)-Epigallocatechin-3-gallate alleviates spatial memory impairment in APP/PS1 mice by restoring IRS-1 signaling defects in the hippocampus. Mol. Cell. Biochem. 2013, 380, 211–218. [Google Scholar] [CrossRef]
- Durairajan, S.S.; Liu, L.F.; Lu, J.H.; Chen, L.L.; Yuan, Q.; Chung, S.K.; Huang, L.; Li, X.S.; Huang, J.D.; Li, M. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 2012, 33, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.N.; Cai, C.Z.; Wu, M.Y.; Su, H.X.; Li, M.; Lu, J.H. Neuroprotective effects of berberine in animal models of Alzheimer’s disease: A systematic review of pre-clinical studies. BMC Complement. Altern. Med. 2019, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.E.; Berg, B.M.; Moore, K.A.; He, B.; Counts, S.E.; Fritz, J.J.; Hu, Y.S.; Lazarov, O.; Lah, J.J.; Mufson, E.J. DHA diet reduces AD pathology in young APPswe/PS1 Delta E9 transgenic mice: Possible gender effects. J. Neurosci. Res. 2010, 88, 1026–1040. [Google Scholar] [PubMed] [Green Version]
- Ren, H.; Luo, C.; Feng, Y.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, H. Omega-3 polyunsaturated fatty acids promote amyloid- β clearance from the brain through mediating the function of the glymphatic system. FASEB J. 2017, 31, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.I.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ -Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: Evidence from meta-analysis. Nutr. J. 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, A.; Teixeira, A.L.; Diniz, B.S. Association of Vitamin D Levels with Incident All-Cause Dementia in Longitudinal Observational Studies: A Systematic Review and Meta-analysis. J. Prev. Alzheimers Dis. 2020, 7, 14–20. [Google Scholar]
- Yang, K.; Chen, J.; Li, X.; Zhou, Y. Vitamin D concentration and risk of Alzheimer disease: A meta-analysis of prospective cohort studies. Medicine 2019, 98, e16804. [Google Scholar] [CrossRef]
- Chai, B.; Gao, F.; Wu, R.; Dong, T.; Gu, C.; Lin, Q.; Zhang, Y. Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: An updated meta-analysis. BMC Neurol. 2019, 19, 284. [Google Scholar] [CrossRef] [PubMed]
- Iseri, L.T.; French, J.H. Magnesium: Nature’s physiologic calcium blocker. Am. Heart J. 1984, 108, 188–193. [Google Scholar] [CrossRef]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 553–569. [Google Scholar]
- Malmendal, A.; Linse, S.; Evenäs, J.; Forsén, S.; Drakenberg, T. Battle for the EF-hands: Magnesium-calcium interference in calmodulin. Biochemistry 1999, 38, 11844–11850. [Google Scholar] [CrossRef]
- Grabarek, Z. Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta 2011, 1813, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, M.; Ninomiya, T.; Ohara, T.; Hirakawa, Y.; Doi, Y.; Hata, J.; Uchida, K.; Shirota, T.; Kitazono, T.; Kiyohara, Y. Self-reported dietary intake of potassium, calcium, and magnesium and risk of dementia in the Japanese: The Hisayama Study. J. Am. Geriatr. Soc. 2012, 60, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Cherbuin, N.; Kumar, R.; Sachdev, P.S.; Anstey, K.J. Dietary Mineral Intake and Risk of Mild Cognitive Impairment: The PATH through Life Project. Front. Aging Neurosci. 2014, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Huang, C.F.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W.; et al. Magnesium oxide use and reduced risk of dementia: A retrospective, nationwide cohort study in Taiwan. Curr. Med. Res. Opin. 2018, 34, 163–169. [Google Scholar] [CrossRef]
- Li, W.; Yu, J.; Liu, Y.; Huang, X.; Abumaria, N.; Zhu, Y.; Huang, X.; Xiong, W.; Ren, C.; Liu, X.G.; et al. Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer’s disease mouse model. Mol. Brain 2014, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Dibaba, D.T.; Xun, P.; He, K. Dietary magnesium intake is inversely associated with serum C-reactive protein levels: Meta-analysis and systematic review. Eur. J. Clin. Nutr. 2014, 68, 510–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simental-Mendia, L.E.; Sahebkar, A.; Rodriguez-Moran, M.; Zambrano-Galvan, G.; Guerrero-Romero, F. Effect of Magnesium Supplementation on Plasma C-reactive Protein Concentrations: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Curr. Pharm. Des. 2017, 23, 4678–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yao, M.; Li, N.; Wang, C.; Zheng, Y.; Cao, X. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 2008, 112, 4961–4970. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ding, X.; Wu, X.; Huang, S. Ketamine inhibits LPS-mediated BV2 microglial inflammation via NMDA receptor blockage. Fundam. Clin. Pharmacol. 2020, 34, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Weinger, J.G.; Lu, Z.L.; Xue, F.; Sadeghpour, S. Efficacy and Safety of MMFS-01, a Synapse Density Enhancer, for Treating Cognitive Impairment in Older Adults: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Alzheimers Dis. 2016, 49, 971–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroolie, T.E.; Chen, K.; Watson, K.T.; Iagaru, A.; Sonni, I.; Snyder, N.; Lee, W.; Reiman, E.M.; Rasgon, N.L. An 8-week open label trial of L-threonic acid magnesium salt in patients with mild to moderate dementia. Pers. Med. Psychiatry 2017, 4–6, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Gao, X.; Na, M.; Kris-Etherton, P.M.; Mitchell, D.C.; Jensen, G.L. Dietary Pattern, Diet Quality, and Dementia: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Alzheimers Dis. 2020, 78, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Guure, C.B.; Ibrahim, N.A.; Adam, M.B.; Said, S.M. Impact of Physical Activity on Cognitive Decline, Dementia, and Its Subtypes: Meta-Analysis of Prospective Studies. Biomed. Res. Int. 2017, 2017, 9016924. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, H.F.; Wan, Y.; Tan, C.C.; Yu, J.T.; Tan, L. Leisure time physical activity and dementia risk: A dose-response meta-analysis of prospective studies. BMJ Open 2017, 7, e014706. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, T.; Kunutsor, S.; Kauhanen, J.; Laukkanen, J.A. Sauna bathing is inversely associated with dementia and Alzheimer’s disease in middle-aged Finnish men. Age Ageing 2017, 46, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Dhana, K.; Evans, D.A.; Rajan, K.B.; Bennett, D.A.; Morris, M.C. Healthy lifestyle and the risk of Alzheimer dementia: Findings from 2 longitudinal studies. Neurology 2020, 95, e374–e383. [Google Scholar] [CrossRef] [PubMed]
- Tari, A.R.; Norevik, C.S.; Scrimgeour, N.R.; Kobro-Flatmoen, A.; Storm-Mathisen, J.; Bergersen, L.H.; Wrann, C.D.; Selbæk, G.; Kivipelto, M.; Moreira, J.B.N.; et al. Are the neuroprotective effects of exercise training systemically mediated? Prog. Cardiovasc. Dis. 2019, 62, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.L.; Castillo- García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Barnes, J.N.; Corkery, A.T. Exercise Improves Vascular Function, but does this Translate to the Brain? Brain Plast. 2018, 4, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, A.P.; Minett, G.M.; Gibson, O.R.; Kerr, G.K.; Stewart, I.B. Could Heat Therapy Be an Effective Treatment for Alzheimer’s and Parkinson’s Diseases? A Narrative Review. Front. Physiol. 2019, 10, 1556. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef]
- Lev-Vachnish, Y.; Cadury, S.; Rotter-Maskowitz, A.; Feldman, N.; Roichman, A.; Illouz, T.; Varvak, A.; Nicola, R.; Madar, R.; Okun, E. L-Lactate Promotes Adult Hippocampal Neurogenesis. Front. Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Barua, S.; Jeong, Y.J.; Lee, J.E. Adiponectin: The Potential Regulator and Therapeutic Target of Obesity and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6419. [Google Scholar] [CrossRef]
- McCarty, M.F. GCN2 and FGF21 are likely mediators of the protection from cancer, autoimmunity, obesity, and diabetes afforded by vegan diets. Med. Hypotheses 2014, 83, 365–371. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. The moderate essential amino acid restriction entailed by low-protein vegan diets may promote vascular health by stimulating FGF21 secretion. Horm. Mol. Biol. Clin. Investig. 2016, 30. [Google Scholar] [CrossRef]
- Castaño-Martinez, T.; Schumacher, F.; Schumacher, S.; Kochlik, B.; Weber, D.; Grune, T.; Biemann, R.; McCann, A.; Abraham, K.; Weikert, C.; et al. Methionine restriction prevents onset of type 2 diabetes in NZO mice. FASEB J. 2019, 33, 7092–7102. [Google Scholar] [CrossRef] [PubMed]
- Giem, P.; Beeson, W.L.; Fraser, G.E. The incidence of dementia and intake of animal products: Preliminary findings from the Adventist Health Study. Neuroepidemiology 1993, 12, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, S.T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.Y.; Yao, W.B.; Gao, X.D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.C.; Chan, K.H. Potential Neuroprotective Effects of Adiponectin in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 9, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrie, H.C.; Ogunniyi, A.; Hall, K.S.; Baiyewu, O.; Unverzagt, F.W.; Gureje, O.; Gao, S.; Evans, R.M.; Ogunseyinde, A.O.; Adeyinka, A.O.; et al. Incidence of dementia and Alzheimer disease in 2 communities: Yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana. JAMA 2001, 285, 739–747. [Google Scholar] [CrossRef]
- Hendrie, H.C.; Murrell, J.; Gao, S.; Unverzagt, F.W.; Ogunniyi, A.; Hall, K.S. International studies in dementia with particular emphasis on populations of African origin. Alzheimer Dis. Assoc. Disord. 2006, 20, S42–S46. [Google Scholar] [CrossRef] [Green Version]
- Parrella, E.; Maxim, T.; Maialetti, F.; Zhang, L.; Wan, J.; Wei, M.; Cohen, P.; Fontana, L.; Longo, V.D. Protein restriction cycles reduce IGF-1 and phosphorylated Tau, and improve behavioral performance in an Alzheimer’s disease mouse model. Aging Cell 2013, 12, 257–268. [Google Scholar] [CrossRef]
- Ruan, Y.; Tang, J.; Guo, X.; Li, K.; Li, D. Dietary Fat Intake and Risk of Alzheimer’s Disease and Dementia: A Meta-Analysis of Cohort Studies. Curr. Alzheimer Res. 2018, 15, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef]
- Desale, S.E.; Chinnathambi, S. Role of dietary fatty acids in microglial polarization in Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF- κB signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Liang, Y.; Zhang, X.; Xu, J.; Lin, J.; Zhang, R.; Kang, K.; Liu, C.; Zhao, C.; Zhao, M. Low-Density Lipoprotein Cholesterol and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2020, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Proitsi, P.; Lupton, M.K.; Velayudhan, L.; Newhouse, S.; Fogh, I.; Tsolaki, M.; Daniilidou, M.; Pritchard, M.; Kloszewska, I.; Soininen, H.; et al. Genetic predisposition to increased blood cholesterol and triglyceride lipid levels and risk of Alzheimer disease: A Mendelian randomization analysis. PLoS Med. 2014, 11, e1001713. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Finan, C.; Schmidt, A.F.; Burgess, S.; Hingorani, A.D. Lipid lowering and Alzheimer disease risk: A mendelian randomization study. Ann. Neurol. 2020, 87, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, H.; Gong, T.; Chen, W.; Mao, S.; Kong, Y.; Yu, J.; Sun, J. Anti-neuroinflammatory Effect of Short-Chain Fatty Acid Acetate against Alzheimer’s Disease via Upregulating GPR41 and Inhibiting ERK/JNK/NF- κB. J. Agric. Food Chem. 2020, 68, 7152–7161. [Google Scholar] [CrossRef]
- Anstey, K.J.; Mack, H.A.; Cherbuin, N. Alcohol consumption as a risk factor for dementia and cognitive decline: Meta-analysis of prospective studies. Am. J. Geriatr. Psychiatry 2009, 17, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, H.; Wan, Y.; Tan, C.; Li, J.; Tan, L.; Yu, J.T. Alcohol consumption and dementia risk: A dose-response meta-analysis of prospective studies. Eur. J. Epidemiol. 2017, 32, 31–42. [Google Scholar] [CrossRef]
- Riss, J.; Décordé, K.; Sutra, T.; Delage, M.; Baccou, J.C.; Jouy, N.; Brune, J.P.; Oréal, H.; Cristol, J.P.; Rouanet, J.M. Phycobiliprotein C-phycocyanin from Spirulina platensis is powerfully responsible for reducing oxidative stress and NADPH oxidase expression induced by an atherogenic diet in hamsters. J. Agric. Food Chem. 2007, 55, 7962–7967. [Google Scholar] [CrossRef]
- McCarty, M.F. NADPH Oxidase Activity in Cerebral Arterioles Is a Key Mediator of Cerebral Small Vessel Disease-Implications for Prevention. Healthcare 2015, 3, 233–251. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F. Supplementation with Phycocyanobilin, Citrulline, Taurine, and Supranutritional Doses of Folic Acid and Biotin-Potential for Preventing or Slowing the Progression of Diabetic Complications. Healthcare 2017, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Lin, L.; Yang, X.; Pan, Q.; Cheng, X. Antidiabetic potential of phycocyanin: Effects on KKAy mice. Pharm. Biol. 2013, 51, 539–544. [Google Scholar] [CrossRef] [PubMed]



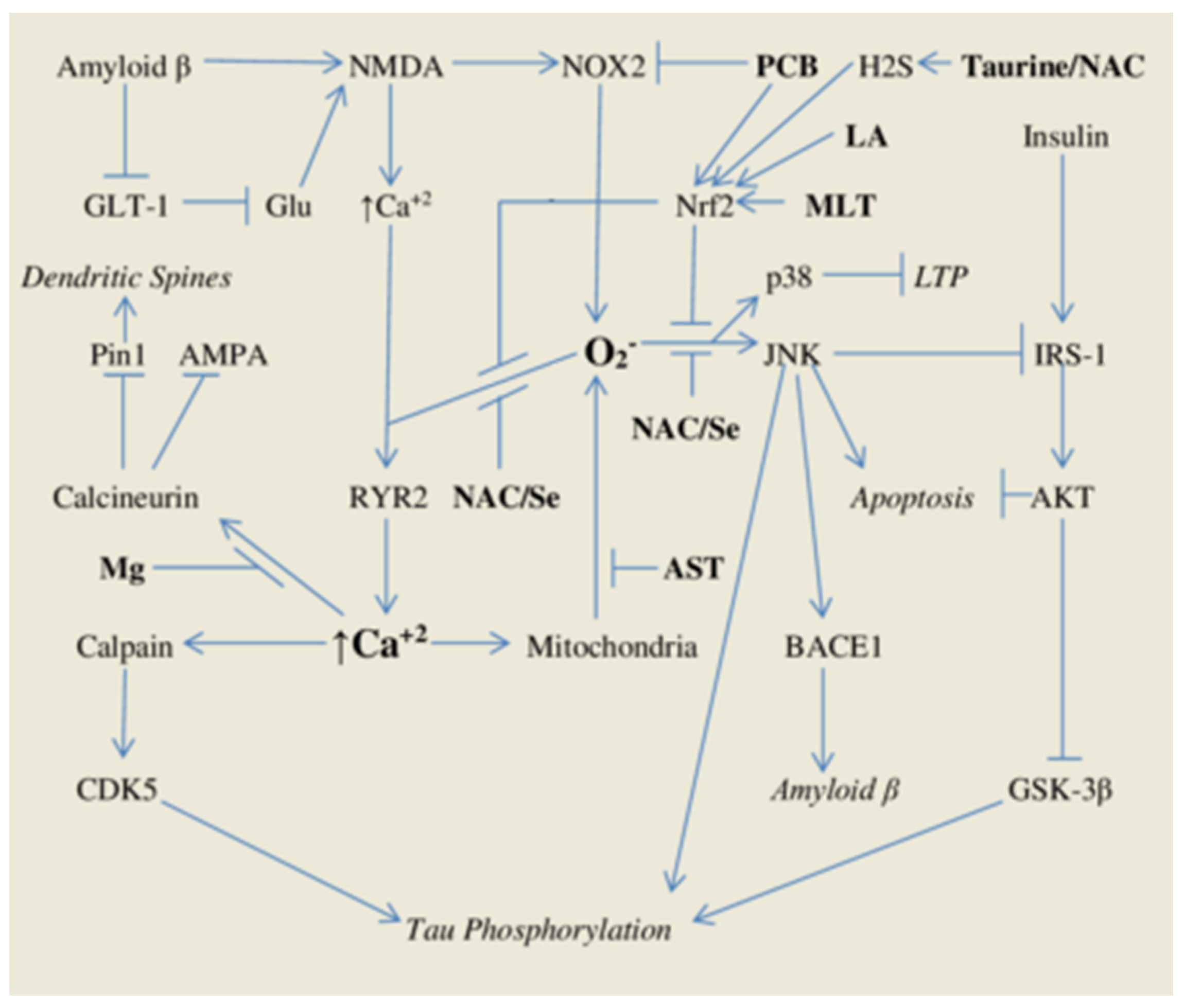{kind=link}
{kind=link}
| Nutraceuticals | Function | Typical Supplemental Dose/Day |
|---|---|---|
| Phycocyanobilin | Algal chromophore antioxidant | 5–15 g spirulina |
| Lipoic Acid | Metabolic cofactor, phase two inducer | 600 mg X 2–3 |
| Melatonin | Neurohormone, phase two inducer | 3–20 mg at bedtime |
| Taurine | Antioxidant/osmoregulatory cofactor | 1–2 g X 2 |
| N-Acetylcysteine | Supplemental source of L-cysteine | 600 mg X 2–3 |
| Selenium | Essential mineral | 50–100 mcg |
| Astaxanthin | Natural carotenoid antioxidant | 12–20 mg |
| Biotin | B vitamin, activator of guanylate cyclase | 10–30 mg |
| Magnesium | Essential mineral | 100–400 mg |
| Citrulline | Precursor/delivery form for arginine | 2 g X 2 |
| DHA | Long-chain omega-3 fatty acid | 100–1000 mg |
| Vitamin D | Vitamin with anti-inflammatory activity | 1000–5000 IU |
| Berberine | Phytochemical which activates AMPK | 500 mg X 2–3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarty, M.F.; DiNicolantonio, J.J.; Lerner, A. A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis—And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder. Int. J. Mol. Sci. 2021, 22, 2140. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042140
McCarty MF, DiNicolantonio JJ, Lerner A. A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis—And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder. International Journal of Molecular Sciences. 2021; 22(4):2140. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042140
Chicago/Turabian StyleMcCarty, Mark F., James J. DiNicolantonio, and Aaron Lerner. 2021. "A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis—And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder" International Journal of Molecular Sciences 22, no. 4: 2140. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042140